
Gene Therapy Developments for Pompe Disease

 ,
, 
Abstract
:1. Introduction
2. Current Standard of Care for Pompe Disease
Enzyme Replacement Therapy
3. Gene Therapy for Pompe Disease
3.1. AAV Vector-Mediated Gene Therapy in Pompe Disease
3.1.1. Muscle Directed Delivery
3.1.2. Liver Directed AAV Gene Therapy
3.1.3. CNS Directed AAV Gene Therapy
3.2. Lentiviral Mediated HSPC Gene Therapy in Lysosomal Storage Disorders
3.2.1. Lentiviral Vector HSPC Gene Therapy for Pompe Disease Treatment
3.2.2. In Vivo Lentiviral Gene Therapy for Pompe Disease
3.3. Alternative Applications to Modulate GAA Mutations and Disease Correction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van der Ploeg, A.T.; Reuser, A.J. Pompe’s disease. Lancet 2008, 372, 1342–1353. [Google Scholar] [CrossRef]
- Chien, Y.-H.; Hwu, W.-L.; Lee, N.-C. Pompe Disease: Early Diagnosis and Early Treatment Make a Difference. Pediatr. Neonatol. 2013, 54, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebbink, B.J.; Poelman, E.; Plug, I.; Lequin, M.H.; Van Doorn, P.A.; Aarsen, F.K.; Van Der Ploeg, A.T.; Hout, J.M.V.D. Cognitive decline in classic infantile Pompe disease: An underacknowledged challenge. Neurology 2016, 86, 1260–1261. [Google Scholar] [CrossRef] [PubMed]
- Raben, N.; Roberts, A.; Plotz, P.H. Role of autophagy in the pathogenesis of Pompe disease. Acta Myol. 2007, 26, 45–48. [Google Scholar]
- DeRuisseau, L.R.; Fuller, D.D.; Qiu, K.; DeRuisseau, K.C.; Donnelly, W.H.; Mah, C.; Reier, P.J.; Byrne, B.J. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc. Natl. Acad. Sci. USA 2009, 106, 9419–9424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.; Desai, A.K.; Kazi, Z.; Corey, K.; Austin, S.; Hobson-Webb, L.D.; Case, L.E.; Jones, H.N.; Kishnani, P.S. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol. Genet. Metab. 2017, 120, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Nicolino, M.; Byrne, B.; Wraith, J.E.; Leslie, N.; Mandel, H.; Freyer, D.R.; Arnold, G.L.; Pivnick, E.K.; Ottinger, C.; Robinson, P.H.; et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet. Med. 2009, 11, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Raben, N.; Fukuda, T.; Gilbert, A.; de Jong, D.; Thurberg, B.L.; Mattaliano, R.J.; Meikle, P.; Hopwood, J.J.; Nagashima, K.; Nagaraju, K.; et al. Replacing acid α-glucosidase in Pompe disease: Recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. Mol. Ther. 2005, 11, 48–56. [Google Scholar] [CrossRef]
- Ebbink, B.J.; Poelman, E.; Aarsen, F.K.; Plug, I.; Régal, L.; Muentjes, C.; Beek, N.A.M.E.V.D.; Lequin, M.H.; Van Der Ploeg, A.T.; Hout, J.M.P.V.D. Classic infantile Pompe patients approaching adulthood: A cohort study on consequences for the brain. Dev. Med. Child Neurol. 2018, 60, 579–586. [Google Scholar] [CrossRef]
- Burrow, T.A.; Grabowski, G.A. Velaglucerase alfa in the treatment of Gaucher disease type 1. Clin. Investig. 2011, 1, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Do, H.V.; Khanna, R.; Gotschall, R. Challenges in treating Pompe disease: An industry perspective. Ann. Transl. Med. 2019, 7, 291. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.; Porto, C.; Tarallo, A.; Vicinanza, M.; Rossi, B.; Polishchuk, E.; Donaudy, F.; Andria, G.; De Matteis, M.A.; Parenti, G. Abnormal mannose-6-phosphate receptor trafficking impairs recombinant alpha-glucosidase uptake in Pompe disease fibroblasts. Pathogenetics 2008, 1, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreland, R.J.; Higgins, S.; Zhou, A.; VanStraten, P.; Cauthron, R.D.; Brem, M.; McLarty, B.J.; Kudo, M.; Canfield, W.M. Species-specific differences in the processing of acid α-glucosidase are due to the amino acid identity at position 201. Gene 2012, 491, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Wisselaar, H.A.; Kroos, M.A.; Hermans, M.M.; Van Beeumen, J.; Reuser, A.J. Structural and functional changes of lysosomal acid alpha-glucosidase during intracellular transport and maturation. J. Biol. Chem. 1993, 268, 2223–2231. [Google Scholar] [CrossRef]
- Kornfeld, S. Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu. Rev. Biochem. 1992, 61, 307–330. [Google Scholar] [CrossRef]
- Desnick, R.J.; Schuchman, E.H. Enzyme replacement and enhancement therapies: Lessons from lysosomal disorders. Nat. Rev. Genet. 2002, 3, 954–966. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, X.; McVie-Wylie, A.; Jiang, C.; Thurberg, B.L.; Raben, N.; Mattaliano, R.J.; Cheng, S.H. Carbohydrate-remodelled acid α-glucosidase with higher affinity for the cation-independent mannose 6-phosphate receptor demonstrates improved delivery to muscles of Pompe mice. Biochem. J. 2005, 389, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Jiang, J.-L.; Gumlaw, N.K.; Zhang, J.; Bercury, S.D.; Ziegler, R.J.; Lee, K.; Kudo, M.; Canfield, W.M.; Edmunds, T.; et al. Glycoengineered Acid α-Glucosidase with Improved Efficacy at Correcting the Metabolic Aberrations and Motor Function Deficits in a Mouse Model of Pompe Disease. Mol. Ther. 2009, 17, 954–963. [Google Scholar] [CrossRef]
- Diaz-Manera, J.; Kishnani, P.S.; Kushlaf, H.; Ladha, S.; Mozaffar, T.; Straub, V.; Toscano, A.; van der Ploeg, A.T.; Berger, K.I.; Clemens, P.R.; et al. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late-onset Pompe disease (COMET): A phase 3, randomised, multicentre trial. Lancet Neurol. 2021, 20, 1012–1026. [Google Scholar] [CrossRef]
