
The Class I HDAC Inhibitor Valproic Acid Strongly Potentiates Gemcitabine Efficacy in Pancreatic Cancer by Immune System Activation

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Compounds
2.2. Cell Proliferation Assay
2.3. Colony-Forming Assay
2.4. Real-Time Quantitative PCR
2.5. Mice
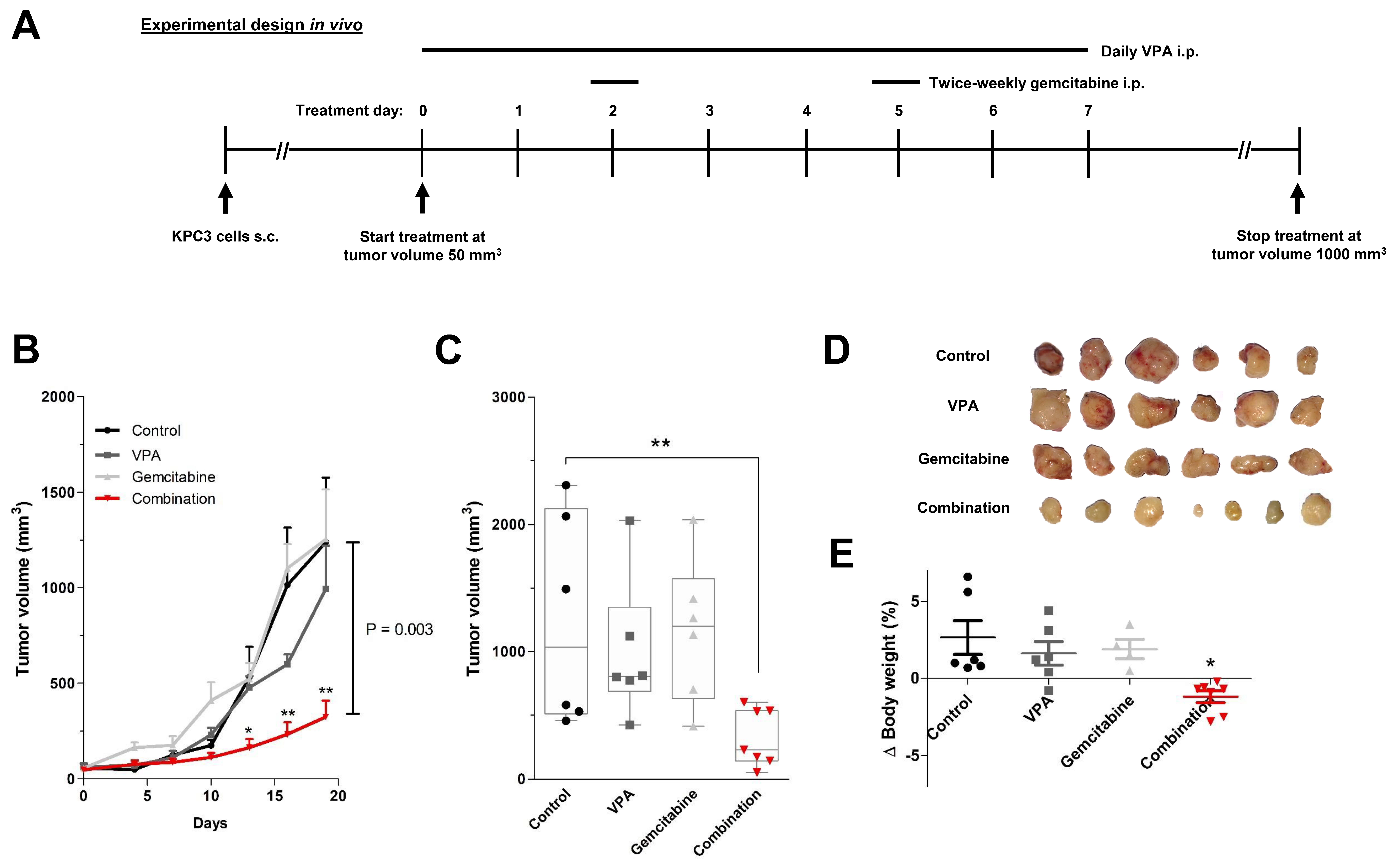
2.6. In Vivo Experiments
2.7. Necropsy Procedures
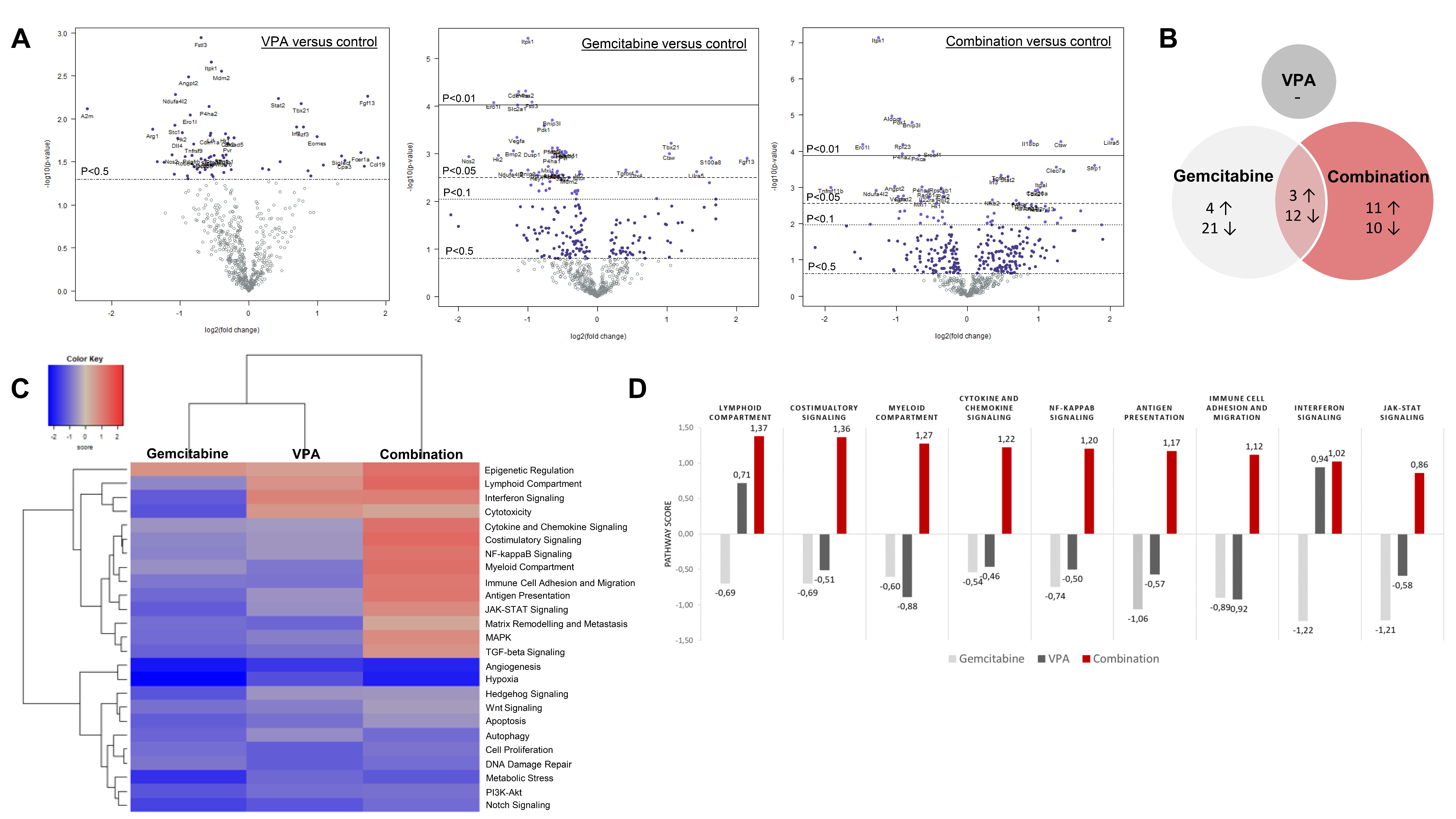
2.8. NanoString Analysis
2.9. Statistical Analysis
3. Results
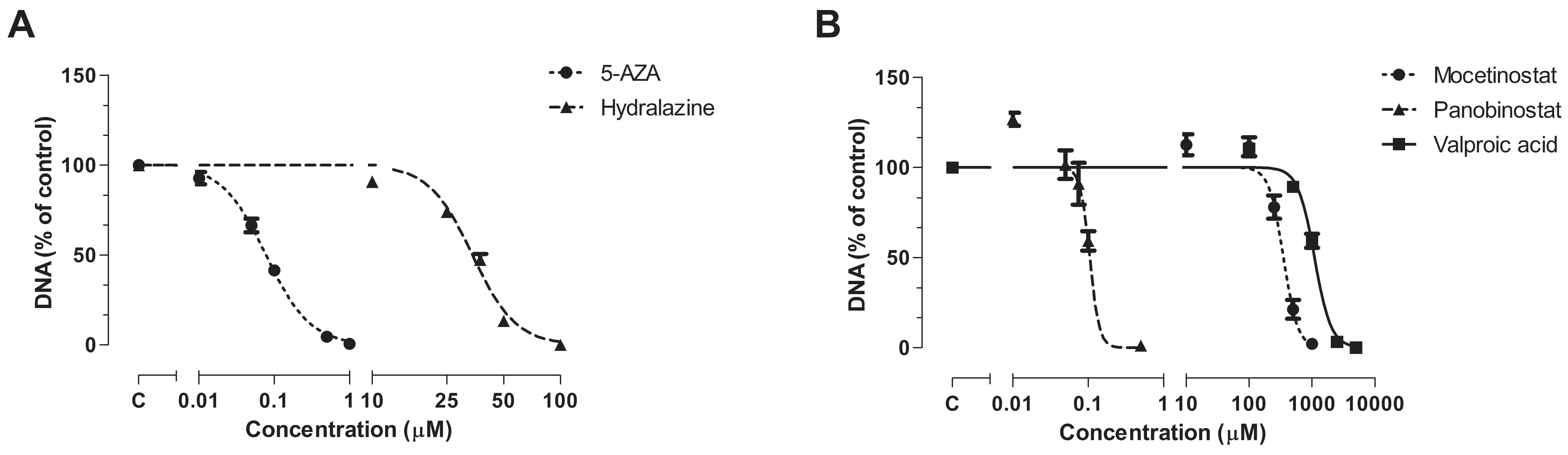
3.1. Effect of Epi-Drug Monotherapy In Vitro
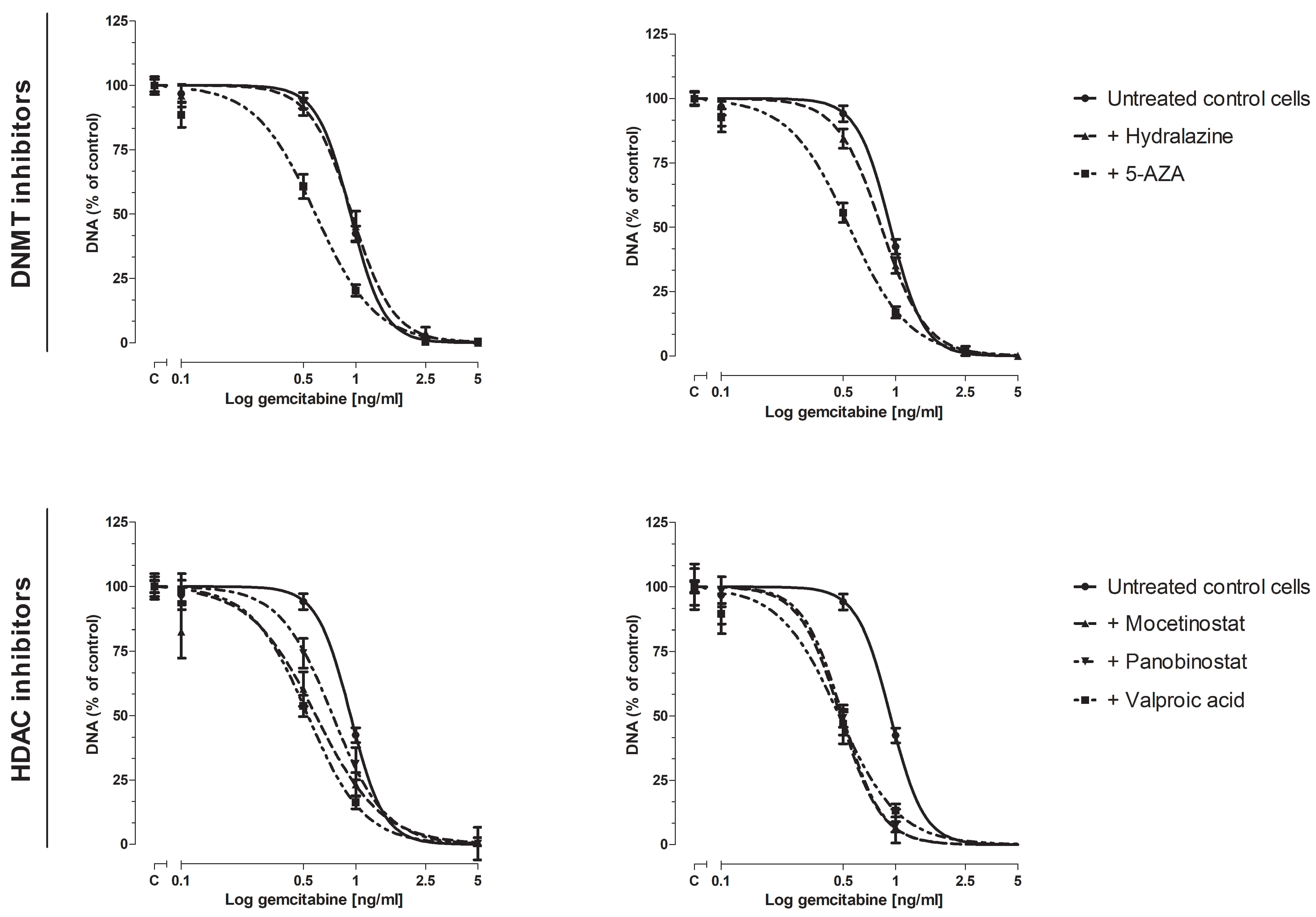
3.2. Effect of Epi-Drugs on Gemcitabine Sensitivity In Vitro
3.2.1. Cell Proliferation Assay
3.2.2. Colony-Forming Assay
3.3. Effect of epi-Drugs on the Expression of Genes Involving Gemcitabine Uptake and Metabolism
3.4. In Vivo Validation of VPA Combined with Gemcitabine in Immune-Competent Mice
3.5. PanCancer IO 360 Gene Expression Panel
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Gnerlich, J.L.; Luka, S.R.; Deshpande, A.D.; Dubray, B.J.; Weir, J.S.; Carpenter, D.H.; Brunt, E.M.; Strasberg, S.M.; Hawkins, W.G.; Linehan, D.C. Microscopic margins and patterns of treatment failure in resected pancreatic adenocarcinoma. Arch. Surg. 2012, 147, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Hishinuma, S.; Ogata, Y.; Tomikawa, M.; Ozawa, I.; Hirabayashi, K.; Igarashi, S. Patterns of recurrence after curative resection of pancreatic cancer, based on autopsy findings. J. Gastrointest Surg. 2006, 10, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- de Sousa Cavalcante, L.; Monteiro, G. Gemcitabine: Metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur. J. Pharmacol. 2014, 741, 8–16. [Google Scholar] [CrossRef]
- Marechal, R.; Mackey, J.R.; Lai, R.; Demetter, P.; Peeters, M.; Polus, M.; Cass, C.E.; Young, J.; Salmon, I.; Deviere, J.; et al. Human equilibrative nucleoside transporter 1 and human concentrative nucleoside transporter 3 predict survival after adjuvant gemcitabine therapy in resected pancreatic adenocarcinoma. Clin. Cancer Res. 2009, 15, 2913–2919. [Google Scholar] [CrossRef]
