
In Situ Gene Expression in Native Cryofixed Bone Tissue

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Rat Animal Model
2.2. Slides Coating
2.3. Embedding and Cryosectioning of Entire Femur of the Rat
2.4. Histological Staining
2.5. Immunofluorescence
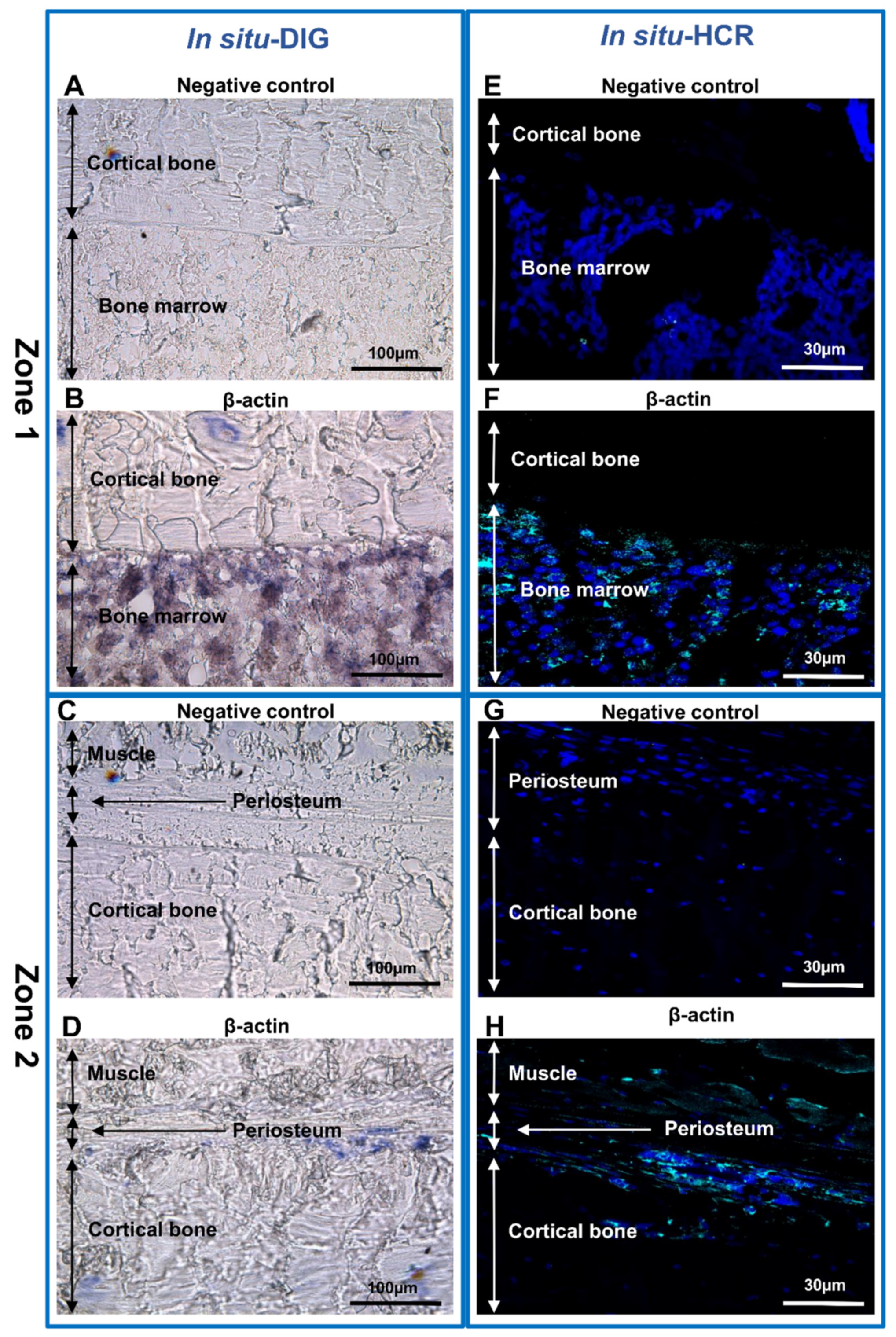
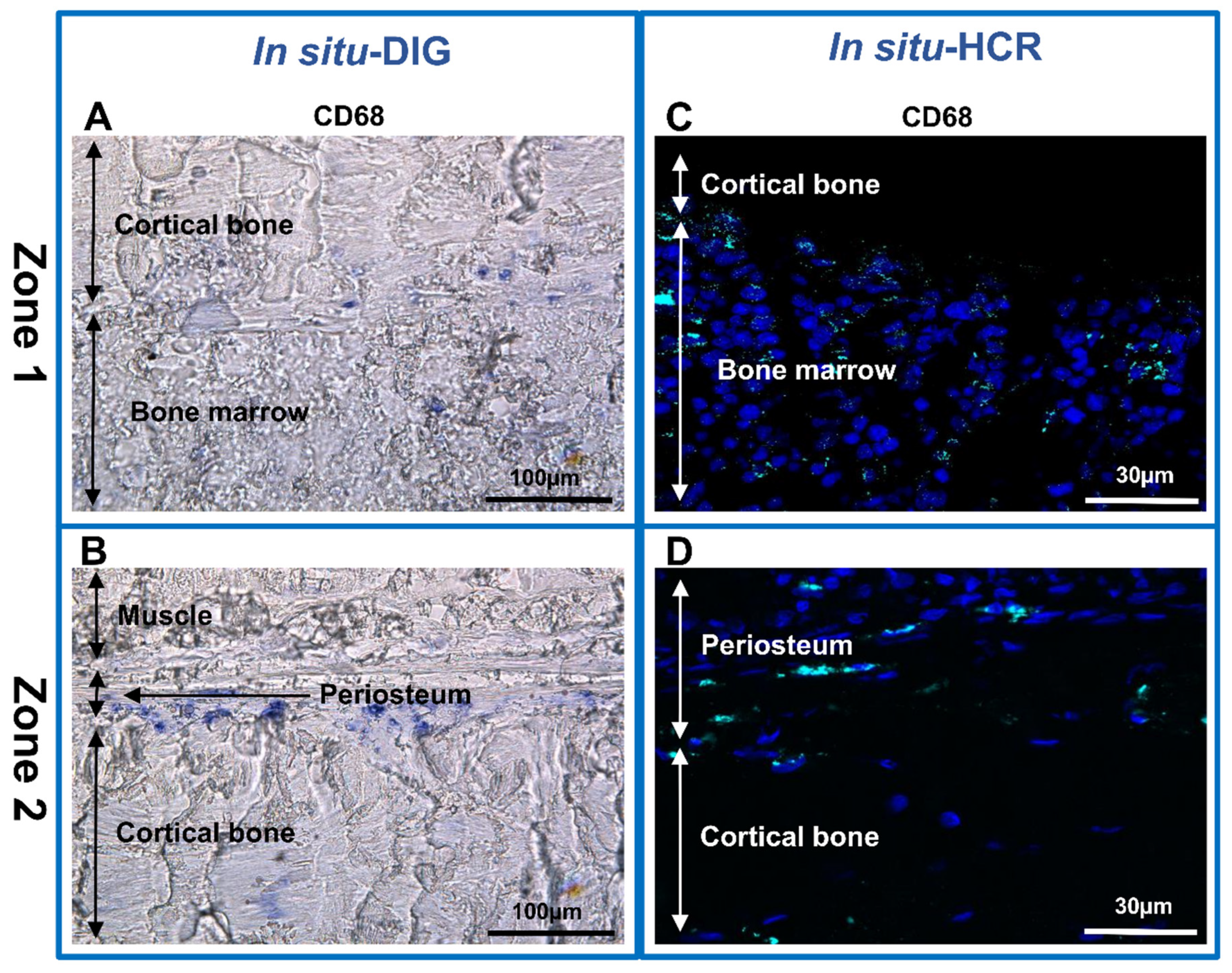
2.6. In Situ-DIG Hybridization
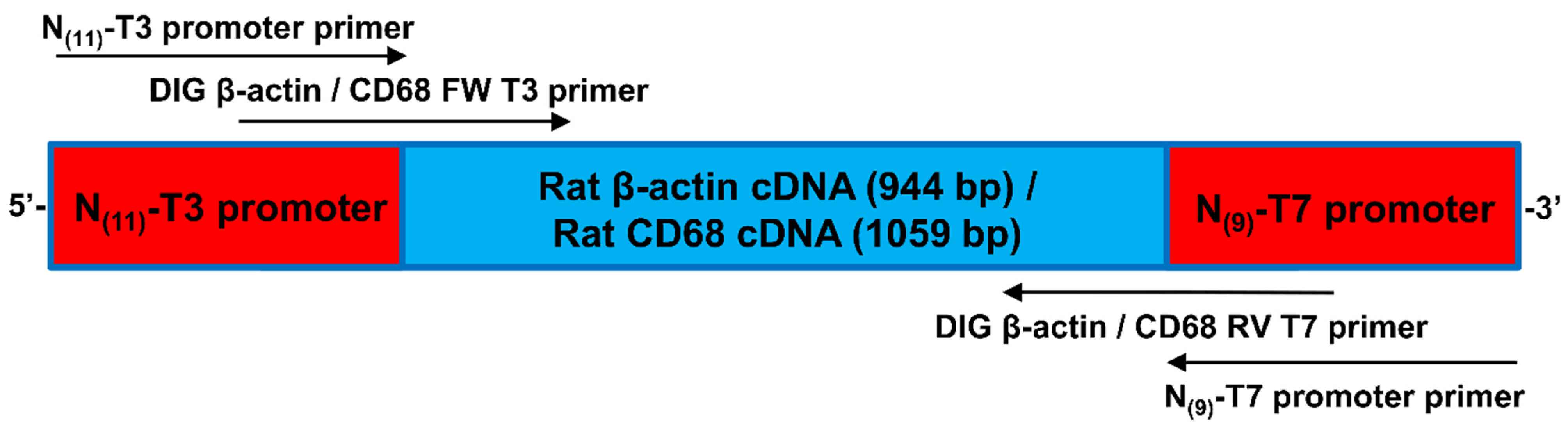
2.6.1. cRNA Probes for In Situ-DIG Hybridization
2.6.2. Fixation and Pretreatment of Sections for In Situ-DIG Hybridization
2.6.3. Prehybridization and Hybridization for In Situ-DIG
2.6.4. Detection for In Situ-DIG
2.7. In Situ-HCR Hybridization
2.7.1. β-Actin Probe Design for In Situ-HCR Hybridization
2.7.2. Fixation and Pretreatment of Sections for In Situ-HCR Hybridization
2.7.3. Prehybridization and Hybridization for In Situ-HCR
2.7.4. Detection for In Situ-HCR
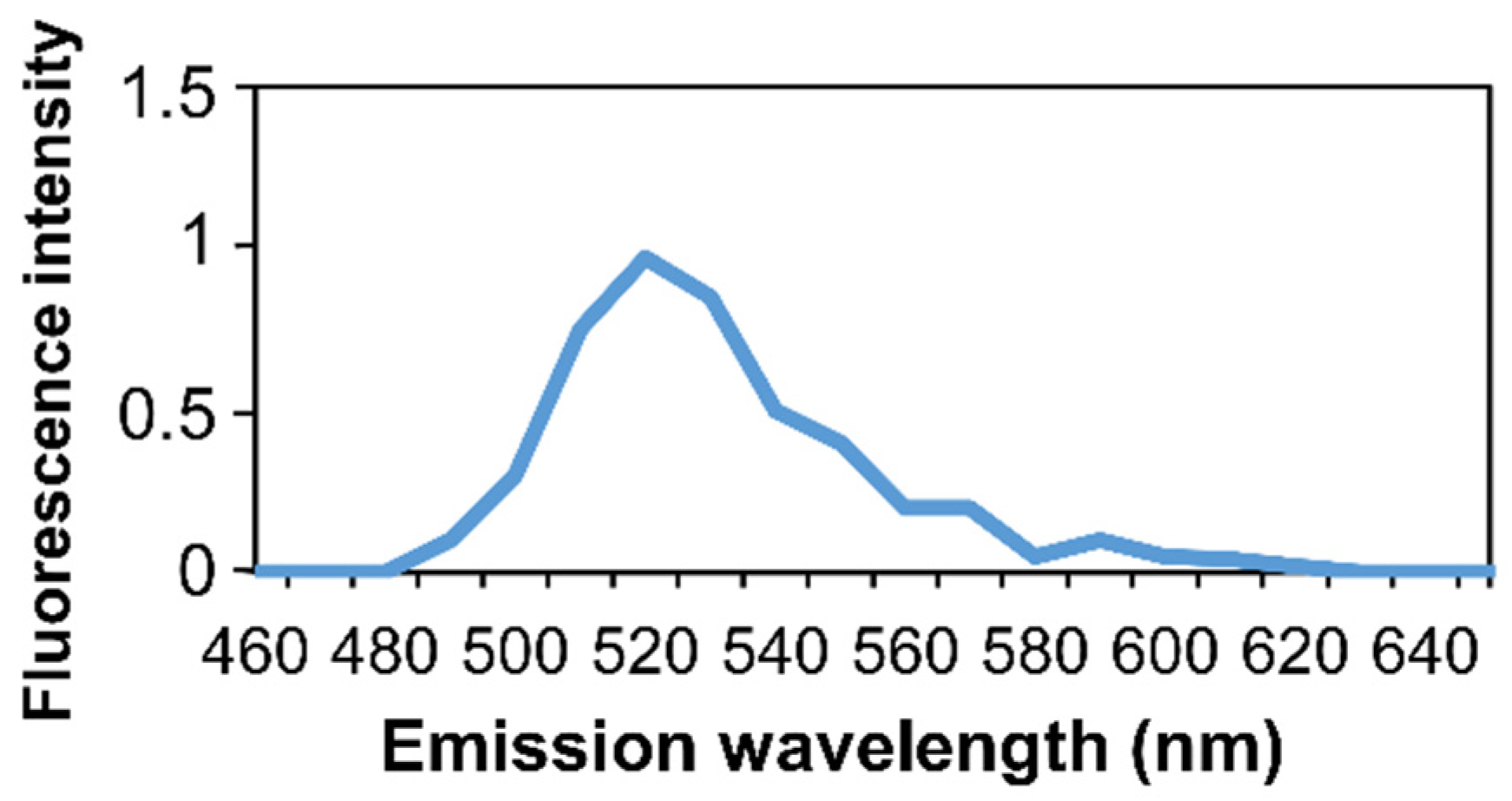
2.8. Microspectrofluorimetry
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Kaur, S.; Raggatt, L.J.; Batoon, L.; Hume, D.A.; Levesque, J.-P.; Pettit, A.R. Role of Bone Marrow Macrophages in Controlling Homeostasis and Repair in Bone and Bone Marrow Niches. Semin. Cell Dev. Biol. 2017, 61, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwood, N.J. Macrophage Polarization and Bone Formation: A Review. Clin. Rev. Allergy Immunol. 2016, 51, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Tang, Q.; Zhong, Y.; Wei, Y.; He, L.; Wu, Y.; Wu, J.; Liao, J. Biomaterial-Based Strategies for Maxillofacial Tumour Therapy and Bone Defect Regeneration. Int. J. Oral Sci. 2021, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.; Akhavan, N.S.; Mullins, A.P.; Arjmandi, B.H. Macrophage Polarization and Osteoporosis: A Review. Nutrients 2020, 12, 2999. [Google Scholar] [CrossRef]