- Parenti, G.; Fecarotta, S.; la Marca, G.; Rossi, B.; Ascione, S.; Donati, M.A.; Morandi, L.O.; Ravaglia, S.; Pichiecchio, A.; Ombrone, D.; et al. A Chaperone Enhances Blood α-Glucosidase Activity in Pompe Disease Patients Treated with Enzyme Replacement Therapy. Mol. Ther. 2014, 22, 2004–2012. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Lun, Y.; Frascella, M.; Garcia, A.; Soska, R.; Nair, A.; Ponery, A.S.; Schilling, A.; Feng, J.; Tuske, S.; et al. Improved efficacy of a next-generation ERT in murine Pompe disease. JCI Insight 2019, 4, e125358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoser, B.; Roberts, M.; Byrne, B.J.; Sitaraman, S.; Jiang, H.; Laforêt, P.; Toscano, A.; Castelli, J.; Díaz-Manera, J.; Goldman, M.; et al. Safety and efficacy of cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo in late-onset Pompe disease (PROPEL): An international, randomised, double-blind, parallel-group, phase 3 trial. Lancet Neurol. 2021, 20, 1027–1037. [Google Scholar] [CrossRef]
- Pennycooke, M.; Chaudary, N.; Shuralyova, I.; Zhang, Y.; Coe, I.R. Differential Expression of Human Nucleoside Transporters in Normal and Tumor Tissue. Biochem. Biophys. Res. Commun. 2001, 280, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Austin, G.L.; Shaffer, R.; Armstrong, D.D.; Gentry, M.S. Antibody-Mediated Enzyme Therapeutics and Applications in Glycogen Storage Diseases. Trends Mol. Med. 2019, 25, 1094–1109. [Google Scholar] [CrossRef]
- Tanaka, S.; Yoshioka, A.; Kinoshita, M.; Kida, S.; Imakiire, A.; Takenaka, S.; Morimoto, H.; Yamamoto, R.; Minami, K.; Sonoda, H.; et al. A novel approach to CNS dysfunctionof Pompe disease with a fusion protein consisting of anti-transferrin receptor antibody and GAA enzyme. Mol. Genet. Metab. 2020, 129, S150–S151. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Goldenberg, P.C.; DeArmey, S.L.; Heller, J.; Benjamin, D.; Young, S.; Bali, D.; Smith, S.A.; Li, J.S.; Mandel, H.; et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol. Genet. Metab. 2010, 99, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Raben, N.; Nagaraju, K.; Lee, A.; Lu, N.; Rivera, Y.; Jatkar, T.; Hopwood, J.J.; Plotz, P.H. Induction of tolerance to a recombinant human enzyme, acid alpha-glucosidase, in enzyme deficient knockout mice. Transgenic Res. 2003, 12, 171–178. [Google Scholar] [CrossRef]
- Costa-Verdera, H.; Collaud, F.; Riling, C.R.; Sellier, P.; Nordin, J.M.L.; Preston, G.M.; Cagin, U.; Fabregue, J.; Barral, S.; Moya-Nilges, M.; et al. Hepatic expression of GAA results in enhanced enzyme bioavailability in mice and non-human primates. Nat. Commun. 2021, 12, 6393. [Google Scholar] [CrossRef]
- Raben, N.; Nagaraju, K.; Lee, E.; Kessler, P.; Byrne, B.; Lee, L.; LaMarca, M.; King, C.; Ward, J.; Sauer, B.; et al. Targeted Disruption of the Acid α-Glucosidase Gene in Mice Causes an Illness with Critical Features of Both Infantile and Adult Human Glycogen Storage Disease Type II. J. Biol. Chem. 1998, 273, 19086–19092. [Google Scholar] [CrossRef] [Green Version]
- Bijvoet, A.G.; Van De Kamp, E.H.; Kroos, M.A.; Ding, J.-H.; Yang, B.Z.; Visser, P.; Bakker, C.E.; Verbeet, M.P.; Oostra, B.A.; Reuser, A.J.; et al. Generalized glycogen storage and cardiomegaly in a knockout mouse model of Pompe disease. Hum. Mol. Genet. 1998, 7, 53–62. [Google Scholar] [CrossRef]
- Amalfitano, A.; McVie-Wylie, A.J.; Hu, H.; Dawson, T.; Raben, N.; Plotz, P.; Chen, Y.T. Systemic correction of the muscle disorder glycogen storage disease type II after hepatic targeting of a modified adenovirus vector encoding human acid-α-glucosidase. Proc. Natl. Acad. Sci. USA 1999, 96, 8861–8866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.-D.; Chen, Y.-T.; Bird, A.; Amalfitano, A.; Koeberl, D.D. Long-term correction of glycogen storage disease type II with a hybrid Ad-AAV vector. Mol. Ther. 2003, 7, 193–201. [Google Scholar] [CrossRef]
- Sun, B.; Chen, Y.-T.; Bird, A.; Xu, F.; Hou, Y.-X.; Amalfitano, A.; Koeberl, D.D. Packaging of an AAV vector encoding human acid α-glucosidase for gene therapy in glycogen storage disease type II with a modified hybrid adenovirus-AAV vector. Mol. Ther. 2003, 7, 467–477. [Google Scholar] [CrossRef]
- Athanasopoulos, T.; Munye, M.M.; Yáñez-Muñoz, R.J. Nonintegrating Gene Therapy Vectors. Hematol. Clin. N. Am. 2017, 31, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, B.; Jayandharan, G.R. Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr. Gene Ther. 2014, 14, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Overcoming the Host Immune Response to Adeno-Associated Virus Gene Delivery Vectors: The Race Between Clearance, Tolerance, Neutralization, and Escape. Annu. Rev. Virol. 2017, 4, 511–534. [Google Scholar] [CrossRef]
- Hamilton, H.; Gomos, J.; Berns, K.I.; Falck-Pedersen, E. Adeno-Associated Virus Site-Specific Integration and AAVS1 Disruption. J. Virol. 2004, 78, 7874–7882. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.H. Adeno-associated virus integration: Virus versus vector. Gene Ther. 2008, 15, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.N.; Everett, J.K.; Kafle, S.; Roche, A.M.; Raymond, H.E.; Leiby, J.; Wood, C.; Assenmacher, C.-A.; Merricks, E.P.; Long, C.T.; et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021, 39, 47–55. [Google Scholar] [CrossRef]
- Fraites, T.J.; Schleissing, M.R.; Shanely, R.; Walter, G.A.; Cloutier, D.A.; Zolotukhin, I.; Pauly, D.F.; Raben, N.; Plotz, P.H.; Powers, S.K.; et al. Correction of the Enzymatic and Functional Deficits in a Model of Pompe Disease Using Adeno-associated Virus Vectors. Mol. Ther. 2002, 5 Pt 1, 571–578. [Google Scholar] [CrossRef]
- Mellies, U.; Stehling, F.; Dohna-Schwake, C.; Ragette, R.; Teschler, H.; Voit, T. Respiratory failure in Pompe disease: Treatment with noninvasive ventilation. Neurology 2005, 64, 1465–1467. [Google Scholar] [CrossRef] [PubMed]
