
TDP-43 Pathology and Prionic Behavior in Human Cellular Models of Alzheimer’s Disease Patients

 ,
,  , ,
, ,  and
and 
Abstract
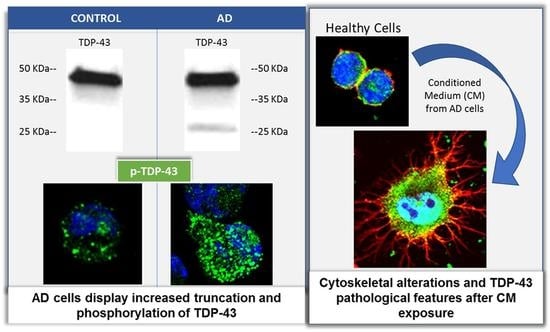
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Subjects
2.3. Isolation of Peripheral Blood Mononuclear Cells and Establishment of Lymphoblastoid Cell Lines
2.4. Cell Culture
2.5. Analysis of mRNA Levels by Quantitative Real-Time PCR
2.6. Inmunological Assays
2.6.1. Cell Extracts
2.6.2. Western Blot Analysis
2.6.3. Confocal Laser Scanning Microscopy
2.7. Conditioned Medium Experiments
2.8. Extracellular Vesicles Isolation and Characterization
2.9. Statistical Analysis
3. Results
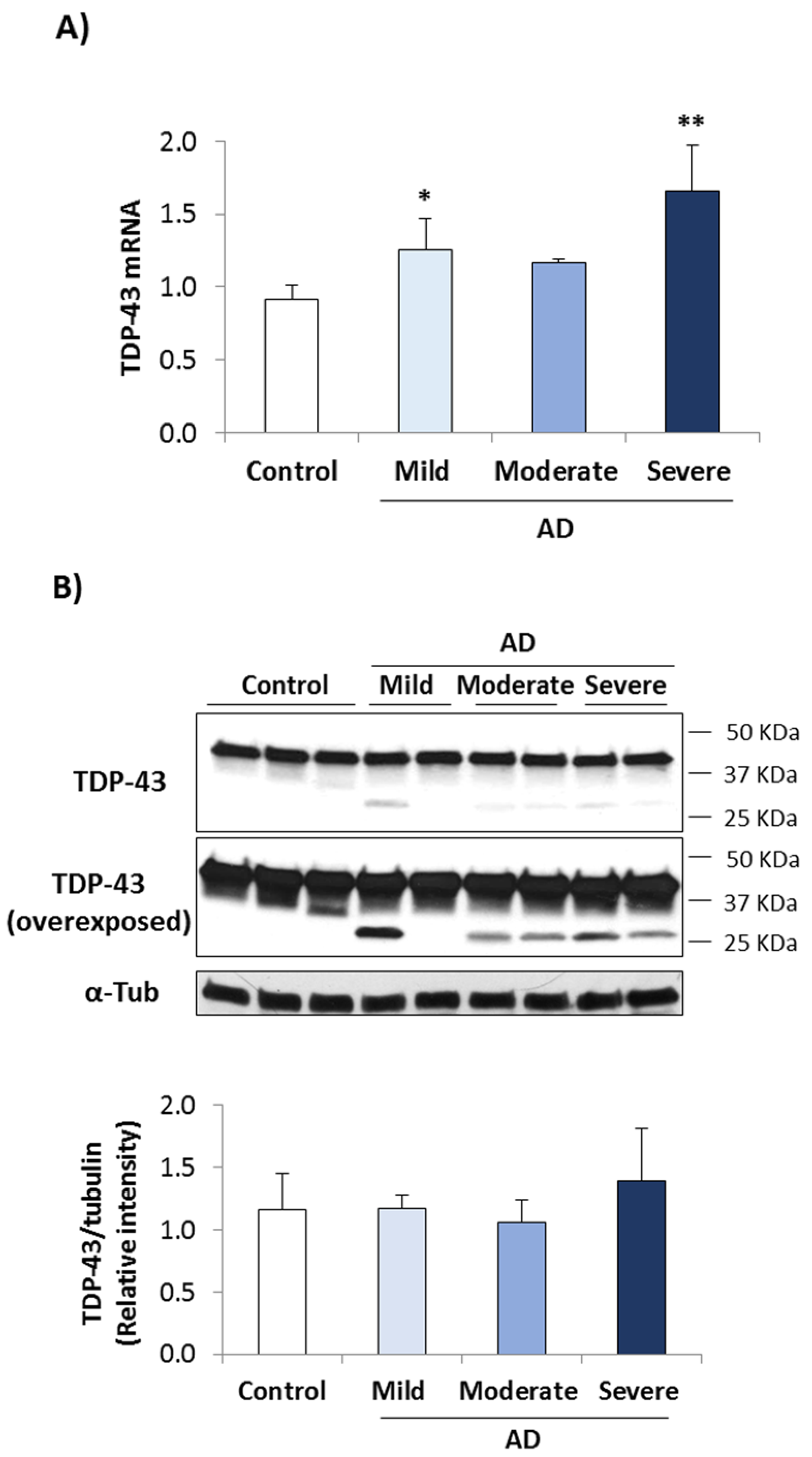
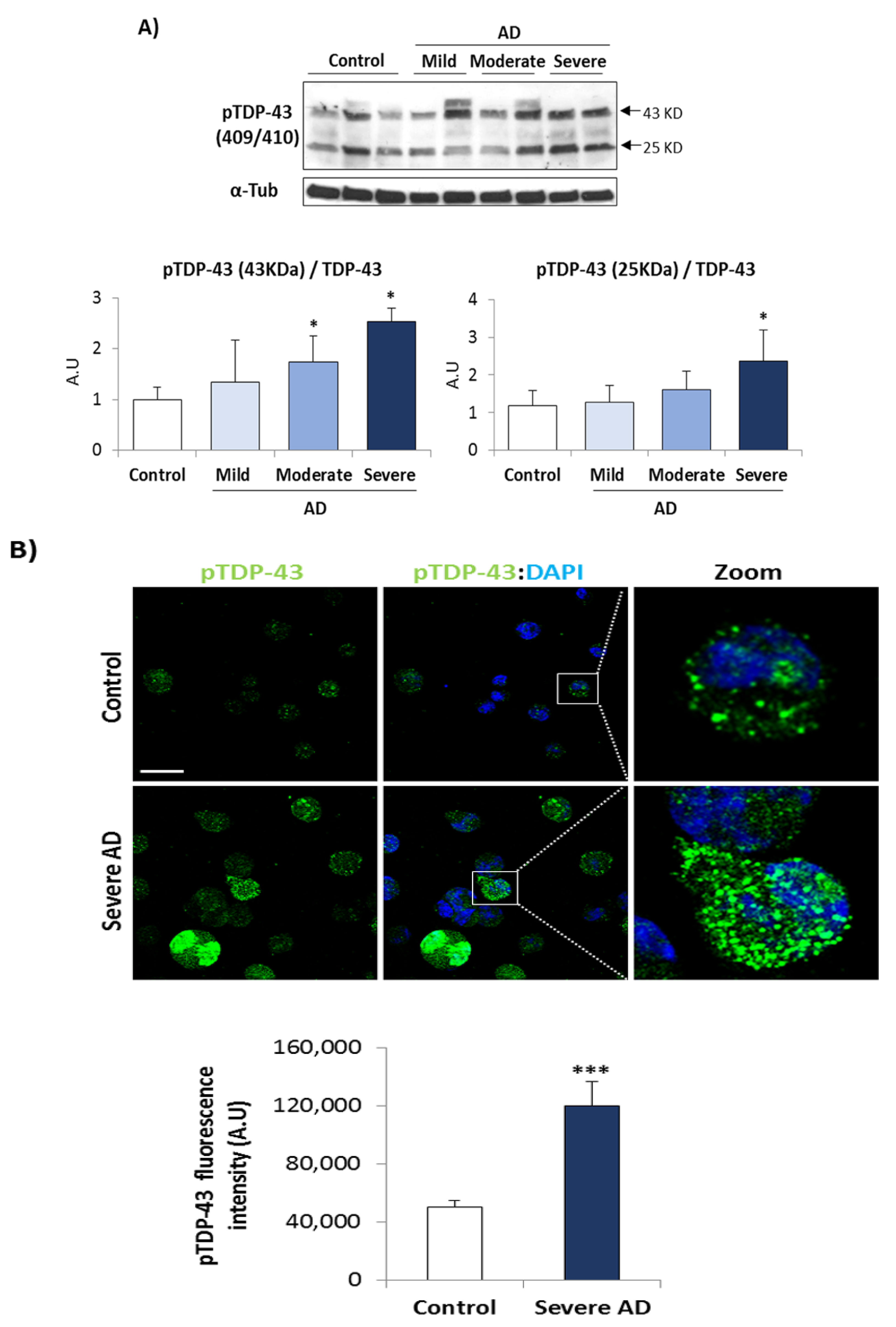
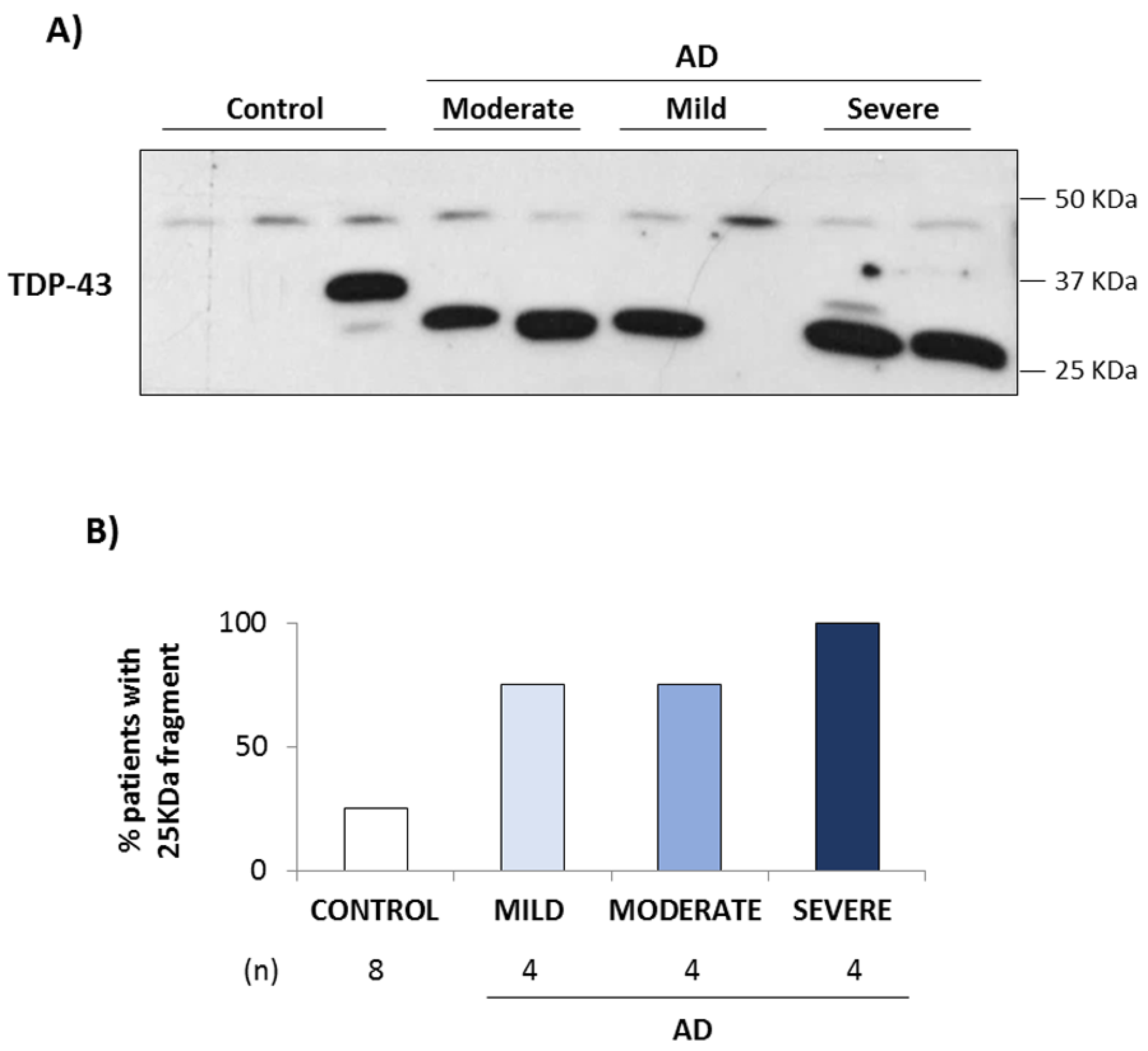
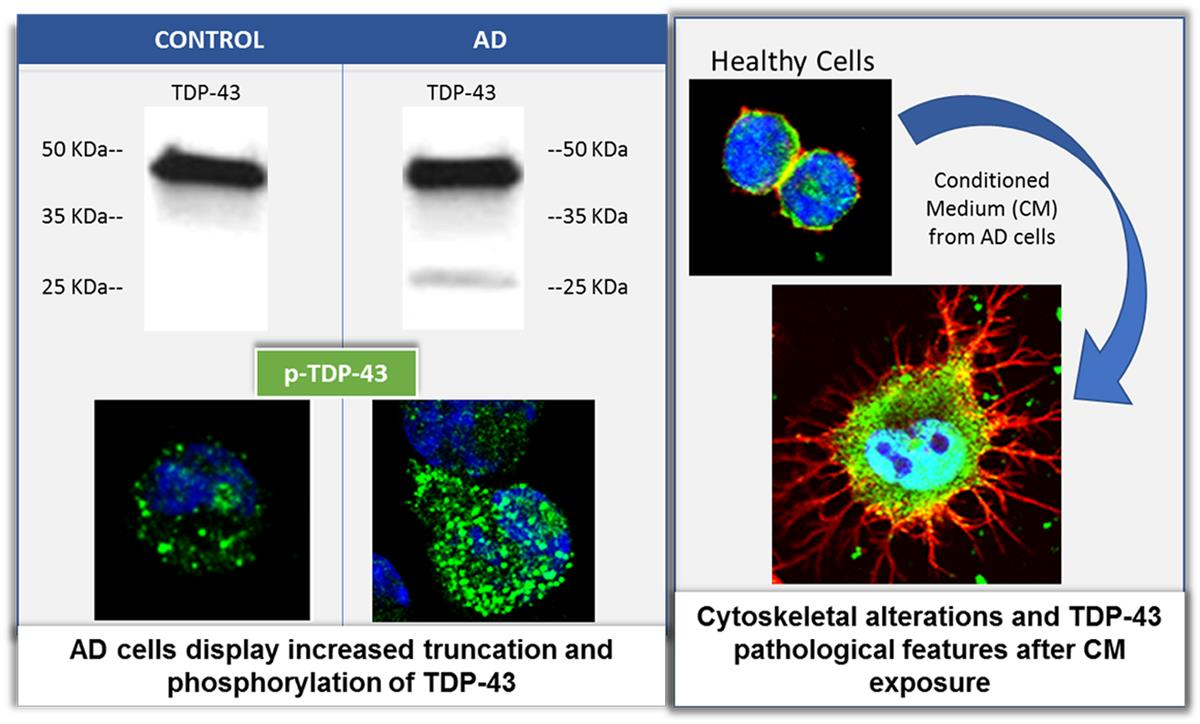
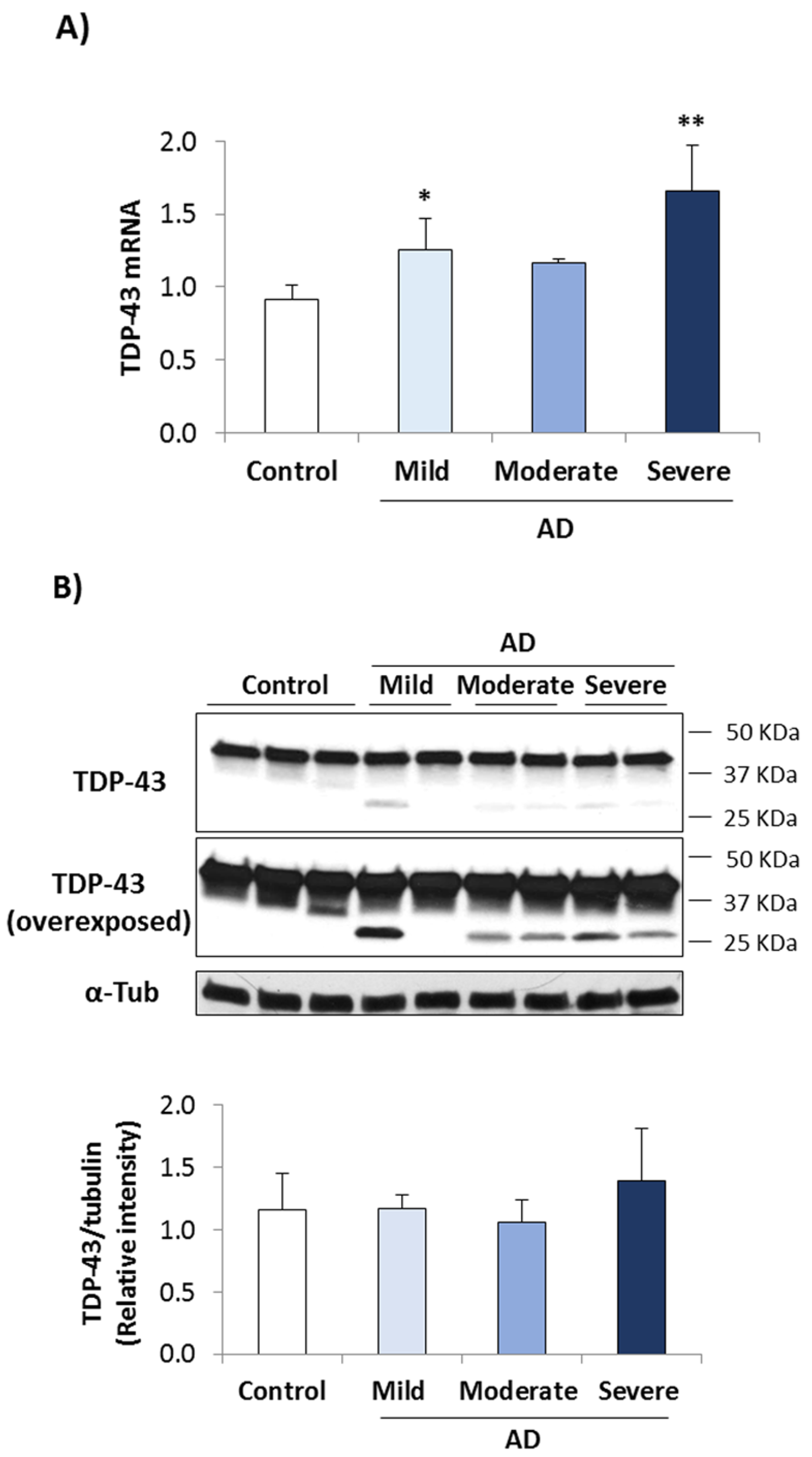
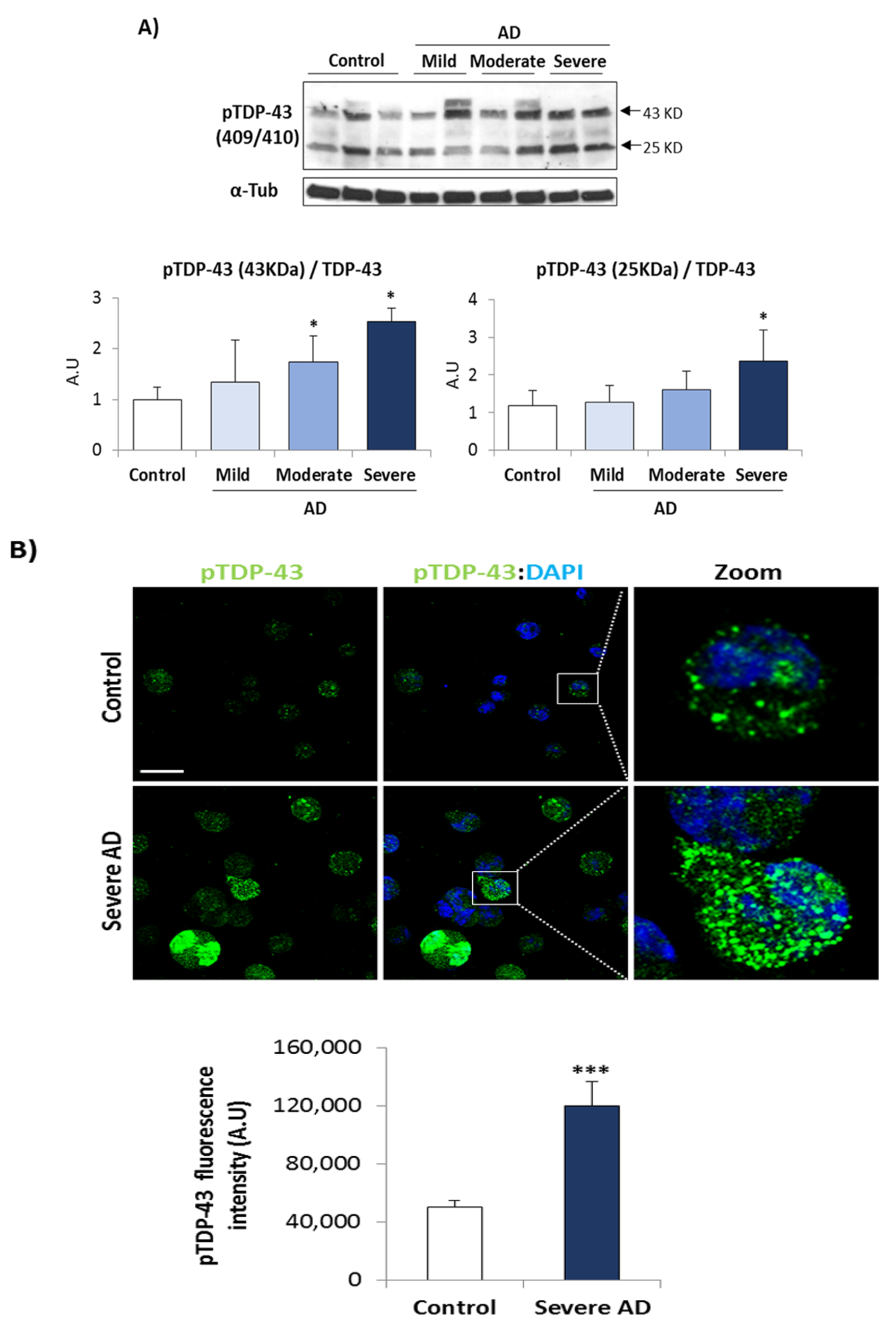
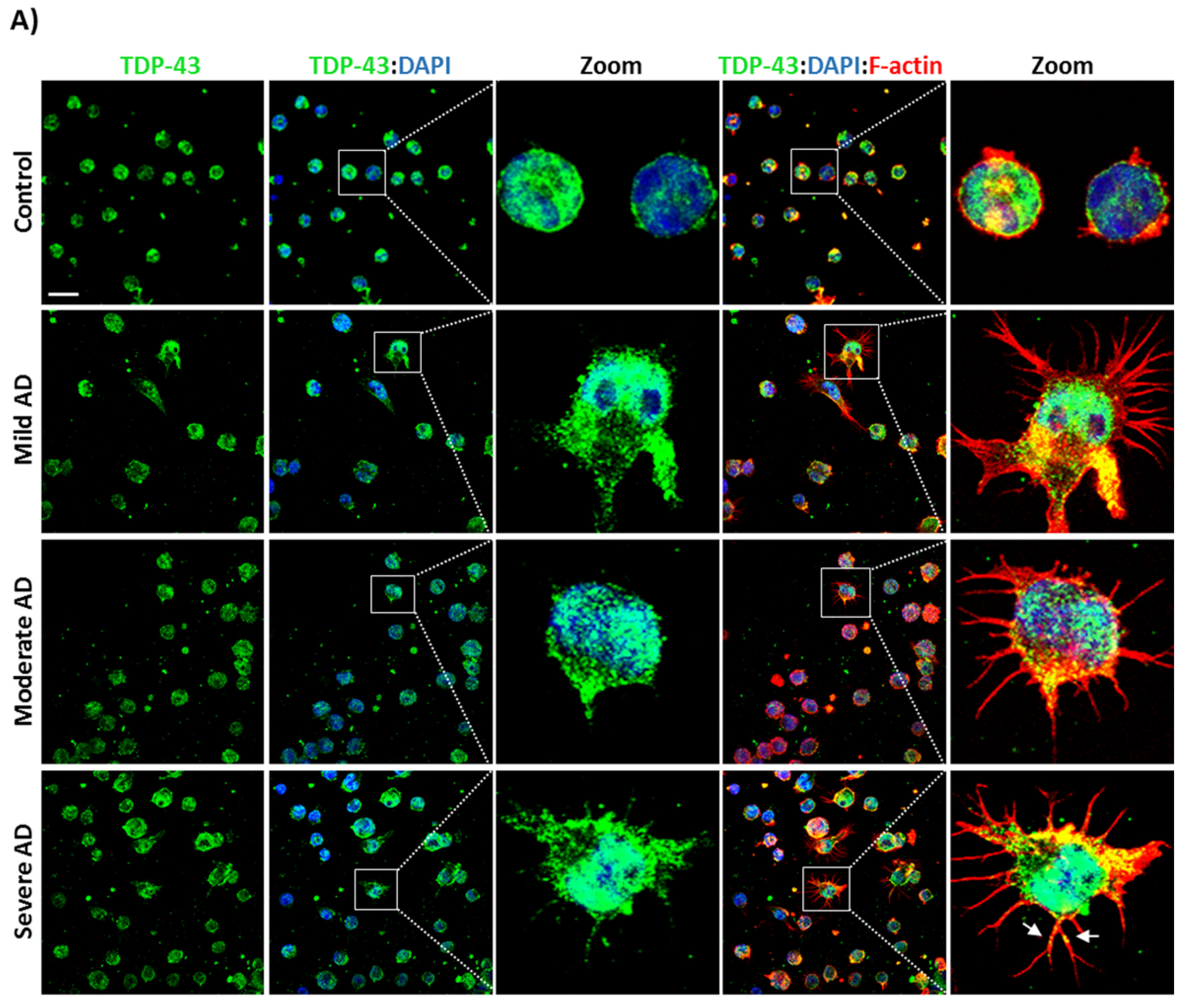
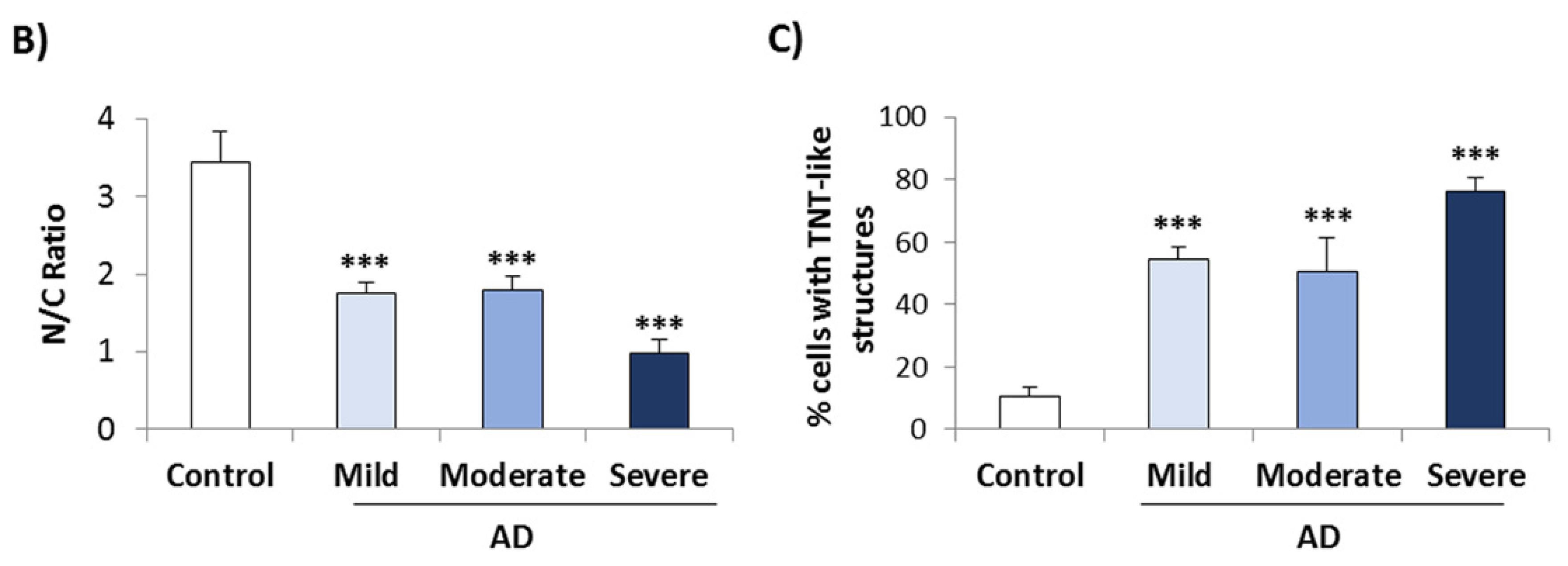
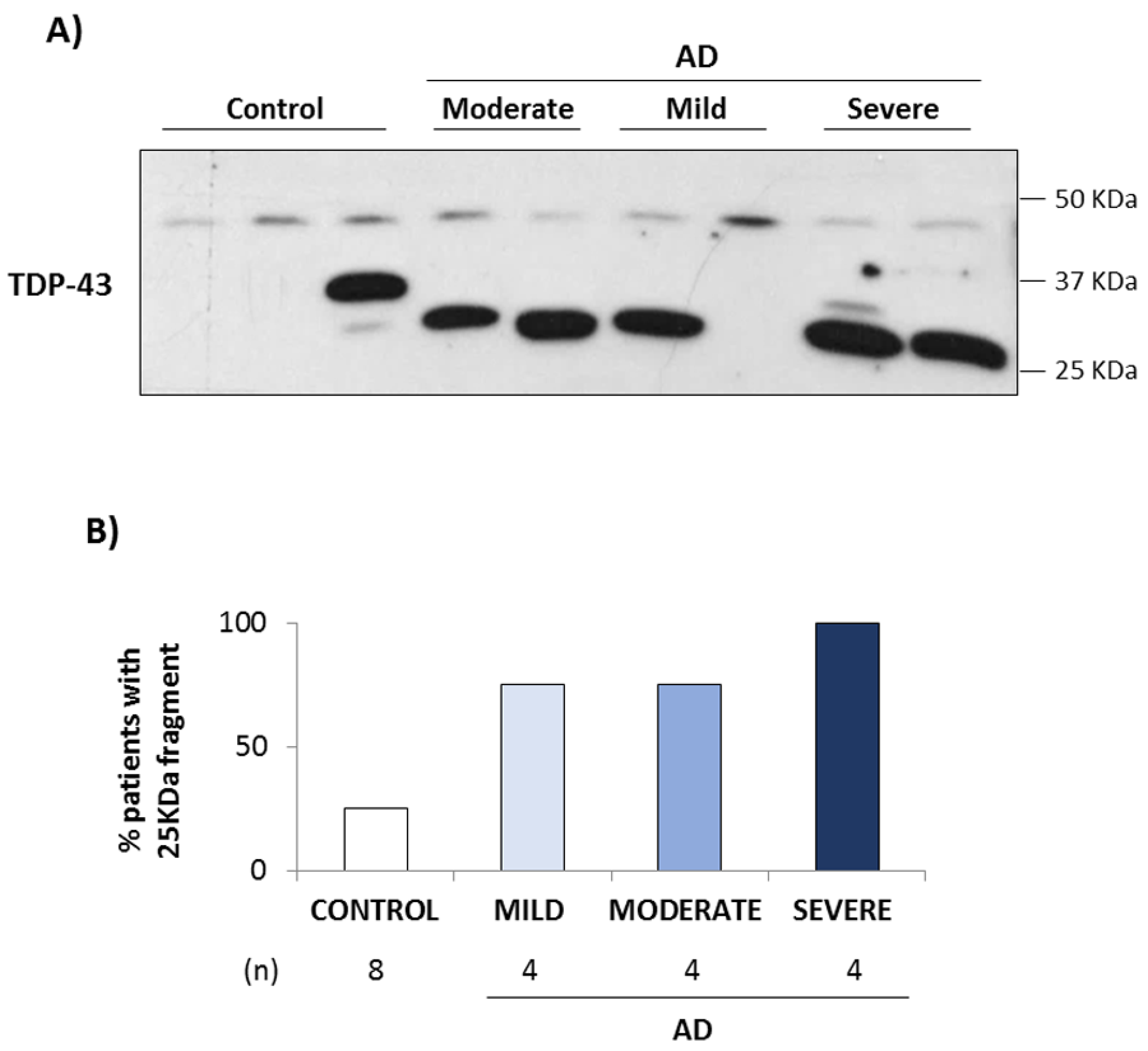
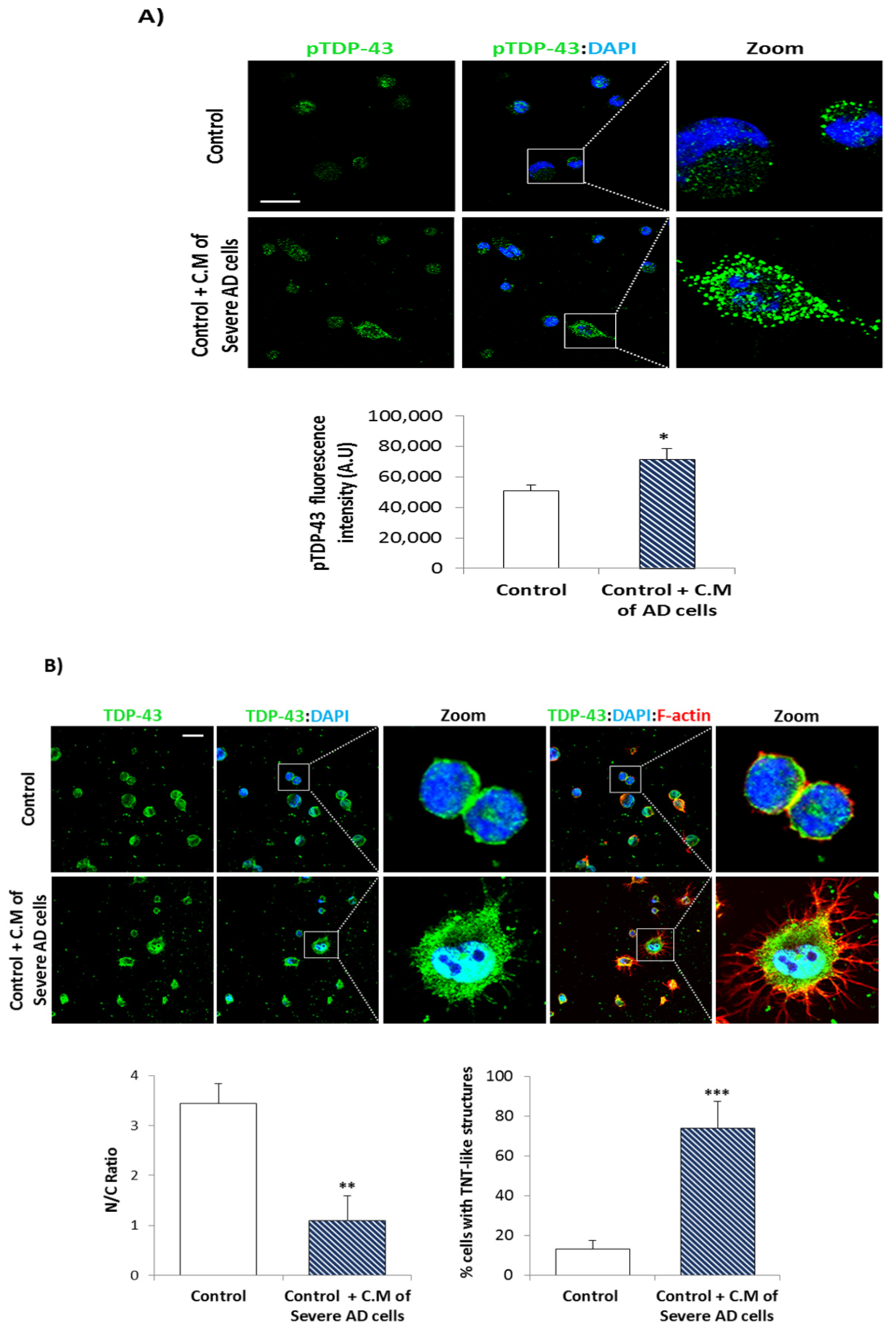
3.1. TDP-43 Pathology in AD Lymphoblasts
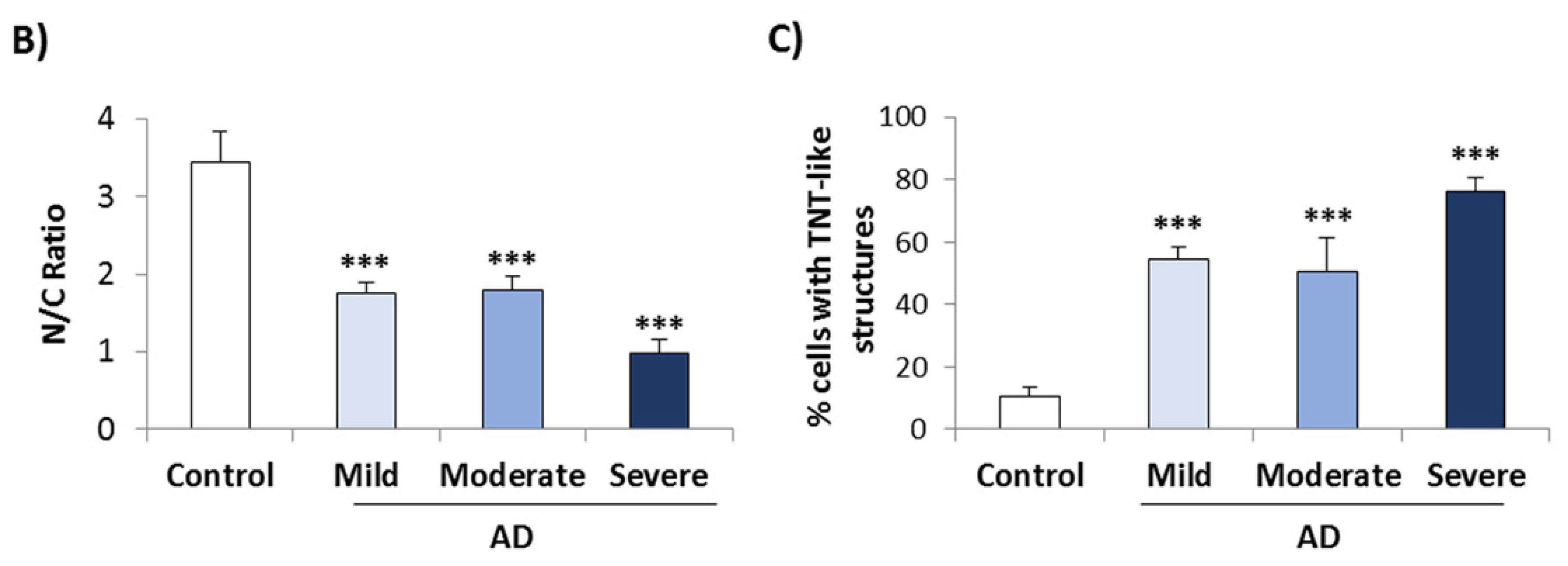
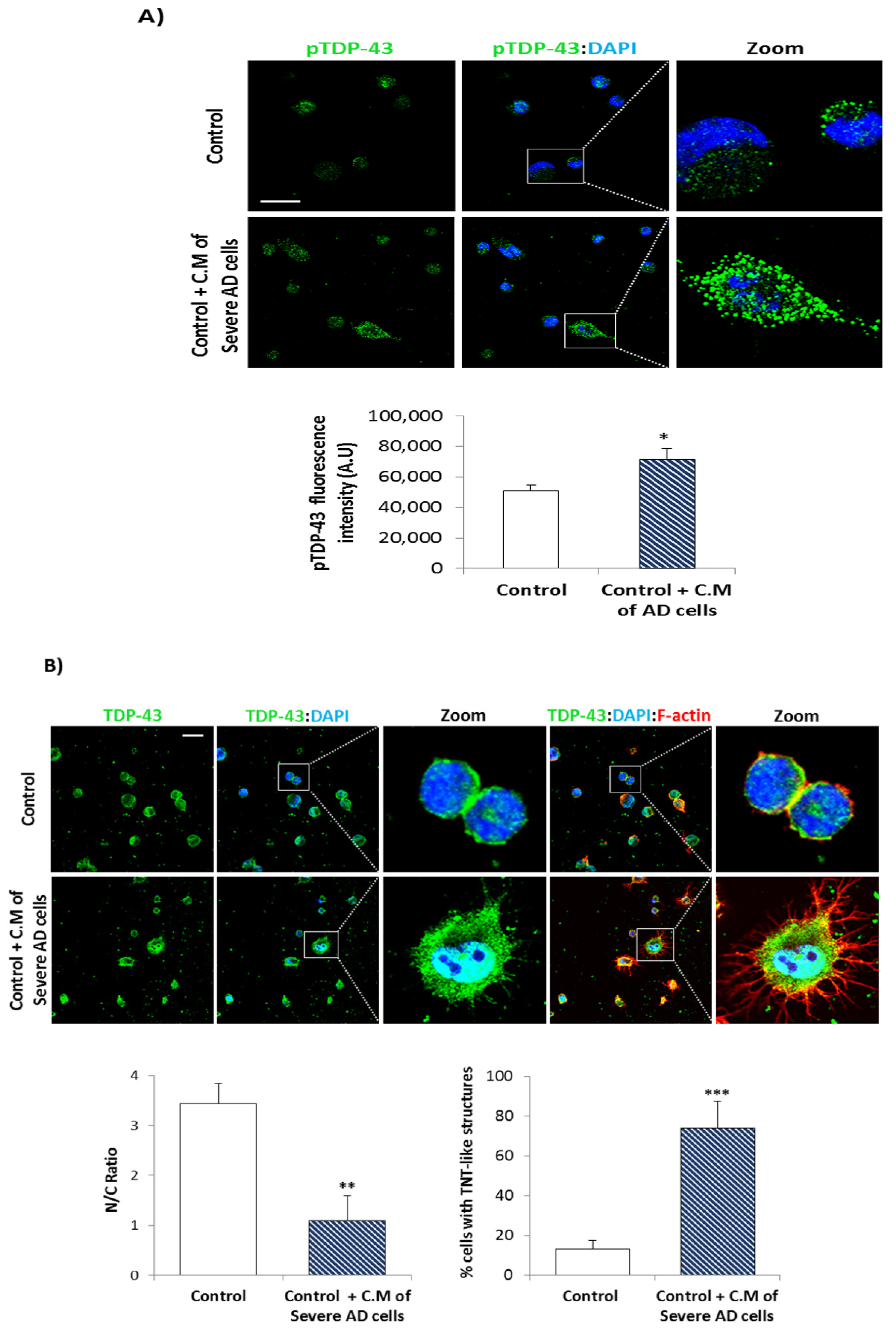
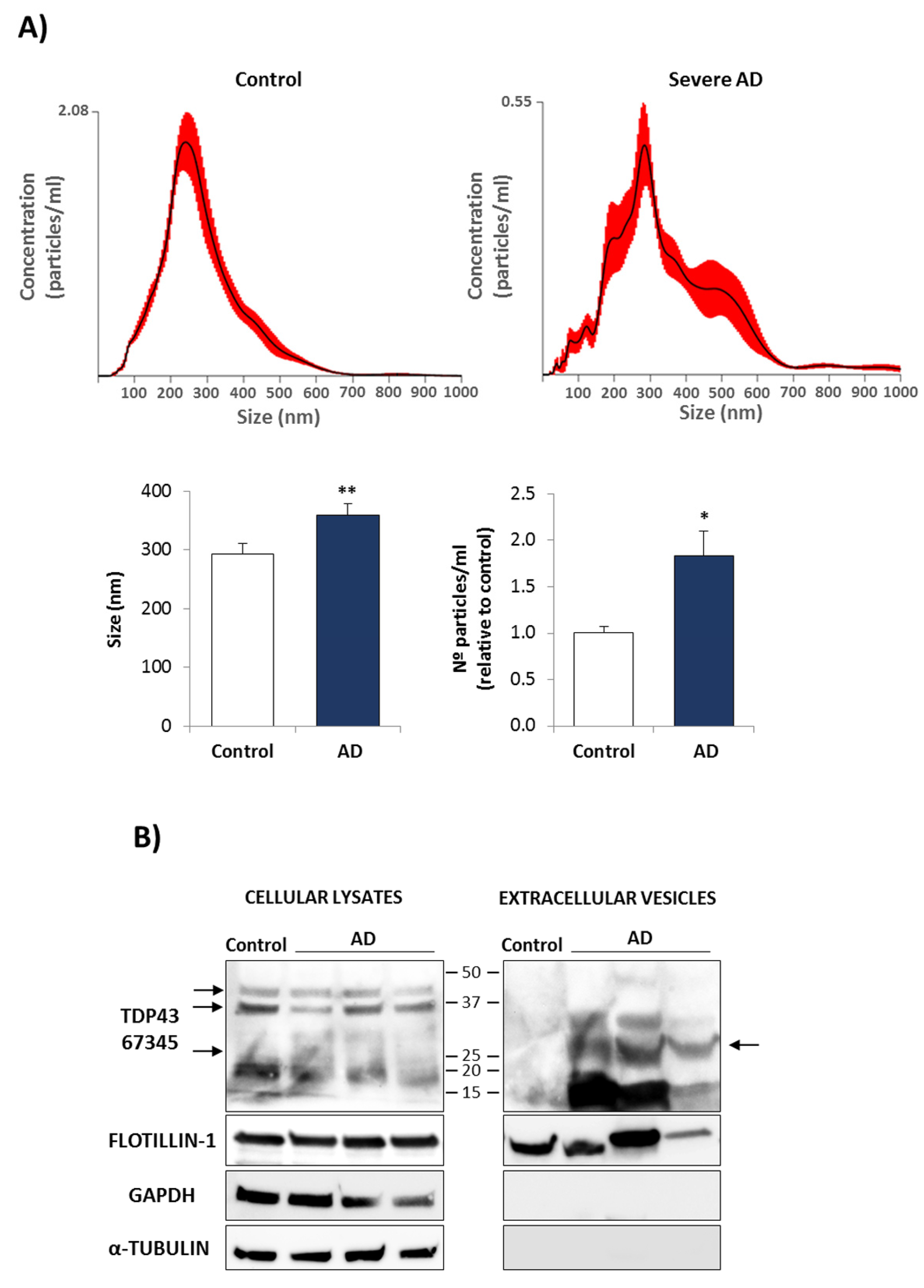
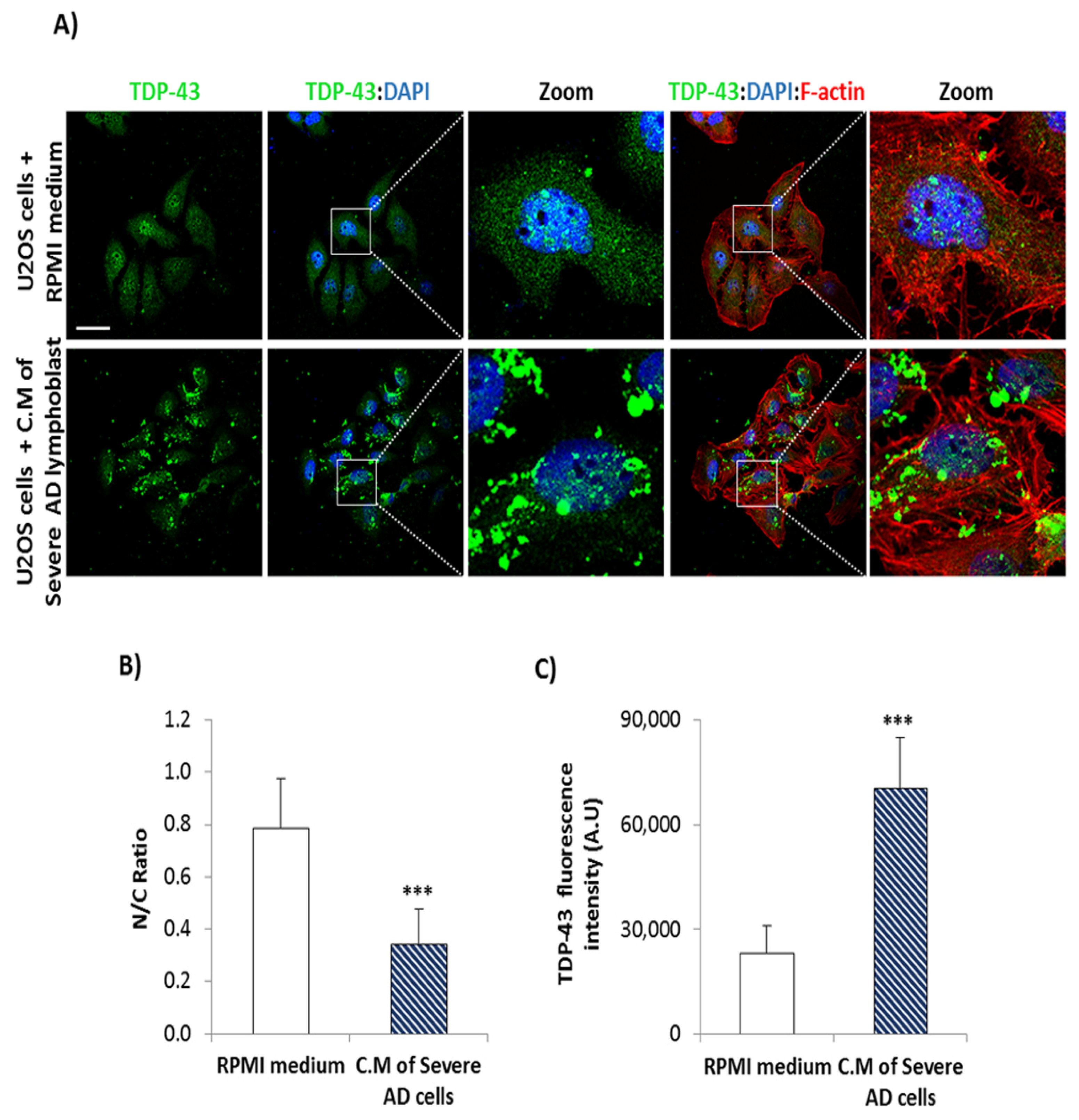
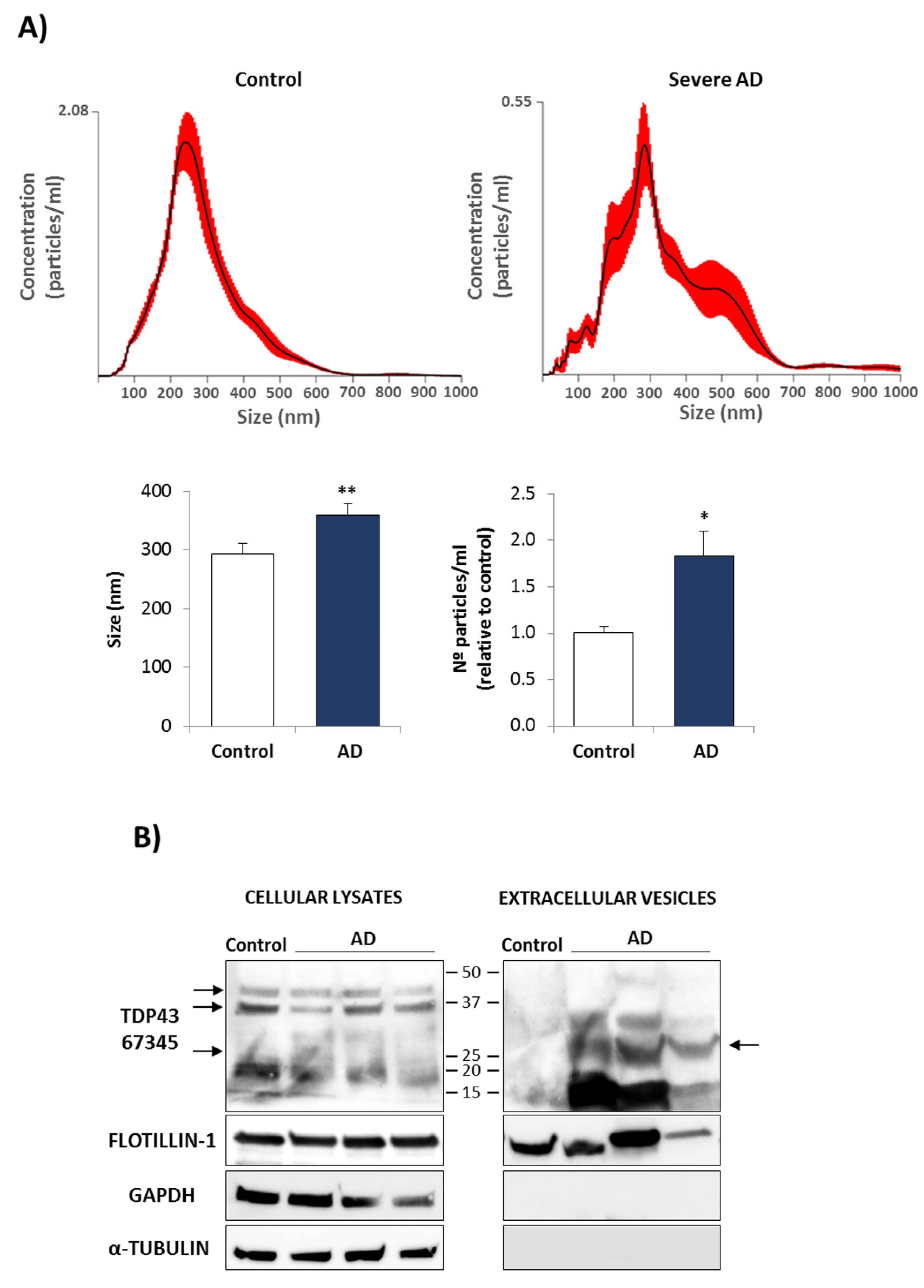
3.2. Conditional Medium from AD Lymphoblast Is Enriched in TDP-43 Fragments
4. Discussion
5. Conclusions
6. Limitations and Future Studies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement. 2013, 9, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, S.; Serio, G.; Scarpazza, C.; D’Ausilio, A.; Borgomaneri, S. Frozen in (e)motion: How reactive motor inhibition is influenced by the emotional content of stimuli in healthy and psychiatric populations. Behav. Res. Ther. 2021, 146, 103963. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Van Cauwenberghe, C.; van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Hannon, E.; Shireby, G.L.; Brookes, K.; Attems, J.; Sims, R.; Cairns, N.J.; Love, S.; Thomas, A.J.; Morgan, K.; Francis, P.T.; et al. Genetic risk for Alzheimer’s disease influences neuropathology via multiple biological pathways. Brain Commun. 2020, 2, fcaa167. [Google Scholar] [CrossRef]