- Weitzmann, M.N. Bone and the Immune System. Toxicol. Pathol. 2017, 45, 911–924. [Google Scholar] [CrossRef]
- Batoon, L.; Millard, S.M.; Raggatt, L.J.; Pettit, A.R. Osteomacs and Bone Regeneration. Curr. Osteoporos. Rep. 2017, 15, 385–395. [Google Scholar] [CrossRef]
- Lebaudy, E.; Fournel, S.; Lavalle, P.; Vrana, N.E.; Gribova, V. Recent Advances in Antiinflammatory Material Design. Adv. Healthc. Mater. 2021, 10, e2001373. [Google Scholar] [CrossRef]
- Eggold, J.T.; Rankin, E.B. Erythropoiesis, EPO, Macrophages, and Bone. Bone 2019, 119, 36–41. [Google Scholar] [CrossRef]
- Pajarinen, J.; Lin, T.; Gibon, E.; Kohno, Y.; Maruyama, M.; Nathan, K.; Lu, L.; Yao, Z.; Goodman, S.B. Mesenchymal Stem Cell-Macrophage Crosstalk and Bone Healing. Biomaterials 2019, 196, 80–89. [Google Scholar] [CrossRef]
- Sun, Y.; Li, J.; Xie, X.; Gu, F.; Sui, Z.; Zhang, K.; Yu, T. Macrophage-Osteoclast Associations: Origin, Polarization, and Subgroups. Front. Immunol. 2021, 12, 778078. [Google Scholar] [CrossRef]
- Chang, M.K.; Raggatt, L.-J.; Alexander, K.A.; Kuliwaba, J.S.; Fazzalari, N.L.; Schroder, K.; Maylin, E.R.; Ripoll, V.M.; Hume, D.A.; Pettit, A.R. Osteal Tissue Macrophages Are Intercalated throughout Human and Mouse Bone Lining Tissues and Regulate Osteoblast Function in Vitro and in Vivo. J. Immunol. 2008, 181, 1232–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miron, R.J.; Bosshardt, D.D. OsteoMacs: Key Players around Bone Biomaterials. Biomaterials 2016, 82, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Batoon, L.; Millard, S.M.; Wullschleger, M.E.; Preda, C.; Wu, A.C.-K.; Kaur, S.; Tseng, H.-W.; Hume, D.A.; Levesque, J.-P.; Raggatt, L.J.; et al. CD169(+) Macrophages Are Critical for Osteoblast Maintenance and Promote Intramembranous and Endochondral Ossification during Bone Repair. Biomaterials 2019, 196, 51–66. [Google Scholar] [CrossRef] [Green Version]