- Menzella, F.; Codeluppi, L.; Lusuardi, M.; Galeone, C.; Valzania, F.; Facciolongo, N. Acute respiratory failure as presentation of late-onset Pompe disease complicating the diagnostic process as a labyrinth: A case report. Multidiscip. Respir. Med. 2018, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Mah, C.; Fraites, T.J.; Cresawn, K.O.; Zolotukhin, I.; Lewis, M.A.; Byrne, B.J. A New Method for Recombinant Adeno-associated Virus Vector Delivery to Murine Diaphragm. Mol. Ther. 2004, 9, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Mah, C.S.; Falk, D.J.; Germain, S.A.; Kelley, J.S.; Lewis, M.A.; Cloutier, D.A.; DeRuisseau, L.; Conlon, T.J.; Cresawn, K.O.; Fraites, T.J., Jr.; et al. Gel-mediated Delivery of AAV1 Vectors Corrects Ventilatory Function in Pompe Mice With Established Disease. Mol. Ther. 2010, 18, 502–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conlon, T.J.; Erger, K.; Porvasnik, S.; Cossette, T.; Roberts, C.; Combee, L.; Islam, S.; Kelley, J.; Cloutier, D.; Clement, N.; et al. Preclinical Toxicology and Biodistribution Studies of Recombinant Adeno-Associated Virus 1 Human Acid α-Glucosidase. Hum. Gene Ther. Clin. Dev. 2013, 24, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Brooks, A.R.; Harkins, R.N.; Wang, P.; Qian, H.S.; Liu, P.; Rubanyi, G.M. Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J. Gene Med. 2004, 6, 395–404. [Google Scholar] [CrossRef]
- Mah, C.; Pacak, C.A.; Cresawn, K.O.; DeRuisseau, L.R.; Germain, S.; Lewis, M.A.; Cloutier, D.A.; Fuller, D.D.; Byrne, B.J. Physiological Correction of Pompe Disease by Systemic Delivery of Adeno-associated Virus Serotype 1 Vectors. Mol. Ther. 2007, 15, 501–507. [Google Scholar] [CrossRef]
- Smith, B.K.; Martin, A.D.; Lawson, L.A.; Vernot, V.; Marcus, J.; Islam, S.; Shafi, N.; Corti, M.; Collins, S.W.; Byrne, B.J. Inspiratory muscle conditioning exercise and diaphragm gene therapy in Pompe disease: Clinical evidence of respiratory plasticity. Exp. Neurol. 2017, 287 Pt 2, 216–224. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Zhang, H.; Franco, L.M.; Brown, T.; Bird, A.; Schneider, A.; Koeberl, D.D. Correction of glycogen storage disease type II by an adeno-associated virus vector containing a muscle-specific promoter. Mol. Ther. 2005, 11, 889–898. [Google Scholar] [CrossRef]
- Falk, D.J.; Soustek, M.S.; Todd, A.G.; Mah, C.S.; Cloutier, D.A.; Kelley, J.S.; Clement, N.; Fuller, D.D.; Byrne, B.J. Comparative impact of AAV and enzyme replacement therapy on respiratory and cardiac function in adult Pompe mice. Mol. Ther.-Methods Clin. Dev. 2015, 2, 15007. [Google Scholar] [CrossRef]
- Paulin, D.; Li, Z. Desmin: A major intermediate filament protein essential for the structural integrity and function of muscle. Exp. Cell Res. 2004, 301, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Corti, M.; Cleaver, B.; Clément, N.; Conlon, T.J.; Faris, K.J.; Wang, G.; Benson, J.; Tarantal, A.F.; Fuller, D.; Herzog, R.W.; et al. Evaluation of Readministration of a Recombinant Adeno-Associated Virus Vector Expressing Acid Alpha-Glucosidase in Pompe Disease: Preclinical to Clinical Planning. Hum. Gene Ther. Clin. Dev. 2015, 26, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggers, M.; Vannoy, C.H.; Huang, J.; Purushothaman, P.; Brassard, J.; Fonck, C.; Meng, H.; Prom, M.J.; Lawlor, M.W.; Cunningham, J.; et al. Muscle-directed gene therapy corrects Pompe disease and uncovers species-specific GAA immunogenicity. EMBO Mol. Med. 2022, 14, e13968. [Google Scholar] [CrossRef]
- Tiegs, G.; Lohse, A.W. Immune tolerance: What is unique about the liver. J. Autoimmun. 2010, 34, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Franco, L.M.; Sun, B.; Yang, X.; Bird, A.; Zhang, H.; Schneider, A.; Brown, T.; Young, S.P.; Clay, T.M.; Amalfitano, A.; et al. Evasion of Immune Responses to Introduced Human Acid α-Glucosidase by Liver-Restricted Expression in Glycogen Storage Disease Type II. Mol. Ther. 2005, 12, 876–884. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, B.; Osada, T.; Rodriguiz, R.; Yang, X.Y.; Luo, X.; Kemper, A.R.; Clay, T.M.; Koeberl, D.D. Immunodominant Liver-Specific Expression Suppresses Transgene-Directed Immune Responses in Murine Pompe Disease. Hum. Gene Ther. 2012, 23, 460–472. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Bird, A.; Young, S.P.; Kishnani, P.S.; Chen, Y.-T.; Koeberl, D.D. Enhanced Response to Enzyme Replacement Therapy in Pompe Disease after the Induction of Immune Tolerance. Am. J. Hum. Genet. 2007, 81, 1042–1049. [Google Scholar] [CrossRef] [Green Version]
- Han, S.-O.; Ronzitti, G.; Arnson, B.; Leborgne, C.; Li, S.; Mingozzi, F.; Koeberl, D. Low-Dose Liver-Targeted Gene Therapy for Pompe Disease Enhances Therapeutic Efficacy of ERT via Immune Tolerance Induction. Mol. Ther.-Methods Clin. Dev. 2017, 4, 126–136. [Google Scholar] [CrossRef]
- Colella, P.; Sellier, P.; Verdera, H.C.; Puzzo, F.; van Wittenberghe, L.; Guerchet, N.; Daniele, N.; Gjata, B.; Marmier, S.; Charles, S.; et al. AAV Gene Transfer with Tandem Promoter Design Prevents Anti-transgene Immunity and Provides Persistent Efficacy in Neonate Pompe Mice. Mol. Ther.-Methods Clin. Dev. 2019, 12, 85–101. [Google Scholar] [CrossRef] [Green Version]
- Puzzo, F.; Colella, P.; Biferi, M.G.; Bali, D.; Paulk, N.K.; Vidal, P.; Collaud, F.; Simon-Sola, M.; Charles, S.; Hardet, R.; et al. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid α-glucosidase. Sci. Transl. Med. 2017, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Zhang, H.; Benjamin, D.K., Jr.; Brown, T.; Bird, A.; Young, S.P.; McVie-Wylie, A.; Chen, Y.-T.; Koeberl, D.D. Enhanced Efficacy of an AAV Vector Encoding Chimeric, Highly Secreted Acid α-Glucosidase in Glycogen Storage Disease Type II. Mol. Ther. 2006, 14, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Cagin, U.; Puzzo, F.; Gómez, M.J.; Moya-Nilges, M.; Sellier, P.; Abad, C.; Van Wittenberghe, L.; Daniele, N.; Guerchet, N.; Gjata, B.; et al. Rescue of Advanced Pompe Disease in Mice with Hepatic Expression of Secretable Acid α-Glucosidase. Mol. Ther. 2020, 28, 2056–2072. [Google Scholar] [CrossRef] [PubMed]