- Carreiras, M.C.; Mendes, E.; Perry, M.J.; Francisco, A.P.; Marco-Contelles, J. The multifactorial nature of Alzheimer’s disease for developing potential therapeutics. Curr. Top. Med. Chem. 2013, 13, 1745–1770. [Google Scholar] [CrossRef]
- Lloret, A.; Monllor, P.; Esteve, D.; Cervera-Ferri, A.; Lloret, M.-A. Obesity as a risk factor for Alzheimer’s disease: Implication of leptin and glutamate. Front. Neurosci. 2019, 13, 508. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Seo, H.I.; Cha, H.Y.; Yang, Y.J.; Kwon, S.H.; Yang, S.J. Diabetes and Alzheimer’s disease: Mechanisms and nutritional aspects. Clin. Nutr. Res. 2018, 7, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; van Giau, V. Type 3 diabetes and its role implications in Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef] [PubMed]
- Yanker, B.A. New clues to Alzheimer’s disease: Unraveling the roles of amyloid and tau. Nat. Med. 1996, 2, 850–852. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J. Alzheimer’s Dis. 2001, 3, 75–80. [Google Scholar] [CrossRef]
- Jellinger, K.A. Alzheimer 100—Highlights in the history of Alzheimer research. J. Neural Transm. 2006, 113, 1603–1623. [Google Scholar] [CrossRef]
- Jellinger, K.A. Neuropathological subtypes of Alzheimer’s disease. Acta Neuropathol. 2012, 123, 153–154. [Google Scholar] [CrossRef]
- Amador-Ortiz, C.; Lin, W.L.; Ahmed, Z.; Personett, D.; Davies, P.; Duara, R.; Graff-Radford, N.R.; Hutton, M.L.; Dickson, D.W. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol. 2007, 61, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Mackenzie, I.R.A.; Hasegawa, M.; Nonoka, T.; Niizato, K.; Tsuchiya, K.; Iritani, S.; Onaya, M.; Akiyama, H. Phosphorylated TDP-43 in Alzheimer’s disease and dementia with lewy bodies. Acta Neuropathol. 