
Amyloid Disassembly: What Can We Learn from Chaperones?

Abstract
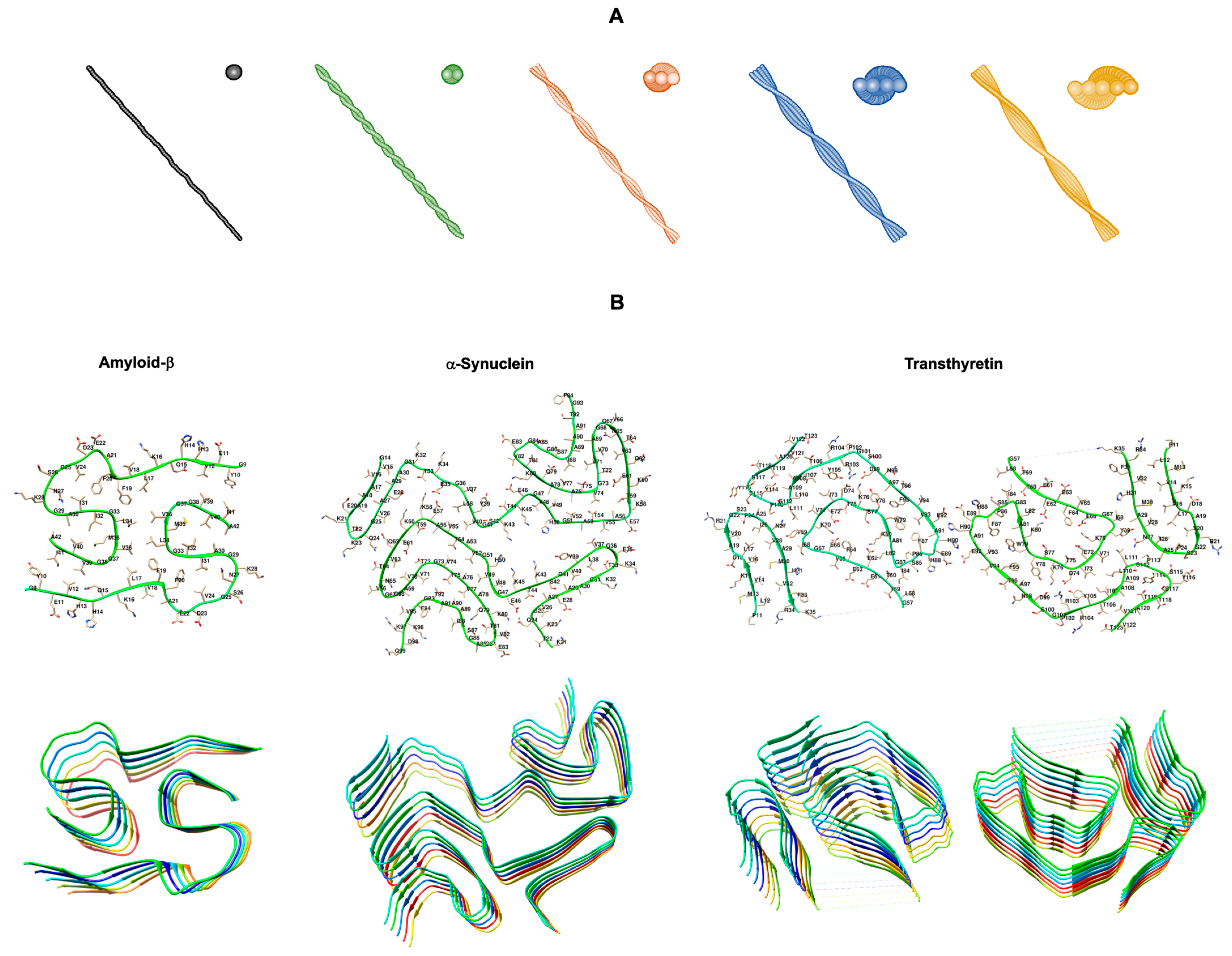
1. Introduction