- Ronzitti, G.; Bortolussi, G.; van Dijk, R.; Collaud, F.; Charles, S.; Leborgne, C.; Vidal, P.; Martin, S.; Gjata, B.; Sola, M.S.; et al. A translationally optimized AAV-UGT1A1 vector drives safe and long-lasting correction of Crigler-Najjar syndrome. Mol. Ther.-Methods Clin. Dev. 2016, 3, 16049. [Google Scholar] [CrossRef] [PubMed]
- Baik, A.D.; Calafati, P.; Zhang, X.; Aaron, N.A.; Mehra, A.; Moller-Tank, S.; Miloscio, L.; Praggastis, M.; Giovannone, N.; Pan, C.; et al. Cell type-selective targeted delivery of a recombinant lysosomal enzyme for enzyme therapies. Mol. Ther. 2021, 29, 3512–3524. [Google Scholar] [CrossRef] [PubMed]
- Byrne, B.J.; Fuller, D.D.; Smith, B.K.; Clement, N.; Coleman, K.; Cleaver, B.; Vaught, L.; Falk, D.J.; McCall, A.; Corti, M. Pompe disease gene therapy: Neural manifestations require consideration of CNS directed therapy. Ann. Transl. Med. 2019, 7, 290. [Google Scholar] [CrossRef]
- Broomfield, A.; Fletcher, J.; Hensman, P.; Wright, R.; Prunty, H.; Pavaine, J.; Jones, S.A. Rapidly Progressive White Matter Involvement in Early Childhood: The Expanding Phenotype of Infantile Onset Pompe? JIMD Rep. 2017, 39, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Qiu, K.; Falk, D.J.; Reier, P.J.; Byrne, B.J.; Fuller, D.D. Spinal Delivery of AAV Vector Restores Enzyme Activity and Increases Ventilation in Pompe Mice. Mol. Ther. 2012, 20, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2008, 27, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Hordeaux, J.; Wang, Q.; Katz, N.; Buza, E.L.; Bell, P.; Wilson, J.M. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol. Ther. 2018, 26, 664–668. [Google Scholar] [CrossRef]
- Al-Zaidy, S.; Pickard, A.S.; Kotha, K.; Alfano, L.N.; Lowes, L.; Paul, G.; Church, K.; Lehman, K.; Sproule, D.M.; Dabbous, O.; et al. Health outcomes in spinal muscular atrophy type 1 following AVXS-101 gene replacement therapy. Pediatr. Pulmonol. 2019, 54, 179–185. [Google Scholar] [CrossRef]
- Lowes, L.P.; Alfano, L.N.; Arnold, W.D.; Shell, R.; Prior, T.W.; McColly, M.; Lehman, K.J.; Church, K.; Sproule, D.M.; Nagendran, S.; et al. Impact of Age and Motor Function in a Phase 1/2A Study of Infants With SMA Type 1 Receiving Single-Dose Gene Replacement Therapy. Pediatr. Neurol. 2019, 98, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hordeaux, J.; Dubreil, L.; Robveille, C.; Deniaud, J.; Pascal, Q.; Dequéant, B.; Pailloux, J.; Lagalice, L.; Ledevin, M.; Babarit, C.; et al. Long-term neurologic and cardiac correction by intrathecal gene therapy in Pompe disease. Acta Neuropathol. Commun. 2017, 5, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.C.; Hwu, W.L.; Muramatsu, S.I.; Falk, D.J.; Byrne, B.J.; Cheng, C.H.; Shih, N.C.; Chang, K.L.; Tsai, L.K.; Chien, Y.H. A Neuron-Specific Gene Therapy Relieves Motor Deficits in Pompe Disease Mice. Mol. Neurobiol. 2017, 55, 5299–5309. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Fitzpatrick, Z.; Harris, A.F.; Maitland, S.A.; Ferreira, J.S.; Zhang, Y.; Ma, S.; Sharma, R.B.; Gray-Edwards, H.L.; Johnson, J.A.; et al. In Vivo Selection Yields AAV-B1 Capsid for Central Nervous System and Muscle Gene Therapy. Mol. Ther. 2016, 24, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Keeler, A.M.; Zieger, M.; Todeasa, S.H.; McCall, A.L.; Gifford, J.C.; Birsak, S.; Choudhury, S.R.; Byrne, B.J.; Sena-Esteves, M.; Elmallah, M.K. Systemic Delivery of AAVB1-GAA Clears Glycogen and Prolongs Survival in a Mouse Model of Pompe Disease. Hum. Gene Ther. 2019, 30, 57–68. [Google Scholar] [CrossRef]
- ElMallah, M.K.; Falk, D.J.; Nayak, S.; Federico, R.A.; Sandhu, M.S.; Poirier, A.; Byrne, B.J.; Fuller, D.D. Sustained Correction of Motoneuron Histopathology Following Intramuscular Delivery of AAV in Pompe Mice. Mol. Ther. 2014, 22, 702–712. [Google Scholar] [CrossRef]
- Maga, J.A.; Zhou, J.; Kambampati, R.; Peng, S.; Wang, X.; Bohnsack, R.N.; Thomm, A.; Golata, S.; Tom, P.; Dahms, N.M.; et al. Glycosylation-independent Lysosomal Targeting of Acid α-Glucosidase Enhances Muscle Glycogen Clearance in Pompe Mice. J. Biol. Chem. 2013, 288, 1428–1438. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Dalton, J.; Butt, M.; Tracy, K.; Kennedy, D.; Haroldsen, P.; Cahayag, R.; Zoog, S.; O’Neill, C.A.; Tsuruda, L.S. Reveglucosidase alfa (BMN 701), an IGF2-Tagged rhAcid α-Glucosidase, Improves Respiratory Functional Parameters in a Murine Model of Pompe Disease. J. Pharmacol. Exp. Ther. 2016, 360, 313–323. [Google Scholar] [CrossRef]
- LeBowitz, J.H.; Grubb, J.H.; Maga, J.A.; Schmiel, D.H.; Vogler, C.; Sly, W.S. Glycosylation-independent targeting enhances enzyme delivery to lysosomes and decreases storage in mucopolysaccharidosis type VII mice. Proc. Natl. Acad. Sci. USA 2004, 101, 3083–3088. [Google Scholar] [CrossRef] [Green Version]
- Byrne, B.J.; Geberhiwot, T.; Barshop, B.A.; Barohn, R.; Hughes, D.; Bratkovic, D.; Desnuelle, C.; Laforet, P.; Mengel, E.; Roberts, M.; et al. A study on the safety and efficacy of reveglucosidase alfa in patients with late-onset Pompe disease. Orphanet J. Rare Dis. 2017, 12, 144. [Google Scholar] [CrossRef]