2009, 117, 125–136. [Google Scholar] [CrossRef]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.A.; Rademakers, R. The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Opin. Neurol. 2008, 21, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devenney, E.; Vucic, S.; Hodges, J.R.; Kiernan, M.C. Motor neuron disease-frontotemporal dementia: A clinical continuum. Expert Rev. Neurother. 2015, 15, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Burrell, J.R.; Halliday, G.M.; Kril, J.J.; Ittner, L.M.; Götz, J.; Kiernan, M.C.; Hodges, J.R. The frontotemporal dementia-motor neuron disease continuum. Lancet 2016, 388, 919–931. [Google Scholar] [CrossRef]
- Hodges, J.R.; Davies, R.R.; Xuereb, J.H.; Casey, B.; Broe, M.; Bak, T.H.; Kril, J.J.; Halliday, G.M. Clinicopathological correlates in frontotemporal dementia. Ann. Neurol. 2004, 56, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Meneses, A.; Koga, S.; O’Leary, J.; Dickson, D.W.; Bu, G.; Zhao, N. TDP-43 pathology in Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Tomé, S.O.; Vandenberghe, R.; Ospitalieri, S.; van Schoor, E.; Tousseyn, T.; Otto, M.; von Arnim, C.A.F.; Thal, D.R. Distinct molecular patterns of TDP-43 pathology in Alzheimer’s disease: Relationship with clinical phenotypes. Acta Neuropathol. Commun. 2020, 8, 61. [Google Scholar] [CrossRef]
- Josephs, K.A.; Whitwell, J.L.; Knopman, D.S.; Hu, W.T.; Stroh, D.A.; Baker, M.; Rademakers, R.; Boeve, B.F.; Parisi, J.E.; Smith, G.E.; et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 2008, 70, 1850–1857. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, C.; St-Amour, I.; Schneider, J.; Bennett, D.A.; Calon, F. Accumulation of transactive response DNA binding protein 43 in mild cognitive impairment and Alzheimer disease. J. Neuropathol. Exp. Neurol. 2011, 70, 788–798. [Google Scholar] [CrossRef]