- Nakano, Y.; Tanno, S.; Koizumi, K.; Nishikawa, T.; Nakamura, K.; Minoguchi, M.; Izawa, T.; Mizukami, Y.; Okumura, T.; Kohgo, Y. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br. J. Cancer 2007, 96, 457–463. [Google Scholar] [CrossRef]
- Ashida, R.; Nakata, B.; Shigekawa, M.; Mizuno, N.; Sawaki, A.; Hirakawa, K.; Arakawa, T.; Yamao, K. Gemcitabine sensitivity-related mRNA expression in endoscopic ultrasound-guided fine-needle aspiration biopsy of unresectable pancreatic cancer. J. Exp. Clin. Cancer Res. 2009, 28, 83. [Google Scholar] [CrossRef]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef]
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; Silva Santos, R.D.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- McCleary-Wheeler, A.L.; Lomberk, G.A.; Weiss, F.U.; Schneider, G.; Fabbri, M.; Poshusta, T.L.; Dusetti, N.J.; Baumgart, S.; Iovanna, J.L.; Ellenrieder, V.; et al. Insights into the epigenetic mechanisms controlling pancreatic carcinogenesis. Cancer Lett. 2013, 328, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Omura, N.; Hong, S.M.; Jaffe, A.; Eshleman, J.; Goggins, M. Genome-wide analysis of promoter methylation associated with gene expression profile in pancreatic adenocarcinoma. Clin. Cancer Res. 2011, 17, 4341–4354. [Google Scholar] [CrossRef] [PubMed]
- Ouaïssi, M.; Sielezneff, I.; Silvestre, R.; Sastre, B.; Bernard, J.P.; Lafontaine, J.S.; Payan, M.J.; Dahan, L.; Pirrò, N.; Seitz, J.F.; et al. High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Ann. Surg. Oncol. 2008, 15, 2318–2328. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, P.; Seidler, B.; Schüler, S.; Schnieke, A.; Göttlicher, M.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009, 58, 1399–1409. [Google Scholar] [CrossRef]
- Ouaïssi, M.; Cabral, S.; Tavares, J.; da Silva, A.C.; Mathieu Daude, F.; Mas, E.; Bernard, J.; Sastre, B.; Lombardo, D.; Ouaissi, A. Histone deacetylase (HDAC) encoding gene expression in pancreatic cancer cell lines and cell sensitivity to HDAC inhibitors. Cancer Biol. Ther. 2008, 7, 523–531. [Google Scholar] [CrossRef] [PubMed]
- van Koetsveld, P.M.; Vitale, G.; de Herder, W.W.; Feelders, R.A.; van der Wansem, K.; Waaijers, M.; van Eijck, C.H.; Speel, E.J.; Croze, E.; van der Lely, A.J.; et al. Potent inhibitory effects of type I interferons on human adrenocortical carcinoma cell growth. J. Clin. Endocrinol. Metab. 2006, 91, 4537–4543. [Google Scholar] [CrossRef][Green Version]