- Doyle, B.M.; Turner, S.M.; Sunshine, M.D.; Doerfler, P.A.; Poirier, A.E.; Vaught, L.A.; Jorgensen, M.L.; Falk, D.J.; Byrne, B.J.; Fuller, D.D. AAV Gene Therapy Utilizing Glycosylation-Independent Lysosomal Targeting Tagged GAA in the Hypoglossal Motor System of Pompe Mice. Mol. Ther.-Methods Clin. Dev. 2019, 15, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Tuske, S.; Yu, T.; Hordeaux, J.; Hung, F.; Feng, J.; Gotschall, R.; Willer, T.; Liu, C.F.; Krampetz, R.; Hoang, Q.Q.; et al. Development of a Novel Gene Therapy for Pompe Disease: Engineered Acid Alpha-Glucosidase Transgene for Improved Expression and Muscle Targeting. In Proceedings of the 22nd Annual Meeting of the American Society of Gene and Cell Therapy, Washington, DC, USA, 29 April–2 May 2019; Elsevier: Amsterdam, The Netherlands, 2019; p. 243. [Google Scholar]
- Hordeaux, J.; Tuske, S.; Yu, T.; So, M.; Tsai, P.; Bell, P.; Do, H.; Wilson, J.M. Combined CNS and Systemic Directed Gene Therapy in a Mouse Model of Pompe Disease with Advanced Disease at Treatment. In Proceedings of the 23rd Annual Meeting of the American Society for Gene and Cell Therapy, Boston, MA, USA, 12 May 2020; Elsevier: Amsterdam, The Netherlands, 2020; p. 401. [Google Scholar]
- Ramezani, A.; Hordeaux, J.; Tuske, S.; Song, C.; Mehta, N.; Tsai, P.; Weimer, J.; So, M.; Bell, P.; Do, H.; et al. Post-Symptomatic Reversal of Muscle Pathology in a Model of Pompe Disease Using Gene Therapy. In Proceedings of the 24th Annual Meeting of the American Society of Gene & Cell Therapy, Virtual. 11–14 May 2021; Elsevier: Amsterdam, The Netherlands, 2021; p. 253. [Google Scholar]
- Kevany, B.M.; Padegimas, L.; Miller, T.J. A novel AAV capsid with improved CNS tropism for treating Pompe disease by intravenous administration. Mol. Genet. Metab. 2019, 126, S83. [Google Scholar] [CrossRef]
- Flotte, T.R. Revisiting the “New” Inflammatory Toxicities of Adeno-Associated Virus Vectors. Hum. Gene Ther. 2020, 31, 398–399. [Google Scholar] [CrossRef]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Shieh, P.B.; Bönnemann, C.G.; Müller-Felber, W.; Blaschek, A.; Dowling, J.J.; Kuntz, N.L.; Seferian, A.M. Re: “Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy” by Wilson and Flotte. Hum. Gene Ther. 2020, 31, 787. [Google Scholar] [CrossRef]
- Calcedo, R.; Morizono, H.; Wang, L.; McCarter, R.; He, J.; Jones, D.; Batshaw, M.L.; Wilson, J.M. Adeno-Associated Virus Antibody Profiles in Newborns, Children, and Adolescents. Clin. Vaccine Immunol. 2011, 18, 1586–1588. [Google Scholar] [CrossRef] [Green Version]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of Serum IgG and Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 5, 6, 8, and 9 in the Healthy Population: Implications for Gene Therapy Using AAV Vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef]
- Calcedo, R.; Vandenberghe, L.H.; Gao, G.; Lin, J.; Wilson, J.M. Worldwide Epidemiology of Neutralizing Antibodies to Adeno-Associated Viruses. J. Infect. Dis. 2009, 199, 381–390. [Google Scholar] [CrossRef]
- Hamilton, B.A.; Wright, J.F. Challenges Posed by Immune Responses to AAV Vectors: Addressing Root Causes. Front. Immunol. 2021, 12, 675897. [Google Scholar] [CrossRef]
- Zhu, J.; Huang, X.; Yang, Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J. Clin. Investig. 2009, 119, 2388–2398. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, T.B.; Shirley, J.L.; Zolotukhin, I.; Li, X.; Kaisho, T.; Xiao, W.; Kumar, S.R.P.; Herzog, R.W. Effect of CpG Depletion of Vector Genome on CD8+ T Cell Responses in AAV Gene Therapy. Front. Immunol. 2021, 12, 672449. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.K.; Wang, S.K.; Chu, C.J.; Copland, D.A.; Letizia, A.J.; Verdera, H.C.; Chiang, J.J.; Sethi, M.; Wang, M.K.; Neidermyer, W.J.; et al. Engineering adeno-associated viral vectors to evade innate immune and inflammatory responses. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Braun, C.J.; Witzel, M.; Paruzynski, A.; Boztug, K.; Von Kalle, C.; Schmidt, M.; Klein, C. Gene therapy for Wiskott-Aldrich Syndrome-Long-term reconstitution and clinical benefits, but increased risk for leukemogenesis. Rare Dis. 2014, 2, e947749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, C.J.; Boztug, K.; Paruzynski, A.; Witzel, M.; Schwarzer, A.; Rothe, M.; Modlich, U.; Beier, R.; Göhring, G.; Steinemann, D.; et al. Gene Therapy for Wiskott-Aldrich Syndrome—Long-Term Efficacy and Genotoxicity. Sci. Transl. Med. 2014, 6, 227ra33. [Google Scholar] [CrossRef]
- Stirnadel-Farrant, H.; Kudari, M.; Garman, N.; Imrie, J.; Chopra, B.; Giannelli, S.; Gabaldo, M.; Corti, A.; Zancan, S.; Aiuti, A.; et al. Gene therapy in rare diseases: The benefits and challenges of developing a patient-centric registry for Strimvelis in ADA-SCID. Orphanet J. Rare Dis. 2018, 13, 49. [Google Scholar] [CrossRef]
- Aiuti, A.; Cattaneo, F.; Galimberti, S.; Benninghoff, U.; Cassani, B.; Callegaro, L.; Scaramuzza, S.; Andolfi, G.; Mirolo, M.; Brigida, I.; et al. Gene Therapy for Immunodeficiency Due to Adenosine Deaminase Deficiency. N. Engl. J. Med. 2009, 360, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, H.B.; Cooray, S.; Gilmour, K.C.; Parsley, K.L.; Zhang, F.; Adams, S.; Bjorkegren, E.; Bayford, J.; Brown, L.; Davies, E.G.; et al. Hematopoietic Stem Cell Gene Therapy for Adenosine Deaminase–Deficient Severe Combined Immunodeficiency Leads to Long-Term Immunological Recovery and Metabolic Correction. Sci. Transl. Med. 2011, 3, 97ra80. [Google Scholar] [CrossRef]