- Wilson, R.S.; Yu, L.; Trojanowski, J.Q.; Chen, E.-Y.; Boyle, P.A.; Bennett, D.A.; Schneider, J.A. TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol. 2013, 70, 1418. [Google Scholar] [CrossRef]
- Warraich, S.T.; Yang, S.; Nicholson, G.A.; Blair, I.P. TDP-43: A DNA and RNA binding protein with roles in neurodegenerative diseases. Int. J. Biochem. Cell Biol. 2010, 42, 1606–1609. [Google Scholar] [CrossRef] [PubMed]
- Ratti, A.; Buratti, E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 2016, 138 (Suppl. 1), 95–111. [Google Scholar] [CrossRef] [PubMed]
- Palomo, V.; Tosat-Bitrian, C.; Nozal, V.; Nagaraj, S.; Martin-Requero, A.; Martinez, A. TDP-43: A key therapeutic target beyond amyotrophic lateral sclerosis. ACS Chem. Neurosci. 2019, 10, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2008, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonaka, T.; Kametani, F.; Arai, T.; Akiyama, H.; Hasegawa, M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet. 2009, 18, 3353–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolomé, F.; de Las Cuevas, N.; Muñoz, Ú.; Bermejo, F.; Martín-Requero, Á. Impaired apoptosis in lymphoblasts from Alzheimer’s disease patients: Cross-talk of Ca2+/Calmodulin and ERK1/2 signaling pathways. Cell. Mol. Life Sci. 2007, 64, 1437–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, U.; Bartolomé, F.; Bermejo, F.; Martín-Requero, A. Enhanced proteasome-dependent degradation of the CDK inhibitor P27kip1 in immortalized lymphocytes from Alzheimer’s dementia patients. Neurobiol. Aging 2008, 29, 1474–1484. [Google Scholar] [CrossRef] [Green Version]
- Esteras, N.; Alquézar, C.; de la Encarnación, A.; Martín-Requero, Á. Lymphocytes in Alzheimer’s disease pathology: Altered signaling pathways. Curr. Alzheimer Res. 2016, 13, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Alquezar, C.; Salado, I.G.; de La Encarnación, A.; Pérez, D.I.; Moreno, F.; Gil, C.; de Munain, A.L.; Martínez, A.; Martín-Requero, Á. Targeting TDP-43 phosphorylation by casein kinase-1δ inhibitors: A novel strategy for the treatment of frontotemporal dementia. Mol. Neurodegener. 