- Herrera-Martinez, A.D.; Feelders, R.A.; de Herder, W.W.; Castano, J.P.; Galvez Moreno, M.A.; Dogan, F.; van Dungen, R.; van Koetsveld, P.; Hofland, L.J. Effects of Ketoconazole on ACTH-Producing and Non-ACTH-Producing Neuroendocrine Tumor Cells. Horm. Cancer 2019, 10, 107–119. [Google Scholar] [CrossRef]
- Blaauboer, A.; Booy, S.; van Koetsveld, P.M.; Karels, B.; Dogan, F.; van Zwienen, S.; van Eijck, C.H.J.; Hofland, L.J. Interferon-beta enhances sensitivity to gemcitabine in pancreatic cancer. BMC Cancer 2020, 20, 913. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, B.; Cai, D.; Zou, X. Therapeutic potential of histone deacetylase inhibitors in pancreatic cancer. Cancer Lett. 2014, 347, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Donadelli, M.; Costanzo, C.; Faggioli, L.; Scupoli, M.T.; Moore, P.S.; Bassi, C.; Scarpa, A.; Palmieri, M.; Trichostatin, A. An inhibitor of histone deacetylases, strongly suppresses growth of pancreatic adenocarcinoma cells. Mol. Carcinog. 2003, 38, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, T.; Wakimoto, N.; Yin, D.; Gery, S.; Kawamata, N.; Takai, N.; Komatsu, N.; Chumakov, A.; Imai, Y.; Koeffler, H.P. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int. J. Cancer 2007, 121, 656–665. [Google Scholar] [CrossRef]
- Kaminskas, E.; Farrell, A.T.; Wang, Y.C.; Sridhara, R.; Pazdur, R. FDA drug approval summary: Azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist 2005, 10, 176–182. [Google Scholar] [CrossRef]
- Agrawal, K.; Das, V.; Vyas, P.; Hajdúch, M. Nucleosidic DNA demethylating epigenetic drugs—A comprehensive review from discovery to clinic. Pharm. Ther. 2018, 188, 45–79. [Google Scholar] [CrossRef]
- Pan, C.H.; Chang, Y.F.; Lee, M.S.; Wen, B.C.; Ko, J.C.; Liang, S.K.; Liang, M.C. Vorinostat enhances the cisplatin-mediated anticancer effects in small cell lung cancer cells. BMC Cancer 2016, 16, 857. [Google Scholar] [CrossRef]