- Altan-Bonnet, G.; Mukherjee, R. Cytokine-Mediated Communication: A Quantitative Appraisal of Immune Complexity. Nat. Rev. Immunol. 2019, 19, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, R.; Liu, C.; Ma, R.; Wang, L.; Chen, B.; Li, L.; Niu, J.; Zhao, D.; Mo, F.; et al. Evaluation of Decalcification Techniques for Rat Femurs Using HE and Immunohistochemical Staining. Biomed. Res. Int. 2017, 2017, 9050754. [Google Scholar] [CrossRef] [PubMed]
- Nikovics, K.; Morin, H.; Riccobono, D.; Bendahmane, A.; Favier, A. Hybridization-chain-reaction Is a Relevant Method for in Situ Detection of M2d-like Macrophages in a Mini-pig Model. FASEB J. 2020, 34, 15675–15686. [Google Scholar] [CrossRef] [PubMed]
- Sicherre, E.; Favier, A.-L.; Riccobono, D.; Nikovics, K. Non-Specific Binding, a Limitation of the Immunofluorescence Method to Study Macrophages In Situ. Genes 2021, 12, 649. [Google Scholar] [CrossRef]
- Gupta, R.; Cooper, W.A.; Selinger, C.; Mahar, A.; Anderson, L.; Buckland, M.E.; O’Toole, S.A. Fluorescent In Situ Hybridization in Surgical Pathology Practice. Adv. Anat. Pathol. 2018, 25, 223–237. [Google Scholar] [CrossRef]
- Chu, Y.-H.; Hardin, H.; Zhang, R.; Guo, Z.; Lloyd, R.V. In Situ Hybridization: Introduction to Techniques, Applications and Pitfalls in the Performance and Interpretation of Assays. Semin. Diagn. Pathol. 2019, 36, 336–341. [Google Scholar] [CrossRef]
- Jensen, E. Technical Review: In Situ Hybridization. Anat Rec. 2014, 297, 1349–1353. [Google Scholar] [CrossRef]
- Choi, H.M.T.; Beck, V.A.; Pierce, N.A. Next-Generation in Situ Hybridization Chain Reaction: Higher Gain, Lower Cost, Greater Durability. ACS Nano 2014, 8, 4284–4294. [Google Scholar] [CrossRef] [PubMed]