2016, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Posa, D.; Martínez-González, L.; Bartolomé, F.; Nagaraj, S.; Porras, G.; Martínez, A.; Martín-Requero, Á. Recapitulation of pathological TDP-43 features in immortalized lymphocytes from sporadic ALS patients. Mol. Neurobiol. 2018, 56, 2424–2432. [Google Scholar] [CrossRef]
- Vaca, G.; Martinez-Gonzalez, L.; Fernandez, A.; Rojas-Prats, E.; Porras, G.; Cuevas, E.P.; Gil, C.; Martinez, A.; Martin-Requero, Á. Therapeutic potential of novel cell division cycle kinase 7 inhibitors on TDP-43-related pathogenesis such as frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS). J. Neurochem. 2021, 156, 379–390. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA work group under the auspices of department of health and human services task force on Alzheimer’s disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folstein, M.F.; Folstein, S.E.; Mchugh, P.R. “Mini-mental State” a practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–193. [Google Scholar] [CrossRef]
- Hussain, T.; Mulherkar, R. Lymphoblastoid cell lines: A continuous in vitro source of cells to study carcinogen sensitivity and DNA repair. Int. J. Mol. Cell. Med. 2012, 1, 75–87. [Google Scholar]
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; de Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2021, 92, 86–95. [Google Scholar] [CrossRef]
- Chornenkyy, Y.; Fardo, D.W.; Nelson, P.T. Tau and TDP-43 proteinopathies: Kindred pathologic cascades and genetic pleiotropy. Lab. Investig. 2019, 99, 993–1007. [Google Scholar] [CrossRef]
- Shih, Y.-H.; Tu, L.-H.; Chang, T.-Y.; Ganesan, K.; Chang, W.-W.; Chang, P.-S.; Fang, Y.-S.; Lin, Y.-T.; Jin, L.-W.; Chen, Y.-R. TDP-43 interacts with amyloid-β, inhibits fibrillization, and worsens pathology in a model of Alzheimer’s disease. Nat. Commun. 2020, 11, 5950. [Google Scholar] [CrossRef]