- Xia, C.; Leon-Ferre, R.; Laux, D.; Deutsch, J.; Smith, B.J.; Frees, M.; Milhem, M. Treatment of resistant metastatic melanoma using sequential epigenetic therapy (decitabine and panobinostat) combined with chemotherapy (temozolomide). Cancer Chemother. Pharmacol. 2014, 74, 691–697. [Google Scholar] [CrossRef]
- Voso, M.T.; Santini, V.; Finelli, C.; Musto, P.; Pogliani, E.; Angelucci, E.; Fioritoni, G.; Alimena, G.; Maurillo, L.; Cortelezzi, A.; et al. Valproic acid at therapeutic plasma levels may increase 5-azacytidine efficacy in higher risk myelodysplastic syndromes. Clin. Cancer Res. 2009, 15, 5002–5007. [Google Scholar] [CrossRef]
- Duenas-Gonzalez, A.; Candelaria, M.; Perez-Plascencia, C.; Perez-Cardenas, E.; de la Cruz-Hernandez, E.; Herrera, L.A. Valproic acid as epigenetic cancer drug: Preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat. Rev. 2008, 34, 206–222. [Google Scholar] [CrossRef]
- Krämer, O.H.; Zhu, P.; Ostendorff, H.P.; Golebiewski, M.; Tiefenbach, J.; Peters, M.A.; Brill, B.; Groner, B.; Bach, I.; Heinzel, T.; et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003, 22, 3411–3420. [Google Scholar] [CrossRef]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [PubMed]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.H.; Xu, X.G.; Yan, S.L.; Sun, Z.; Ying, Y.; Wang, B.K.; Tu, Y.X. Depletion of HDAC1, 7 and 8 by Histone Deacetylase Inhibition Confers Elimination of Pancreatic Cancer Stem Cells in Combination with Gemcitabine. Sci. Rep. 2018, 8, 1621. [Google Scholar] [CrossRef]
- Wang, G.; He, J.; Zhao, J.; Yun, W.; Xie, C.; Taub, J.W.; Azmi, A.; Mohammad, R.M.; Dong, Y.; Kong, W.; et al. Class I and class II histone deacetylases are potential therapeutic targets for treating pancreatic cancer. PLoS ONE 2012, 7, e52095. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gao, J.; Man, X.H.; Li, Z.S.; Gong, Y.F. Significance of DNA methyltransferase-1 and histone deacetylase-1 in pancreatic cancer. Oncol. Rep. 2009, 21, 1439–1447. [Google Scholar]
- Schüler, S.; Fritsche, P.; Diersch, S.; Arlt, A.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 attenuates TRAIL-induced apoptosis of pancreatic cancer cells. Mol. Cancer 2010, 9, 80. [Google Scholar] [CrossRef]