- Looi, L.M.; Cheah, P.L. In Situ Hybridisation: Principles and Applications. Malays. J. Pathol. 1992, 14, 69–76. [Google Scholar]
- Veselinyová, D.; Mašlanková, J.; Kalinová, K.; Mičková, H.; Mareková, M.; Rabajdová, M. Selected In Situ Hybridization Methods: Principles and Application. Molecules 2021, 26, 3874. [Google Scholar] [CrossRef] [PubMed]
- Schramm, A.; De Beer, D.; Wagner, M.; Amann, R. Identification and Activities in Situ of Nitrosospira and Nitrospira Spp. as Dominant Populations in a Nitrifying Fluidized Bed Reactor. Appl. Environ. Microbiol. 1998, 64, 3480–3485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikovics, K.; Favier, A.-L. Macrophage Identification In Situ. Biomedicines 2021, 9, 1393. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.M.T.; Calvert, C.R.; Husain, N.; Huss, D.; Barsi, J.C.; Deverman, B.E.; Hunter, R.C.; Kato, M.; Lee, S.M.; Abelin, A.C.T.; et al. Mapping a Multiplexed Zoo of MRNA Expression. Development 2016, 143, 3632–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.M.T.; Schwarzkopf, M.; Fornace, M.E.; Acharya, A.; Artavanis, G.; Stegmaier, J.; Cunha, A.; Pierce, N.A. Third-Generation in Situ Hybridization Chain Reaction: Multiplexed, Quantitative, Sensitive, Versatile, Robust. Development 2018, 145, dev165753. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.M.T.; Schwarzkopf, M.; Pierce, N.A. Multiplexed Quantitative In Situ Hybridization with Subcellular or Single-Molecule Resolution within Whole-Mount Vertebrate Embryos: QHCR and DHCR Imaging (v3.0). Methods Mol. Biol. 2020, 2148, 159–178. [Google Scholar] [CrossRef]