2. Chaperones in Amyloid Disassembly
2.1. Molecular Chaperones in Amyloid Disassembly
2.1.1. Hsp70 Chaperone Network
2.1.2. Hsp90
2.1.3. Tripartite Motif Proteins (TRIMs)
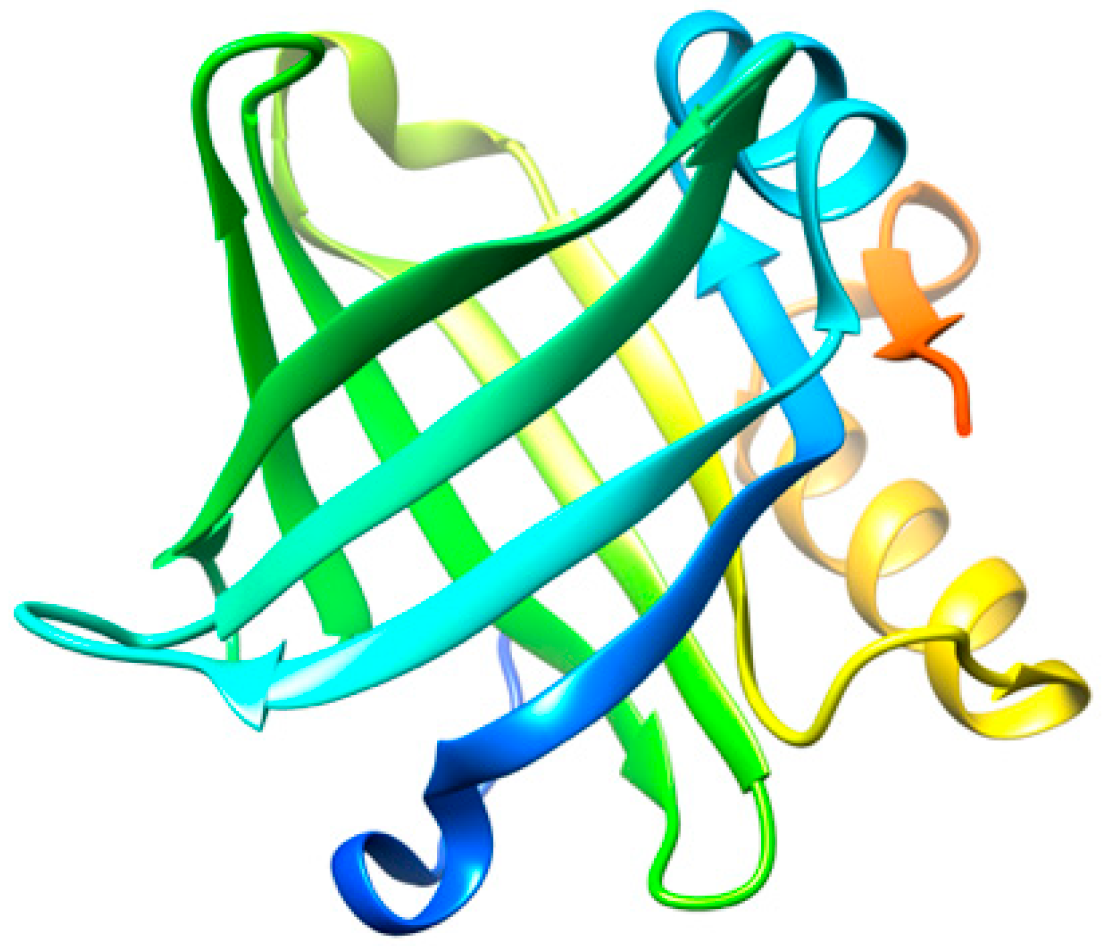
2.1.4. Lipocalin-Prostaglandin D Synthase (L-PGDS)