- Miyake, K.; Yoshizumi, T.; Imura, S.; Sugimoto, K.; Batmunkh, E.; Kanemura, H.; Morine, Y.; Shimada, M. Expression of hypoxia-inducible factor-1alpha, histone deacetylase 1, and metastasis-associated protein 1 in pancreatic carcinoma: Correlation with poor prognosis with possible regulation. Pancreas 2008, 36, e1–e9. [Google Scholar] [CrossRef]
- Ohhashi, S.; Ohuchida, K.; Mizumoto, K.; Fujita, H.; Egami, T.; Yu, J.; Toma, H.; Sadatomi, S.; Nagai, E.; Tanaka, M. Down-regulation of deoxycytidine kinase enhances acquired resistance to gemcitabine in pancreatic cancer. Anticancer Res. 2008, 28, 2205–2212. [Google Scholar] [PubMed]
- Avramis, V.I.; Mecum, R.A.; Nyce, J.; Steele, D.A.; Holcenberg, J.S. Pharmacodynamic and DNA methylation studies of high-dose 1-beta-D-arabinofuranosyl cytosine before and after in vivo 5-azacytidine treatment in pediatric patients with refractory acute lymphocytic leukemia. Cancer Chemother. Pharm. 1989, 24, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.B.; Tong, W.P.; Chou, T.C. Induction of deoxycytidine kinase by 5-azacytidine in an HL-60 cell line resistant to arabinosylcytosine. Mol. Pharmacol. 1991, 39, 250–257. [Google Scholar] [PubMed]
- Peters, G.J.; Hodzic, J.; Ortega, B.; Giovannetti, E.; Adema, A.D.; Broekhuizen, R.; Kaspers, G.J.; Hubeek, I. Methylation specific PCR to characterize methylation of the promoter of deoxycytidine kinase. Nucleosides Nucleotides Nucleic Acids 2010, 29, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.C.; Yoo, C.B.; Kwan, J.M.; Li, T.W.; Liang, G.; Yang, A.S.; Jones, P.A. Comparison of biological effects of non-nucleoside DNA methylation inhibitors versus 5-aza-2’-deoxycytidine. Mol. Cancer Ther. 2005, 4, 1515–1520. [Google Scholar] [CrossRef]
- Candelaria, M.; de la Cruz-Hernandez, E.; Taja-Chayeb, L.; Perez-Cardenas, E.; Trejo-Becerril, C.; Gonzalez-Fierro, A.; Chavez-Blanco, A.; Soto-Reyes, E.; Dominguez, G.; Trujillo, J.E.; et al. DNA methylation-independent reversion of gemcitabine resistance by hydralazine in cervical cancer cells. PLoS ONE 2012, 7, e29181. [Google Scholar] [CrossRef]
- Damaraju, V.L.; Mowles, D.; Yao, S.; Ng, A.; Young, J.D.; Cass, C.E.; Tong, Z. Role of human nucleoside transporters in the uptake and cytotoxicity of azacitidine and decitabine. Nucleosides Nucleotides Nucleic Acids 2012, 31, 236–255. [Google Scholar] [CrossRef]