- Lei, Z.; van Mil, A.; Xiao, J.; Metz, C.H.G.; van Eeuwijk, E.C.M.; Doevendans, P.A.; Sluijter, J.P.G. MMISH: Multicolor MicroRNA in Situ Hybridization for Paraffin Embedded Samples. Biotechnol. Rep. 2018, 18, e00255. [Google Scholar] [CrossRef]
- Kremer, M.; Quintanilla-Martínez, L.; Nährig, J.; von Schilling, C.; Fend, F. Immunohistochemistry in Bone Marrow Pathology: A Useful Adjunct for Morphologic Diagnosis. Virchows Arch. 2005, 447, 920–937. [Google Scholar] [CrossRef]
- Rijntjes, N.V.; Van de Putte, L.B.; Van der Pol, M.; Guelen, P.J. Cryosectioning of Undecalcified Tissues for Immunofluorescence. J. Immunol. Methods 1979, 30, 263–268. [Google Scholar] [CrossRef]
- Kawamoto, T. Light Microscopic Autoradiography for Study of Early Changes in the Distribution of Water-Soluble Materials. J. Histochem. Cytochem. 1990, 38, 1805–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamoto, T. Use of a New Adhesive Film for the Preparation of Multi-Purpose Fresh-Frozen Sections from Hard Tissues, Whole-Animals, Insects and Plants. Arch. Histol. Cytol. 2003, 66, 123–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamoto, T.; Kawamoto, K. Preparation of Thin Frozen Sections from Nonfixed and Undecalcified Hard Tissues Using Kawamoto’s Film Method (2020). Methods Mol. Biol. 2021, 2230, 259–281. [Google Scholar] [CrossRef]
- Salie, R.; Li, H.; Jiang, X.; Rowe, D.W.; Kalajzic, I.; Susa, M. A Rapid, Nonradioactive in Situ Hybridization Technique for Use on Cryosectioned Adult Mouse Bone. Calcif. Tissue Int. 2008, 83, 212–221. [Google Scholar] [CrossRef]
- Kramer, I.; Salie, R.; Susa, M.; Kneissel, M. Studying Gene Expression in Bone by in Situ Hybridization. Methods Mol. Biol. 2012, 816, 305–320. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Q.; Zhang, L.; Fu, X.; Chen, J.; Hong, D. A Modified Tape Transfer Approach for Rapidly Preparing High-Quality Cryosections of Undecalcified Adult Rodent Bones. J. Orthop. Translat. 2021, 26, 92–100. [Google Scholar] [CrossRef]