- Cicalese, M.P.; Ferrua, F.; Castagnaro, L.; Pajno, R.; Barzaghi, F.; Giannelli, S.; Dionisio, F.; Brigida, I.; Bonopane, M.; Casiraghi, M.; et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood 2016, 128, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In Vivo Gene Delivery and Stable Transduction of Nondividing Cells by a Lentiviral Vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Zielske, S.P.; Gerson, S.L. Cytokines, including stem cell factor alone, enhance lentiviral transduction in nondividing human LTCIC and NOD/SCID repopulating cells. Mol. Ther. 2003, 7, 325–333. [Google Scholar] [CrossRef]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Maele, B.; De Rijck, J.; De Clercq, E.; Debyser, Z. Impact of the Central Polypurine Tract on the Kinetics of Human Immunodeficiency Virus Type 1 Vector Transduction. J. Virol. 2003, 77, 4685–4694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zufferey, R.; Donello, J.E.; Trono, D.; Hope, T.J. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J. Virol. 1999, 73, 2886–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkina, R.K.; Walton, R.M.; Chen, M.L.; Li, Q.X.; Planelles, V.; Chen, I.S. High-efficiency gene transfer into CD34+ cells with a human immunodeficiency virus type 1-based retroviral vector pseudotyped with vesicular stomatitis virus envelope glycoprotein G. J. Virol. 1996, 70, 2581–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, J.C.; Friedmann, T.; Driever, W.; Burrascano, M.; Yee, J.K. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: Concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. USA 1993, 90, 8033–8037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- Khan, A.; Barber, D.L.; Huang, J.; Rupar, C.A.; Rip, J.W.; Auray-Blais, C.; Boutin, M.; O’Hoski, P.; Gargulak, K.; McKillop, W.M.; et al. Lentivirus-mediated gene therapy for Fabry disease. Nat. Commun. 2021, 12, 1178. [Google Scholar] [CrossRef]
- Gentner, B.; Tucci, F.; Galimberti, S.; Fumagalli, F.; De Pellegrin, M.; Silvani, P.; Camesasca, C.; Pontesilli, S.; Darin, S.; Ciotti, F.; et al. Hematopoietic Stem- and Progenitor-Cell Gene Therapy for Hurler Syndrome. N. Engl. J. Med. 2021, 385, 1929–1940. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic Stem Cell Gene Therapy with a Lentiviral Vector in X-Linked Adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bougnères, P.; Hacein-Bey-Abina, S.; Labik, I.; Adamsbaum, C.; Castaignède, C.; Bellesme, C.; Schmidt, M. Long-Term Follow-Up of Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. Hum. Gene Ther. 2021, 32, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Eichler, F.; Duncan, C.; Musolino, P.L.; Orchard, P.J.; De Oliveira, S.; Thrasher, A.; Armant, M.; Dansereau, C.; Lund, T.C.; Miller, W.P.; et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017, 377, 1630–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loza, L.I.M.; Yuen, E.C.; McCray, J.P.B. Lentiviral Vectors for the Treatment and Prevention of Cystic Fibrosis Lung Disease. Genes 2019, 10, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saif, M.A.; Bigger, B.W.; Brookes, K.E.; Mercer, J.; Tylee, K.L.; Church, H.J.; Bonney, D.K.; Jones, S.; Wraith, J.E.; Wynn, R.F. Hematopoietic stem cell transplantation improves the high incidence of neutralizing allo-antibodies observed in Hurler’s syndrome after pharmacological enzyme replacement therapy. Haematologica 2012, 97, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Eglitis, M.A.; Mezey, É. Hematopoietic cells differentiate into both microglia and macroglia in the brains of adult mice. Proc. Natl. Acad. Sci. USA 1997, 94, 4080–4085. [Google Scholar] [CrossRef] [Green Version]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [Green Version]
- Biffi, A.; Capotondo, A.; Fasano, S.; Del Carro, U.; Marchesini, S.; Azuma, H.; Malaguti, M.C.; Amadio, S.; Brambilla, R.; Grompe, M.; et al. Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J. Clin. Investig. 2006, 116, 3070–3082. [Google Scholar] [CrossRef]
- Wolf, N.I.; Breur, M.; Plug, B.; Beerepoot, S.; Westerveld, A.S.R.; Van Rappard, D.F.; De Vries, S.I.; Kole, M.H.P.; Vanderver, A.; Van Der Knaap, M.S.; et al. Metachromatic leukodystrophy and transplantation: Remyelination, no cross-correction. Ann. Clin. Transl. Neurol. 2020, 7, 169–180. [Google Scholar] [CrossRef]
- Douillard-Guilloux, G.; Richard, E.; Batista, L.; Caillaud, C. Partial phenotypic correction and immune tolerance induction to enzyme replacement therapy after hematopoietic stem cell gene transfer of α-glucosidase in Pompe disease. J. Gene Med. 2009, 11, 279–287. [Google Scholar] [CrossRef]
- Van Til, N.P.; Stok, M.; Kaya, F.S.F.A.; De Waard, M.C.; Farahbakhshian, E.; Visser, T.P.; Kroos, M.A.; Jacobs, E.H.; Willart, M.A.; Van Der Wegen, P.; et al. Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype. Blood 2010, 115, 5329–5337. [Google Scholar] [CrossRef] [PubMed]
- Stok, M.; de Boer, H.; Huston, M.W.; Jacobs, E.H.; Roovers, O.; Visser, T.P.; Jahr, H.; Duncker, D.J.; van Deel, E.D.; Reuser, A.J.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Corrects Murine Pompe Disease. Mol. Ther.-Methods Clin. Dev. 2020, 17, 1014–1025. [Google Scholar] [CrossRef]
- Bigger, B.; Siapati, E.K.; Mistry, A.; Waddington, S.; Nivsarkar, M.S.; Jacobs, L.; Perrett, R.; Holder, M.; Ridler, C.; Kemball-Cook, G.; et al. Permanent partial phenotypic correction and tolerance in a mouse model of hemophilia B by stem cell gene delivery of human factor IX. Gene Ther. 2005, 13, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Gleitz, H.F.; Liao, A.Y.; Cook, J.R.; Rowlston, S.F.; Forte, G.M.; D’Souza, Z.; O’Leary, C.; Holley, R.J.; Bigger, B.W. Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. EMBO Mol. Med. 2018, 10, e8730. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Stepto, H.; Schneider, C.K. Development of the First World Health Organization Lentiviral Vector Standard: Toward the Production Control and Standardization of Lentivirus-Based Gene Therapy Products. Hum. Gene Ther. Methods 2017, 28, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.B.; Bjorkegren, E.; Parsley, K.; Gilmour, K.; King, D.; Sinclair, J.; Zhang, F.; Giannakopoulos, A.; Adams, S.; Fairbanks, L.D.; et al. Successful Reconstitution of Immunity in ADA-SCID by Stem Cell Gene Therapy Following Cessation of PEG-ADA and Use of Mild Preconditioning. Mol. Ther. 2006, 14, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Equihua, C.A.; Zhang, L.; Knight, S.; Saadeh, H.; Scholz, S.; Carmo, M.; Alonso-Ferrero, M.E.; Blundell, M.P.; Monkeviciute, A.; Schulz, R.; et al. The β-globin locus control region in combination with the EF1alpha short promoter allows enhanced lentiviral vector-mediated erythroid gene expression with conserved multilineage activity. Mol. Ther. 2012, 20, 1400–1409. [Google Scholar] [CrossRef] [Green Version]