- Rius, M.; Stresemann, C.; Keller, D.; Brom, M.; Schirrmacher, E.; Keppler, D.; Lyko, F. Human concentrative nucleoside transporter 1-mediated uptake of 5-azacytidine enhances DNA demethylation. Mol. Cancer Ther. 2009, 8, 225–231. [Google Scholar] [CrossRef]
- Činčárová, L.; Zdráhal, Z.; Fajkus, J. New perspectives of valproic acid in clinical practice. Expert Opin. Investig. Drugs 2013, 22, 1535–1547. [Google Scholar] [CrossRef]
- Wang, Y.; Kuramitsu, Y.; Kitagawa, T.; Tokuda, K.; Baron, B.; Akada, J.; Nakamura, K. The Histone Deacetylase Inhibitor Valproic Acid Sensitizes Gemcitabine-Induced Cytotoxicity in Gemcitabine-Resistant Pancreatic Cancer Cells Possibly Through Inhibition of the DNA Repair Protein Gamma-H2AX. Target Oncol. 2015, 10, 575–581. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Z.; Gao, C.; Wu, S.; Duan, Q.; Wu, H.; Wang, C.; Shen, Q.; Yin, T. Combination chemotherapy of valproic acid (VPA) and gemcitabine regulates STAT3/Bmi1 pathway to differentially potentiate the motility of pancreatic cancer cells. Cell Biosci. 2019, 9, 50. [Google Scholar] [CrossRef]
- Lee, J.W.; Komar, C.A.; Bengsch, F.; Graham, K.; Beatty, G.L. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras(G12D/+);LSL-Trp53(R172H/+);Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr. Protoc. Pharmacol. 2016, 73, 14–39. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.W.; Morton, J.P.; Pinese, M.; Saturno, G.; Jamieson, N.B.; McGhee, E.; Timpson, P.; Leach, J.; McGarry, L.; Shanks, E.; et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: Inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 2015, 7, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.; Frese, K.K.; Bapiro, T.E.; Jacobetz, M.A.; Gopinathan, A.; Miller, J.L.; Rao, S.S.; Demuth, T.; Howat, W.J.; Jodrell, D.I.; et al. Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J. Exp. Med. 2012, 209, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.V.; Parikh, P.; von Pawel, J.; Biesma, B.; Vansteenkiste, J.; Manegold, C.; Serwatowski, P.; Gatzemeier, U.; Digumarti, R.; Zukin, M.; et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 2008, 26, 3543–3551. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ren, Q.; Zuo, W.; Jia, R.; Xie, L.; Lin, R.; Zhao, H.; Chen, J.; Lei, Y.; Wang, P.; et al. Valproic acid exhibits anti-tumor activity selectively against EGFR/ErbB2/ErbB3-coexpressing pancreatic cancer via induction of ErbB family members-targeting microRNAs. J. Exp. Clin. Cancer Res. 2019, 38, 150. [Google Scholar] [CrossRef]
- Shi, P.; Yin, T.; Zhou, F.; Cui, P.; Gou, S.; Wang, C. Valproic acid sensitizes pancreatic cancer cells to natural killer cell-mediated lysis by upregulating MICA and MICB via the PI3K/Akt signaling pathway. BMC Cancer 2014, 14, 370. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Hao, C.L.; Liu, P.; Tian, X.; Wang, L.H.; Zhao, L.; Zhu, C.M. Valproic acid inhibits tumor angiogenesis in mice transplanted with Kasumi-1 leukemia cells. Mol. Med. Rep. 2014, 9, 443–449. [Google Scholar] [CrossRef][Green Version]
- Blaauboer, A.; Sideras, K.; van Eijck, C.H.J.; Hofland, L.J. Type I interferons in pancreatic cancer and development of new therapeutic approaches. Crit. Rev. Oncol. Hematol. 2021, 159, 103204. [Google Scholar] [CrossRef]
- Miyashita, T.; Miki, K.; Kamigaki, T.; Makino, I.; Tajima, H.; Nakanuma, S.; Hayashi, H.; Takamura, H.; Fushida, S.; Ahmed, A.K.; et al. Low-dose valproic acid with low-dose gemcitabine augments MHC class I-related chain A/B expression without inducing the release of soluble MHC class I-related chain A/B. Oncol. Lett. 2017, 14, 5918–5926. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi, S.; Utsunomiya, T.; Imura, S.; Morine, Y.; Ikemoto, T.; Arakawa, Y.; Saito, Y.; Ishikawa, D.; Shimada, M. Effects of valproic acid in combination with S-1 on advanced pancreatobiliary tract cancers: Clinical study phases I/II. Anticancer Res. 2014, 34, 5187–5191. [Google Scholar] [PubMed]
- Ueno, H.; Ioka, T.; Ikeda, M.; Ohkawa, S.; Yanagimoto, H.; Boku, N.; Fukutomi, A.; Sugimori, K.; Baba, H.; Yamao, K.; et al. Randomized phase III study of gemcitabine plus S-1, S-1 alone, or gemcitabine alone in patients with locally advanced and metastatic pancreatic cancer in Japan and Taiwan: GEST study. J. Clin. Oncol. 