- Kosmac, K.; Peck, B.D.; Walton, R.G.; Mula, J.; Kern, P.A.; Bamman, M.M.; Dennis, R.A.; Jacobs, C.A.; Lattermann, C.; Johnson, D.L.; et al. Immunohistochemical Identification of Human Skeletal Muscle Macrophages. Bio-Protocol 2018, 8, e2883. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, A.J.; McNeill, E.; Kapellos, T.S.; Regan-Komito, D.; Norman, S.; Burd, S.; Smart, N.; Machemer, D.E.W.; Stylianou, E.; McShane, H.; et al. Human CD68 Promoter GFP Transgenic Mice Allow Analysis of Monocyte to Macrophage Differentiation in Vivo. Blood 2014, 124, e33–e44. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Killingsworth, M.C.; Myasoedova, V.A.; Orekhov, A.N.; Bobryshev, Y.V. CD68/Macrosialin: Not Just a Histochemical Marker. Lab. Investig. 2017, 97, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Changes in Transcriptome of Macrophages in Atherosclerosis. J. Cell Mol. Med. 2015, 19, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Takahashi, N.; Akatsu, T.; Tanaka, H.; Sasaki, T.; Nishihara, T.; Koga, T.; Martin, T.J.; Suda, T. Origin of Osteoclasts: Mature Monocytes and Macrophages Are Capable of Differentiating into Osteoclasts under a Suitable Microenvironment Prepared by Bone Marrow-Derived Stromal Cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7260–7264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razafimahefa, J.; Gosset, C.; Mongiat-Artus, P.; Andriamampionona, T.F.; Verine, J. Stromal Osseous Metaplasia in Urothelial Carcinoma of the Bladder: A Rare Case Report and Literature Review. Diagn. Pathol. 2019, 14, 75. [Google Scholar] [CrossRef] [Green Version]