- Avendaño-Vázquez, S.E.; Dhir, A.; Bembich, S.; Buratti, E.; Proudfoot, N.; Baralle, F.E. Autoregulation of TDP-43 MRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 2012, 26, 1679–1684. [Google Scholar] [CrossRef] [Green Version]
- Uryu, K.; Nakashima-Yasuda, H.; Forman, M.S.; Kwong, L.K.; Clark, C.M.; Grossman, M.; Miller, B.L.; Kretzschmar, H.A.; Lee, V.M.Y.; Trojanowski, J.Q.; et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol. 2008, 67, 555–564. [Google Scholar] [CrossRef]
- Josephs, K.A.; Whitwell, J.L.; Tosakulwong, N.; Weigand, S.D.; Murray, M.E.; Liesinger, A.M.; Petrucelli, L.; Senjem, M.L.; Ivnik, R.J.; Parisi, J.E.; et al. TAR DNA-binding protein 43 and pathological subtype of Alzheimer’s disease impact clinical features. Ann. Neurol. 2015, 78, 697–709. [Google Scholar] [CrossRef]
- Huang, W.; Zhou, Y.; Tu, L.; Ba, Z.; Huang, J.; Huang, N.; Luo, Y. TDP-43: From Alzheimer’s disease to limbic-predominant age-related TDP-43 encephalopathy. Front. Mol. Neurosci. 2020, 13, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalbano, M.; McAllen, S.; Cascio, F.L.; Sengupta, U.; Garcia, S.; Bhatt, N.; Ellsworth, A.; Heidelman, E.A.; Johnson, O.D.; Doskocil, S.; et al. TDP-43 and tau oligomers in Alzheimer’s disease, amyotrophic lateral sclerosis, and frontotemporal dementia. Neurobiol. Dis. 2020, 146, 105130. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.M.; Khandelwal, P.J.; Stanczyk, B.B.; Rebeck, G.W.; Moussa, C.E.-H. β-amyloid triggers ALS-associated TDP-43 pathology in AD models. Brain Res. 2011, 1386, 191–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolicelli, R.C.; Jawaid, A.; Henstridge, C.M.; Valeri, A.; Merlini, M.; Robinson, J.L.; Lee, E.B.; Rose, J.; Appel, S.; Lee, V.M.-Y.; et al. TDP-43 depletion in microglia promotes amyloid clearance but also induces synapse loss. Neuron 2017, 95, 297–308.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, S.A.; Gan, K.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 expression influences amyloidβ plaque deposition and tau aggregation. Neurobiol. Dis. 2017, 103, 154–162. [Google Scholar] [CrossRef]