2013, 31, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Furuse, J.; Okusaka, T.; Boku, N.; Ohkawa, S.; Sawaki, A.; Masumoto, T.; Funakoshi, A. S-1 monotherapy as first-line treatment in patients with advanced biliary tract cancer: A multicenter phase II study. Cancer Chemother. Pharmacol. 2008, 62, 849–855. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC25 Epi-Drug Treated Cells | IC50 Epi-Drug Treated Cells | |
|---|---|---|
| Untreated control cells | 0.9341 (0.8967–0.9731) | |
| + 5-AZA | 0.5882 (0.5418–0.6385) *** | 0.5426 (0.4986–0.5904) *** |
| + Hydralazine | 0.9513 (0.8752–1.0340) | 0.8298 (0.7721–0.8919) |
| + Mocetinostat | 0.5910 (0.5052–0.6913) *** | 0.4834 (0.4173–0.559) *** |
| + Panobinostat | 0.7390 (0.6570–0.8313) *** | 0.4972 (0.4254–0.5811) *** |
| + VPA | 0.5320 (0.4971–0.5695) *** | 0.4758 (0.4355–0.5198) *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blaauboer, A.; van Koetsveld, P.M.; Mustafa, D.A.M.; Dumas, J.; Dogan, F.; van Zwienen, S.; van Eijck, C.H.J.; Hofland, L.J. The Class I HDAC Inhibitor Valproic Acid Strongly Potentiates Gemcitabine Efficacy in Pancreatic Cancer by Immune System Activation. Biomedicines 2022, 10, 517. https://doi.org/10.3390/biomedicines10030517
Blaauboer A, van Koetsveld PM, Mustafa DAM, Dumas J, Dogan F, van Zwienen S, van Eijck CHJ, Hofland LJ. The Class I HDAC Inhibitor Valproic Acid Strongly Potentiates Gemcitabine Efficacy in Pancreatic Cancer by Immune System Activation. Biomedicines. 2022; 10(3):517. https://doi.org/10.3390/biomedicines10030517
Chicago/Turabian StyleBlaauboer, Amber, Peter M. van Koetsveld, Dana A. M. Mustafa, Jasper Dumas, Fadime Dogan, Suzanne van Zwienen, Casper H. J. van Eijck, and Leo J. Hofland. 2022. "The Class I HDAC Inhibitor Valproic Acid Strongly Potentiates Gemcitabine Efficacy in Pancreatic Cancer by Immune System Activation" Biomedicines 10, no. 3: 517. https://doi.org/10.3390/biomedicines10030517
APA StyleBlaauboer, A., van Koetsveld, P. M., Mustafa, D. A. M., Dumas, J., Dogan, F., van Zwienen, S., van Eijck, C. H. J., & Hofland, L. J. (2022). The Class I HDAC Inhibitor Valproic Acid Strongly Potentiates Gemcitabine Efficacy in Pancreatic Cancer by Immune System Activation. Biomedicines, 10(3), 517. https://doi.org/10.3390/biomedicines10030517