- Piras, G.; Montiel-Equihua, C.; Chan, Y.-K.A.; Wantuch, S.; Stuckey, D.; Burke, D.; Prunty, H.; Phadke, R.; Chambers, D.; Partida-Gaytan, A.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Rescues Clinical Phenotypes in a Murine Model of Pompe Disease. Mol. Ther.-Methods Clin. Dev. 2020, 18, 558–570. [Google Scholar] [CrossRef]
- Liang, Q.; Stok, M.; van Helsdingen, Y.; van der Velden, G.; Jacobs, E.; Duncker, D.; Reuser, A.; van der Ploeg, A.; Vulto, A.; van Til, N.P.; et al. Lentiviral Stem Cell Gene Therapy for Pompe Disease. J. Neuromuscul. Dis. 2015, 2, S64. [Google Scholar] [CrossRef] [Green Version]
- Dogan, Y.; Barese, C.N.; Schindler, J.W.; Yoon, J.K.; Unnisa, Z.; Guda, S.; Jacobs, M.E.; Oborski, C.; Clarke, D.L.; Schambach, A.; et al. Screening of Chimeric GAA Variants in a Preclinical Study of Pompe Disease Results in Candidate Vector for Hematopoietic Stem Cell Gene Therapy. bioRxiv 2021, preprint. [Google Scholar] [CrossRef]
- Kan, S.-H.; Aoyagi-Scharber, M.; Le, S.Q.; Vincelette, J.; Ohmi, K.; Bullens, S.; Wendt, D.J.; Christianson, T.M.; Tiger, P.M.N.; Brown, J.R.; et al. Delivery of an enzyme-IGFII fusion protein to the mouse brain is therapeutic for mucopolysaccharidosis type IIIB. Proc. Natl. Acad. Sci. USA 2014, 111, 14870–14875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capotondo, A.; Milazzo, R.; Politi, L.S.; Quattrini, A.; Palini, A.; Plati, T.; Merella, S.; Nonis, A.; DI Serio, M.S.; Montini, E.; et al. Brain conditioning is instrumental for successful microglia reconstitution following hematopoietic stem cell transplantation. Proc. Natl. Acad. Sci. USA 2012, 109, 15018–15023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittelbronn, M.; Dietz, K.; Schluesener, H.J.; Meyermann, R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001, 101, 249–255. [Google Scholar] [CrossRef]
- Visigalli, I.; Delai, S.; Politi, L.S.; Di Domenico, C.; Cerri, F.; Mrak, E.; D’Isa, R.; Ungaro, D.; Stok, M.; Sanvito, F.; et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood 2010, 116, 5130–5139. [Google Scholar] [CrossRef]
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of Colony Stimulation Factor-1 Receptor Results in Loss of Microglia, Disrupted Brain Development and Olfactory Deficits. PLoS ONE 2011, 6, e26317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Xu, Z.; Xiong, S.; Sun, F.; Qin, G.; Hu, G.; Wang, J.; Zhao, L.; Liang, Y.-X.; Wu, T.; et al. Author Correction: Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat. Neurosci. 2021, 24, 288. [Google Scholar] [CrossRef]
- Xu, Z.; Rao, Y.; Huang, Y.; Zhou, T.; Feng, R.; Xiong, S.; Yuan, T.-F.; Qin, S.; Lu, Y.; Zhou, X.; et al. Efficient Strategies for Microglia Replacement in the Central Nervous System. Cell Rep. 2020, 32, 108041. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [Green Version]
- Giugliani, R.; Giugliani, L.; de Oliveira Poswar, F.; Donis, K.C.; Corte, A.D.; Schmidt, M.; Boado, R.J.; Nestrasil, I.; Nguyen, C.; Chen, S.; et al. Neurocognitive and somatic stabilization in pediatric patients with severe Mucopolysaccharidosis Type I after 52 weeks of intravenous brain-penetrating insulin receptor antibody-iduronidase fusion protein (valanafusp alpha): An open label phase 1-2 trial. Orphanet J. Rare Dis. 2018, 13, 110. [Google Scholar] [CrossRef]
- Ponder, K.P. Immunology of neonatal gene transfer. Curr. Gene Ther. 2007, 7, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Kyosen, S.O.; Iizuka, S.; Kobayashi, H.; Kimura, T.; Fukuda, T.; Shen, J.; Shimada, Y.; Ida, H.; Eto, Y.; Ohashi, T. Neonatal gene transfer using lentiviral vector for murine Pompe disease: Long-term expression and glycogen reduction. Gene Ther. 2009, 17, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantore, A.; Ranzani, M.; Bartholomae, C.C.; Volpin, M.; Della Valle, P.; Sanvito, F.; Sergi, L.S.; Gallina, P.; Benedicenti, F.; Bellinger, D.; et al. Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci. Transl. Med. 2015, 7, 277ra28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani, M.; Annoni, A.; Moalli, F.; Liu, T.; Cesana, D.; Calabria, A.; Bartolaccini, S.; Biffi, M.; Russo, F.; Visigalli, I.; et al. Phagocytosis-shielded lentiviral vectors improve liver gene therapy in nonhuman primates. Sci. Transl. Med. 2019, 11, eaav7325. [Google Scholar] [CrossRef]
- Meneghini, V.; Lattanzi, A.; Tiradani, L.; Bravo, G.; Morena, F.; Sanvito, F.; Calabria, A.; Bringas, J.; Fisher-Perkins, J.M.; Dufour, J.P.; et al. Pervasive supply of therapeutic lysosomal enzymes in the CNS of normal and Krabbe-affected non-human primates by intracerebral lentiviral gene therapy. EMBO Mol. Med. 2016, 8, 489–510. [Google Scholar] [CrossRef]
- Bergsma, A.J.; In ’t Groen, S.L.; Verheijen, F.W.; van der Ploeg, A.T.; Pijnappel, W. From Cryptic Toward Canonical Pre-mRNA Splicing in Pompe Disease: A Pipeline for the Development of Antisense Oligonucleotides. Mol. Ther. Nucleic Acids 2016, 5, e361. [Google Scholar] [CrossRef]