- Gu, J.; Wu, F.; Xu, W.; Shi, J.; Hu, W.; Jin, N.; Qian, W.; Wang, X.; Iqbal, K.; Gong, C.-X.; et al. TDP-43 suppresses tau expression via promoting its MRNA instability. Nucleic Acids Res. 2017, 45, 6177–6193. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.B.; Lee, V.M.-Y.; Trojanowski, J.Q. Gains or losses: Molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 2011, 13, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Martínez-González, L.; Rodríguez-Cueto, C.; Cabezudo, D.; Bartolomé, F.; Andrés-Benito, P.; Ferrer, I.; Gil, C.; Martín-Requero, Á.; Fernández-Ruiz, J.; Martínez, A.; et al. Motor neuron preservation and decrease of in vivo TDP-43 phosphorylation by protein CK-1δ kinase inhibitor treatment. Sci. Rep. 2020, 10, 4449. [Google Scholar] [CrossRef] [Green Version]
- Nozal, V.; Martínez-González, L.; Gomez-Almeria, M.; Gonzalo-Consuegra, C.; Santana, P.; Chaikuad, A.; Pérez-Cuevas, E.; Knapp, S.; Lietha, D.; Ramírez, D.; et al. TDP-43 modulation by tau-tubulin kinase 1 inhibitors: A new avenue for future amyotrophic lateral sclerosis therapy. J. Med. Chem. 2022, 65, 1585–1607. [Google Scholar] [CrossRef]
- Budini, M.; Romano, V.; Quadri, Z.; Buratti, E.; Baralle, F.E. TDP-43 loss of cellular function through aggregation requires additional structural determinants beyond its C-terminal Q/N prion-like domain. Hum. Mol. Genet. 2015, 24, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Gousset, K.; Schiff, E.; Langevin, C.; Marijanovic, Z.; Caputo, A.; Browman, D.T.; Chenouard, N.; de Chaumont, F.; Martino, A.; Enninga, J.; et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 2009, 11, 328–336. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Species | Dilution (WB/IF) | Supplier (Catalog#) |
| TDP-43 | Rabbit | 1:1000/na | Proteintech (10782-2-AP) |
| TDP-43 | Mouse | 1:1000/1:1000 | Proteintech (67345-1-Ig) |