- Gan, F.; Luk, G.D.; Gesell, M.S. Nonradioactive in Situ Hybridization Techniques for Routinely Prepared Pathology Specimens and Cultured Cells. Null 1994, 17, 313–319. [Google Scholar] [CrossRef]
- Hoock, T.C.; Newcomb, P.M.; Herman, I.M. Beta Actin and Its MRNA Are Localized at the Plasma Membrane and the Regions of Moving Cytoplasm during the Cellular Response to Injury. J. Cell Biol. 1991, 112, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taneja, K.L.; Singer, R.H. Detection and Localization of Actin MRNA Isoforms in Chicken Muscle Cells by in Situ Hybridization Using Biotinated Oligonucleotide Probes. J. Cell Biochem. 1990, 44, 241–252. [Google Scholar] [CrossRef]
- Bertram, S.; Schildhaus, H.-U. Fluorescence in situ hybridization for the diagnosis of soft-tissue and bone tumors. Pathologe 2020, 41, 589–605. [Google Scholar] [CrossRef]
- Frickmann, H.; Zautner, A.E.; Moter, A.; Kikhney, J.; Hagen, R.M.; Stender, H.; Poppert, S. Fluorescence in Situ Hybridization (FISH) in the Microbiological Diagnostic Routine Laboratory: A Review. Crit. Rev. Microbiol. 2017, 43, 263–293. [Google Scholar] [CrossRef]
- Tsuneoka, Y.; Funato, H. Modified in Situ Hybridization Chain Reaction Using Short Hairpin DNAs. Front. Mol. Neurosci. 2020, 13, 75. [Google Scholar] [CrossRef]
- Fauch, L.; Palander, A.; Dekker, H.; Schulten, E.A.; Koistinen, A.; Kullaa, A.; Keinänen, M. Narrowband-Autofluorescence Imaging for Bone Analysis. Biomed. Opt. Express 2019, 10, 2367–2382. [Google Scholar] [CrossRef]
- Su, W.; Yang, L.; Luo, X.; Chen, M.; Liu, J. Elimination of Autofluorescence in Archival Formaldehyde-Fixed, Paraffin-Embedded Bone Marrow Biopsies. Arch. Pathol. Lab. Med. 2019, 143, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capasso, L.; D’Anastasio, R.; Guarnieri, S.; Viciano, J.; Mariggiò, M. Bone Natural Autofluorescence and Confocal Laser Scanning Microscopy: Preliminary Results of a Novel Useful Tool to Distinguish between Forensic and Ancient Human Skeletal Remains. Forensic Sci. Int. 2017, 272, 87–96. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resin (R)/Paraffin (P)-Section | Cryo-Section | |
|---|---|---|
| Section preparation | Long method | Short method |
| Toxic substances | Long period | Short period |
| Size of the section | 5 μm | 5 μm |
| Quality of morphology | Good | Medium |
| Application | Histological staining (R, P) Immunolabeling (P) In situ hybridization (P, medium sensitivity) | Histological staining Immunolabeling In situ hybridization (strong sensitivity) Laser microdissection |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikovics, K.; Castellarin, C.; Holy, X.; Durand, M.; Morin, H.; Bendahmane, A.; Favier, A.-L. In Situ Gene Expression in Native Cryofixed Bone Tissue. Biomedicines 2022, 10, 484. https://doi.org/10.3390/biomedicines10020484
Nikovics K, Castellarin C, Holy X, Durand M, Morin H, Bendahmane A, Favier A-L. In Situ Gene Expression in Native Cryofixed Bone Tissue. Biomedicines. 2022; 10(2):484. https://doi.org/10.3390/biomedicines10020484
Chicago/Turabian StyleNikovics, Krisztina, Cédric Castellarin, Xavier Holy, Marjorie Durand, Halima Morin, Abdelhafid Bendahmane, and Anne-Laure Favier. 2022. "In Situ Gene Expression in Native Cryofixed Bone Tissue" Biomedicines 10, no. 2: 484. https://doi.org/10.3390/biomedicines10020484
APA StyleNikovics, K., Castellarin, C., Holy, X., Durand, M., Morin, H., Bendahmane, A., & Favier, A.-L. (2022). In Situ Gene Expression in Native Cryofixed Bone Tissue. Biomedicines, 10(2), 484. https://doi.org/10.3390/biomedicines10020484