- van der Wal, E.; Bergsma, A.J.; Pijnenburg, J.M.; van der Ploeg, A.T.; Pijnappel, W.P. Antisense Oligonucleotides Promote Exon Inclusion and Correct the Common c.-32-13T>G GAA Splicing Variant in Pompe Disease. Mol. Ther.-Nucleic Acids 2017, 7, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Haché, M.; Swoboda, K.J.; Sethna, N.; Farrow-Gillespie, A.; Khandji, A.; Xia, S.; Bishop, K.M. Intrathecal Injections in Children With Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. J. Child Neurol. 2016, 31, 899–906. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E.; Eteplirsen Study Group and Telethon Foundation DMD Italian Network. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef]
- Schaaf, G.J.; van Gestel, T.J.; Brusse, E.; Verdijk, R.M.; de Coo, I.F.; van Doorn, P.A.; van der Ploeg, A.T.; Pijnappel, W.P. Lack of robust satellite cell activation and muscle regeneration during the progression of Pompe disease. Acta Neuropathol. Commun. 2015, 3, 65. [Google Scholar] [CrossRef] [Green Version]
- Schaaf, G.J.; Canibano-Fraile, R.; Van Gestel, T.J.M.; Van Der Ploeg, A.T.; Pijnappel, W.W.M.P. Restoring the regenerative balance in neuromuscular disorders: Satellite cell activation as therapeutic target in Pompe disease. Ann. Transl. Med. 2019, 7, 280. [Google Scholar] [CrossRef]
- Lenhare, L.; Crisol, B.M.; Silva, V.R.R.; Katashima, C.K.; Cordeiro, A.V.; Pereira, K.; Luchessi, A.; da Silva, A.S.R.; Cintra, D.E.; Moura, L.P.; et al. Physical exercise increases Sestrin 2 protein levels and induces autophagy in the skeletal muscle of old mice. Exp. Gerontol. 2017, 97, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwalm, C.; Jamart, C.; Benoit, N.; Naslain, D.; Prémont, C.; Prévet, J.; Van Thienen, R.; Deldicque, L.; Francaux, M. Activation of autophagy in human skeletal muscle is dependent on exercise intensity and AMPK activation. FASEB J. 2015, 29, 3515–3526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellas, N.; Liu, J.; Botham, R.C.; Huisman, G.W. Adapting protein sequences for optimized therapeutic efficacy. Curr. Opin. Chem. Biol. 2021, 64, 38–47. [Google Scholar] [CrossRef]
- Botham, R.C.; Hallows, W.C.; Reddy, C.; Dellas, N.; Homan, D.; Lao, J.; Zhu, C.; Chng, C.; Sero, A.; Miller, M.; et al. Engineering α-glucosidase to improve protein stability and cellular uptake for the potential treatment of Pompe disease. Mol. Genet. Metab. 2021, 132, S20–S21. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Villiger, L.; Grisch-Chan, H.M.; Lindsay, H.; Ringnalda, F.; Pogliano, C.B.; Allegri, G.; Fingerhut, R.; Häberle, J.; Matos, J.; Robinson, M.D.; et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018, 24, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Y.; Kan, S.-H.; Sandfeld, E.K.; Dalton, N.D.; Rangel, A.D.; Chan, Y.; Davis-Turak, J.; Neumann, J.; Wang, R. CRISPR-Cas9 generated Pompe knock-in murine model exhibits early-onset hypertrophic cardiomyopathy and skeletal muscle weakness. Sci. Rep. 2020, 10, 10321. [Google Scholar] [CrossRef]


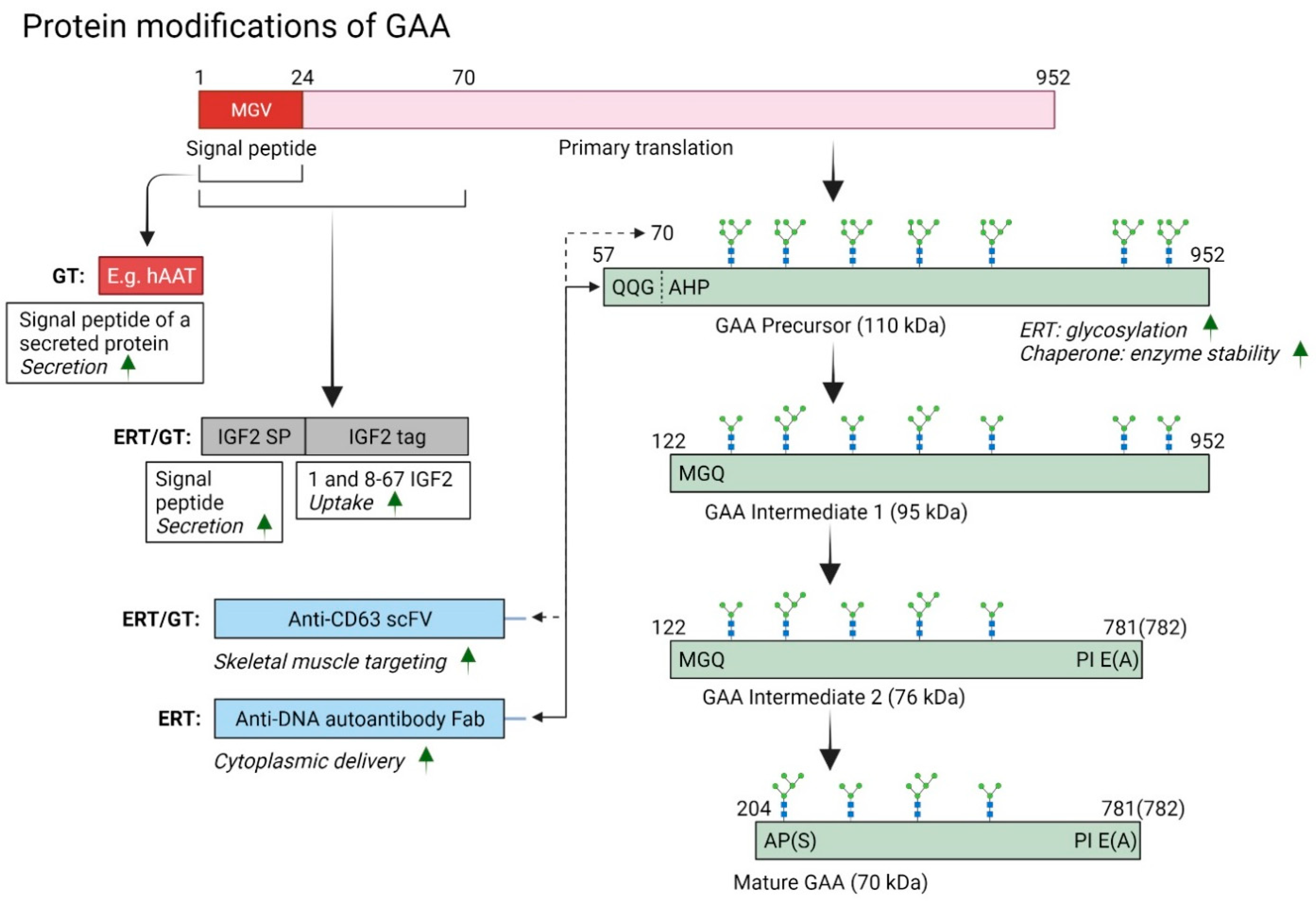

{kind=link}
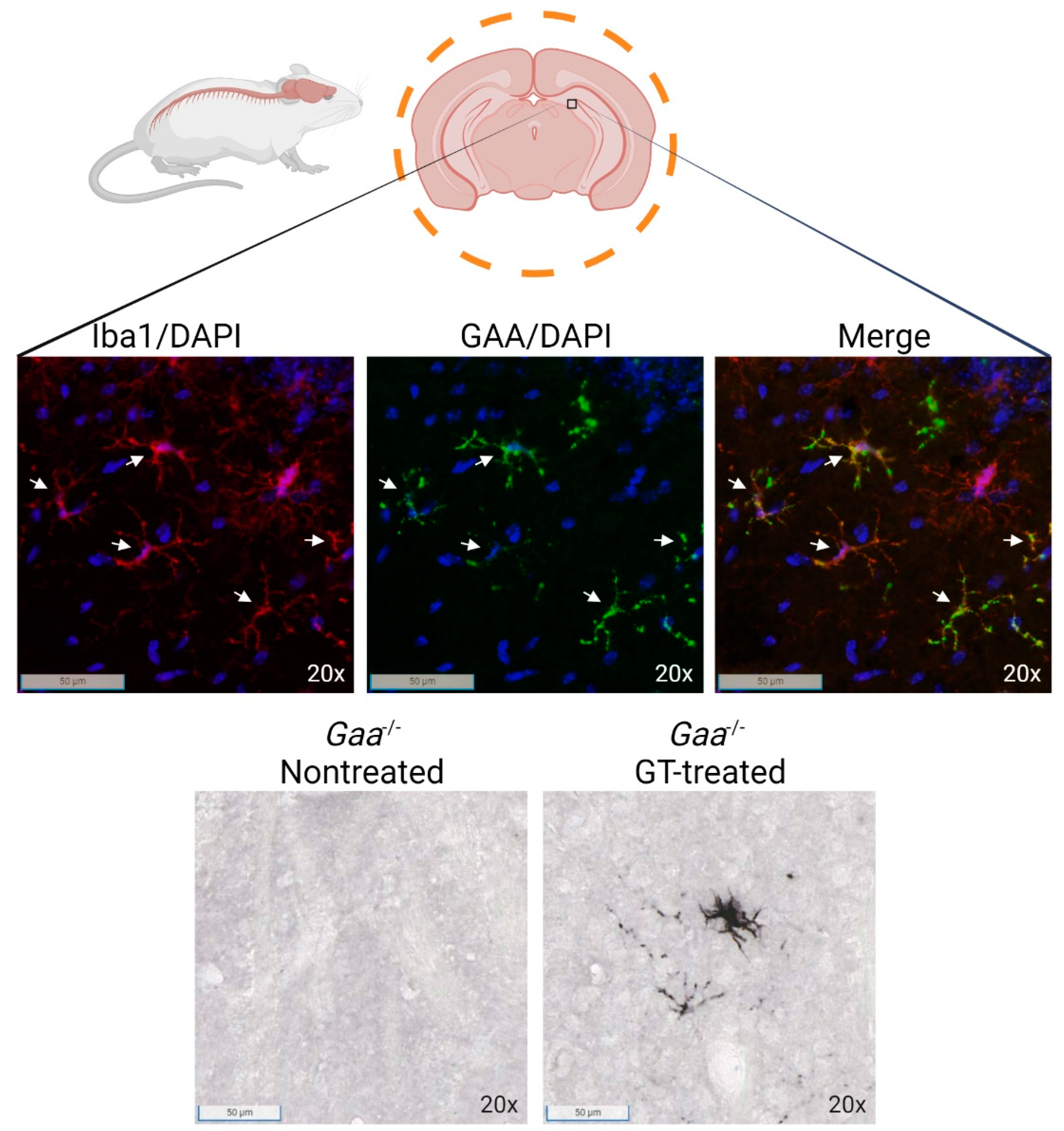
{kind=link}
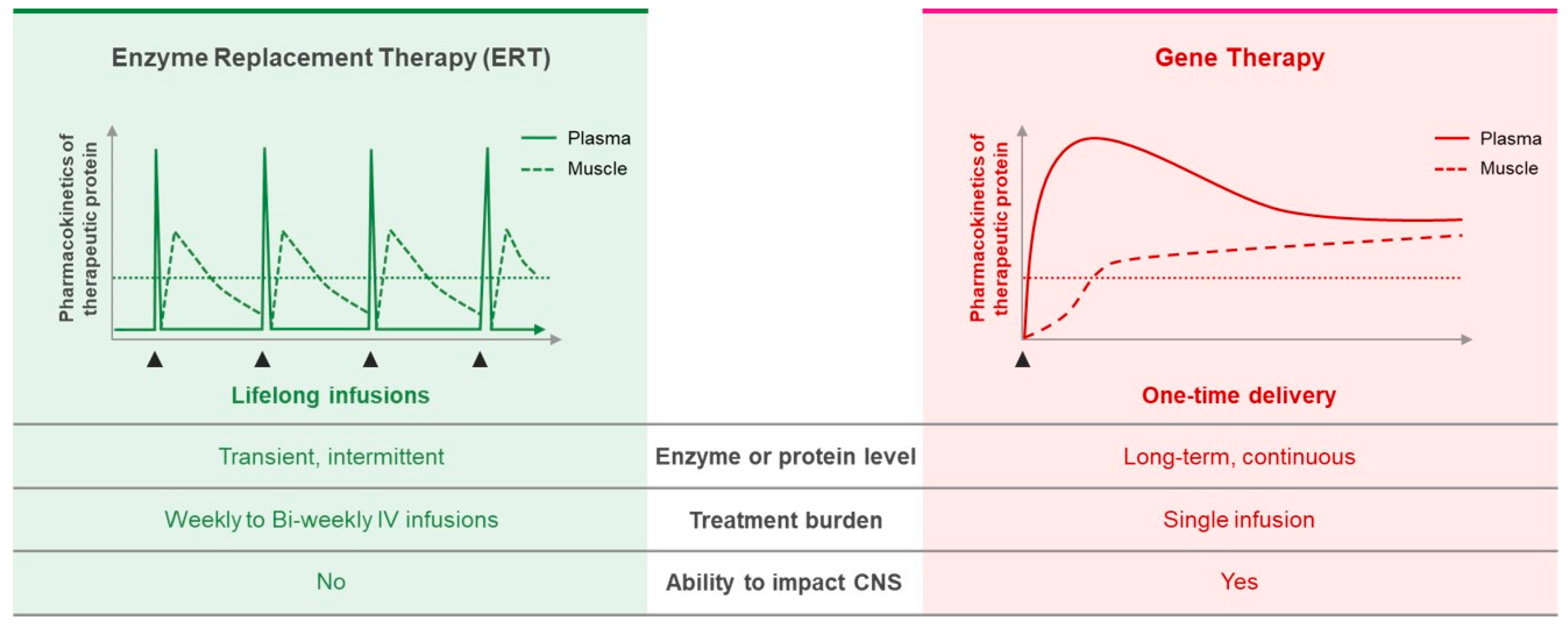
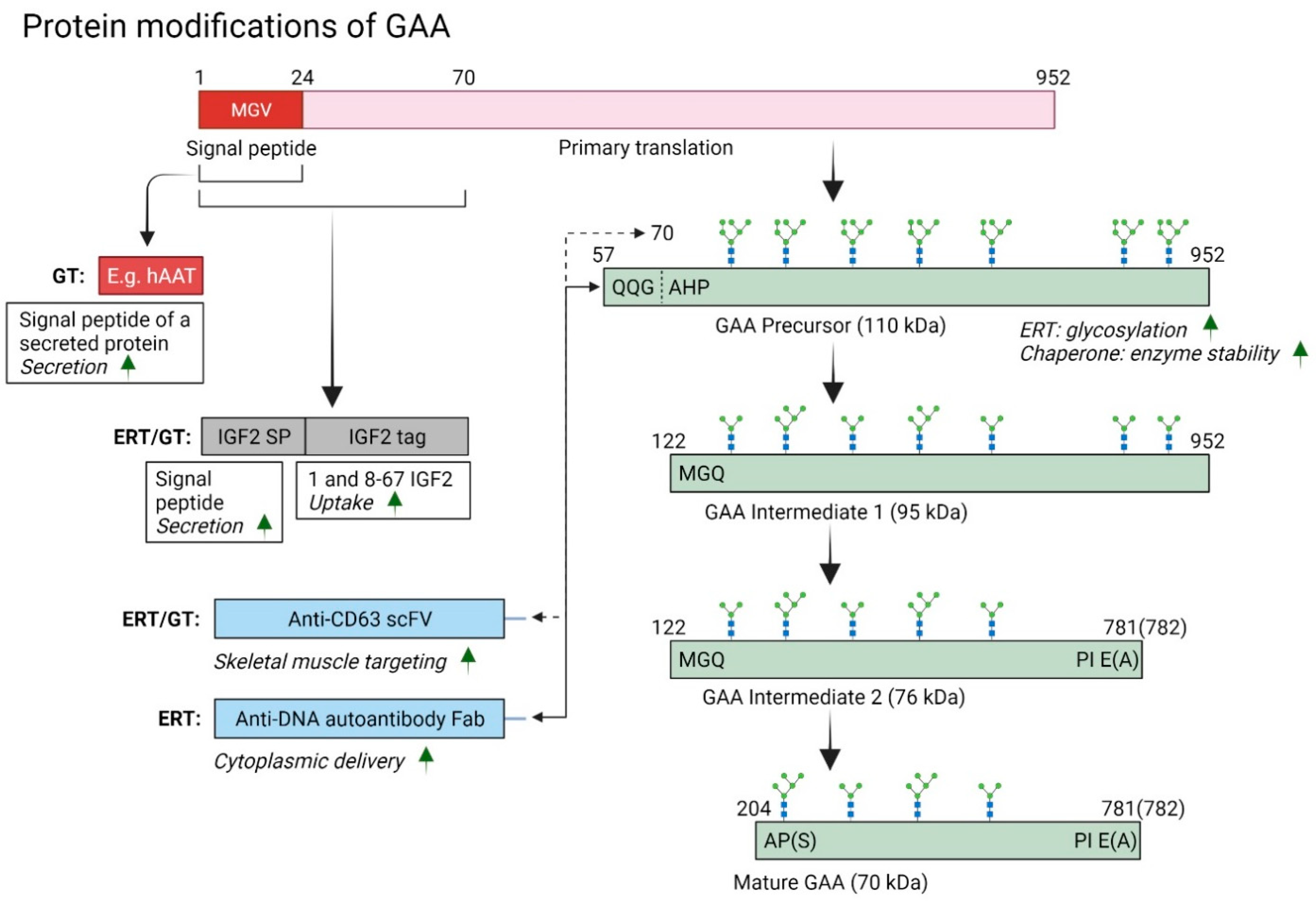{kind=link}
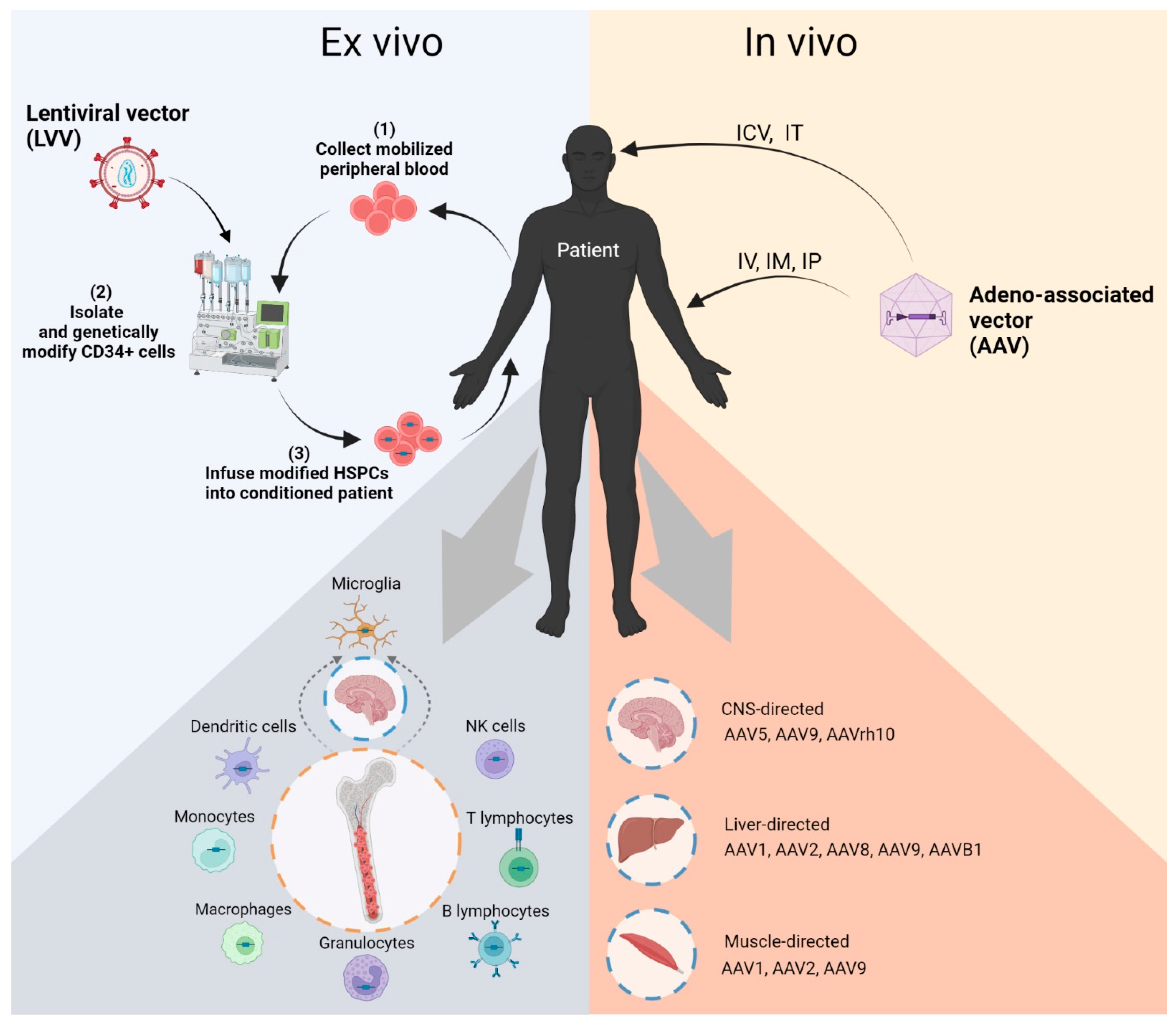
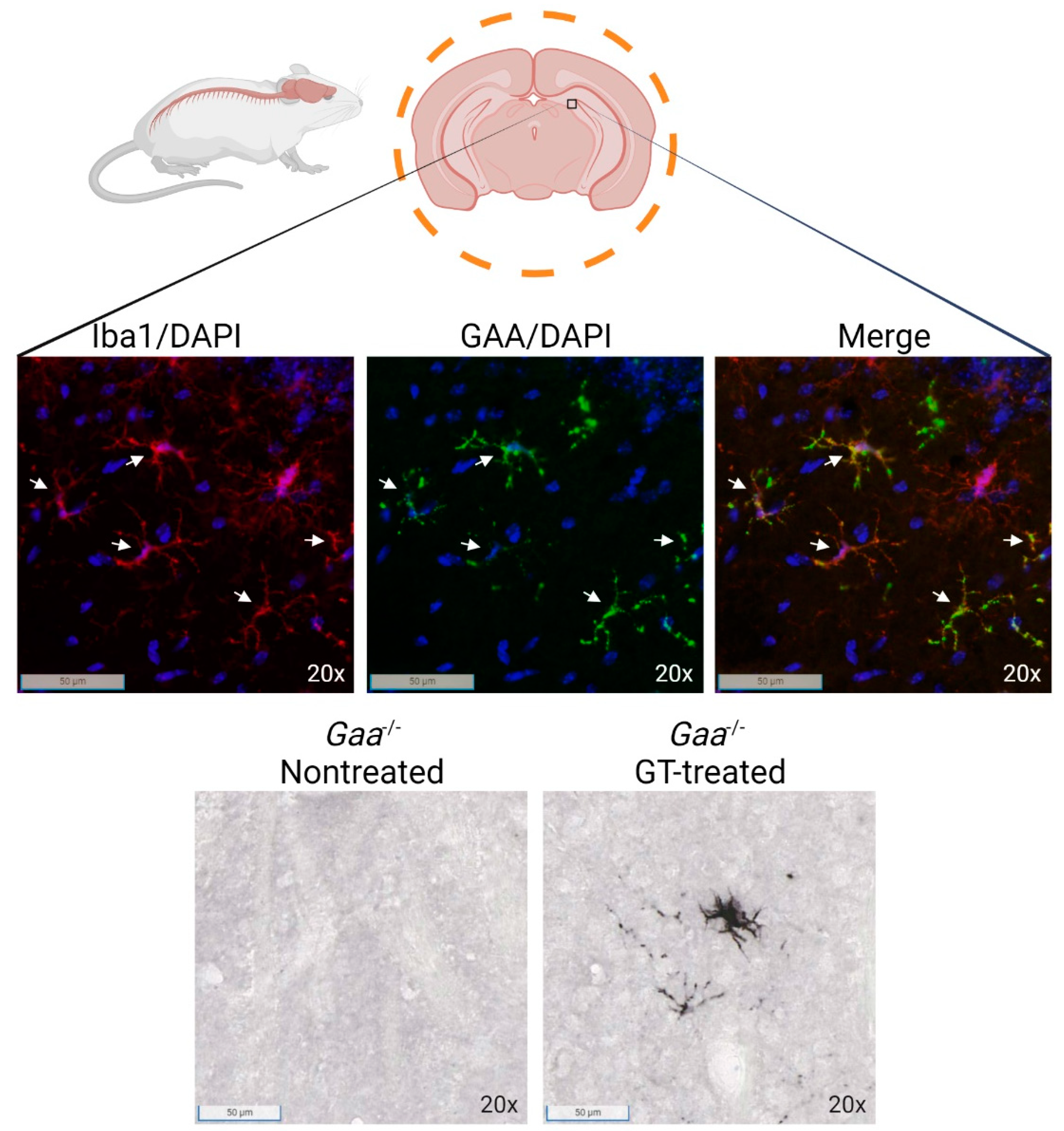{kind=link}
| Preclinical Program | Clinical Stage | Marketed Authorization |
|---|---|---|
| AAV2/8 Gene Therapy delivered Ab-GAA (Regeneron) | AAV2/8 LSPhGAA liver-directed Gene Therapy (LOPD, Phase 1) Bayer/Actus/AskBIO (Actus-101) (NCT03533673) | Myozyme/Lumizyme (IOPD, LOPD) Genzyme (alglucosidase alfa) |
| AAV/Proprietary capsid Gene Therapy (Amicus) | AAV9 muscle-directed Gene Therapy w/immune modulation (LOPD, Phase 1) University of Florida (NCT02240407) | Nexviazyme (LOPD) Genzyme (avalglucosidase alfa) |
| AAV Gene Therapy (Sarepta-licensed from Lacerta) | AAV1/CMV-hGAA muscle-directed Gene Therapy (LOPD, Phase 1/2) University of Florida (NCT00976352) | |
| AAV/Proprietary capsid Gene Therapy (Abeona) | AAV8 liver-directed Gene Therapy (LOPD, Phase 1/2) Audentes (AT845) (NCT04174105) | |
| HSPC LV Gene Therapy (Erasmus MC) | AAV/Proprietary Rh74-derived capsid, liver-directed Gene Therapy (LOPD, Phase 1/2) Spark/Roche (SPK-3006) (NCT04093349) | |
| HSPC LV Gene Therapy AVR-RD-03 (AVROBIO) | Chaperone/ERT (IOPD/LOPD, Phase 3) Amicus (ATB200/AT2221) (NCT03729362) | |
| JR-162; IV (JCR Pharma) J-Brain Cargo platform to cross blood–brain barrier | Nexviazyme (IOPD, Phase 2) Genzyme (avalglucosidase alfa) (NCT03019406) |
| Disease | Product | Recommended Dosage (mg/kg) | Frequency |
|---|---|---|---|
| Fabry | FABRAZYME® (agalsidase beta) | 1 | Every two weeks |
| Gaucher | VPRIV® (velaglucerase alfa) | 60 U/kg (~1.5 mg/kg) * | Every two weeks |
| MPS I | ALDURAZYME® (laronidase) | 0.58 | Once a week |
| MPS II | ELAPRASE® (idursulfase) | 0.5 | Once a week |
| MPS VI | NAGLAZYME® (galsulfase) | 1 | Once a week |
| Pompe | MYOZYME® (alglucosidase alfa) | 20 | Every two weeks |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Unnisa, Z.; Yoon, J.K.; Schindler, J.W.; Mason, C.; van Til, N.P. Gene Therapy Developments for Pompe Disease. Biomedicines 2022, 10, 302. https://doi.org/10.3390/biomedicines10020302
Unnisa Z, Yoon JK, Schindler JW, Mason C, van Til NP. Gene Therapy Developments for Pompe Disease. Biomedicines. 2022; 10(2):302. https://doi.org/10.3390/biomedicines10020302
Chicago/Turabian StyleUnnisa, Zeenath, John K. Yoon, Jeffrey W. Schindler, Chris Mason, and Niek P. van Til. 2022. "Gene Therapy Developments for Pompe Disease" Biomedicines 10, no. 2: 302. https://doi.org/10.3390/biomedicines10020302
APA StyleUnnisa, Z., Yoon, J. K., Schindler, J. W., Mason, C., & van Til, N. P. (2022). Gene Therapy Developments for Pompe Disease. Biomedicines, 10(2), 302. https://doi.org/10.3390/biomedicines10020302