| p(Ser409/410)- TDP-43 | Rabbit | 1:500/1:1000 | Proteintech (22309-1-AP) |
| α-tubulin | Mouse | 1:5000/na | Santa Cruz (23948) |
| GADPH | Rabbit | 1:1000/na | Cell Signal (5174) |
| Flotillin-1 | Rabbit | 1:1000/na | Thermo Fisher (PA5-97756) |
| Secondary Antibody | Immunological Procedure | Dilution | Supplier (Catalog#) |
| Goat anti-mouseIgG HRP conjugate | WB | 1:7000 | Bio-Rad (1706516) |
| Goat anti-rabbitIgG HRP conjugate | WB | 1:7000 | Bio-Rad (1706515) |
| Anti-mouse Alexa 488 | IF | 1:1000 | Molecular Probes (A-11001) |
| Cell Lines | n | Age | Gender | ||
|---|---|---|---|---|---|
| Mean | Range Age | Male | Female | ||
| CONTROL | 8 | 68 ± 12 | 52–83 | 5 | 3 |
| AD | |||||
| Total | 12 | 74 ± 7 | 60–84 | 6 | 6 |
| Mild | 4 | 74 ± 6 | 66–80 | 2 | 2 |
| Moderate | 4 | 79 ± 5 | 73–84 | 2 | 2 |
| Severe | 4 | 70 ± 7 | 60–75 | 2 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuevas, E.P.; Rodríguez-Fernández, A.; Palomo, V.; Martínez, A.; Martín-Requero, Á. TDP-43 Pathology and Prionic Behavior in Human Cellular Models of Alzheimer’s Disease Patients. Biomedicines 2022, 10, 385. https://doi.org/10.3390/biomedicines10020385
Cuevas EP, Rodríguez-Fernández A, Palomo V, Martínez A, Martín-Requero Á. TDP-43 Pathology and Prionic Behavior in Human Cellular Models of Alzheimer’s Disease Patients. Biomedicines. 2022; 10(2):385. https://doi.org/10.3390/biomedicines10020385
Chicago/Turabian StyleCuevas, Eva P., Alberto Rodríguez-Fernández, Valle Palomo, Ana Martínez, and Ángeles Martín-Requero. 2022. "TDP-43 Pathology and Prionic Behavior in Human Cellular Models of Alzheimer’s Disease Patients" Biomedicines 10, no. 2: 385. https://doi.org/10.3390/biomedicines10020385
APA StyleCuevas, E. P., Rodríguez-Fernández, A., Palomo, V., Martínez, A., & Martín-Requero, Á. (2022). TDP-43 Pathology and Prionic Behavior in Human Cellular Models of Alzheimer’s Disease Patients. Biomedicines, 10(2), 385. https://doi.org/10.3390/biomedicines10020385