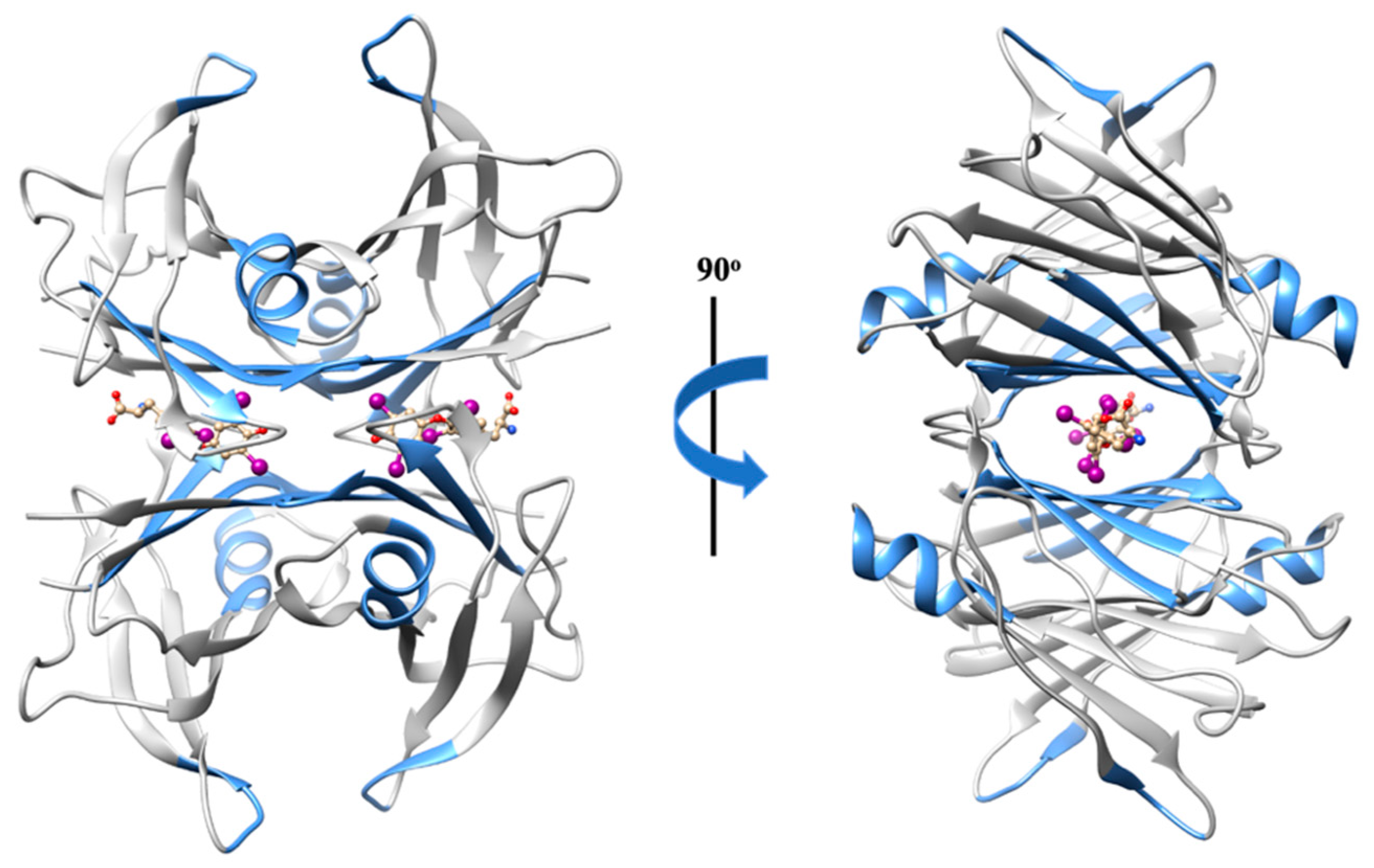
2.1.5. Transthyretin (TTR)
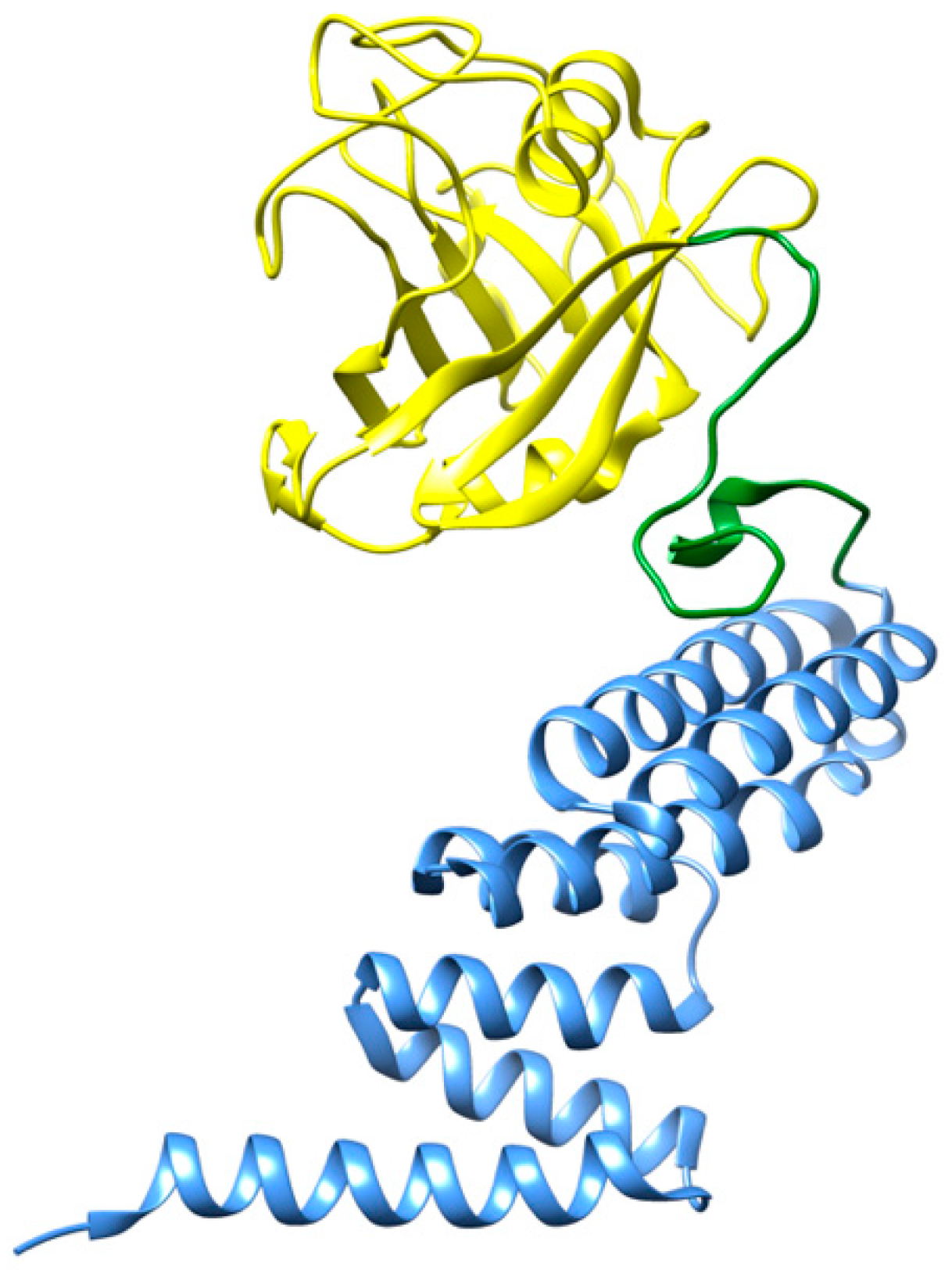
2.1.6. Cyclophilin 40 (CyP40)
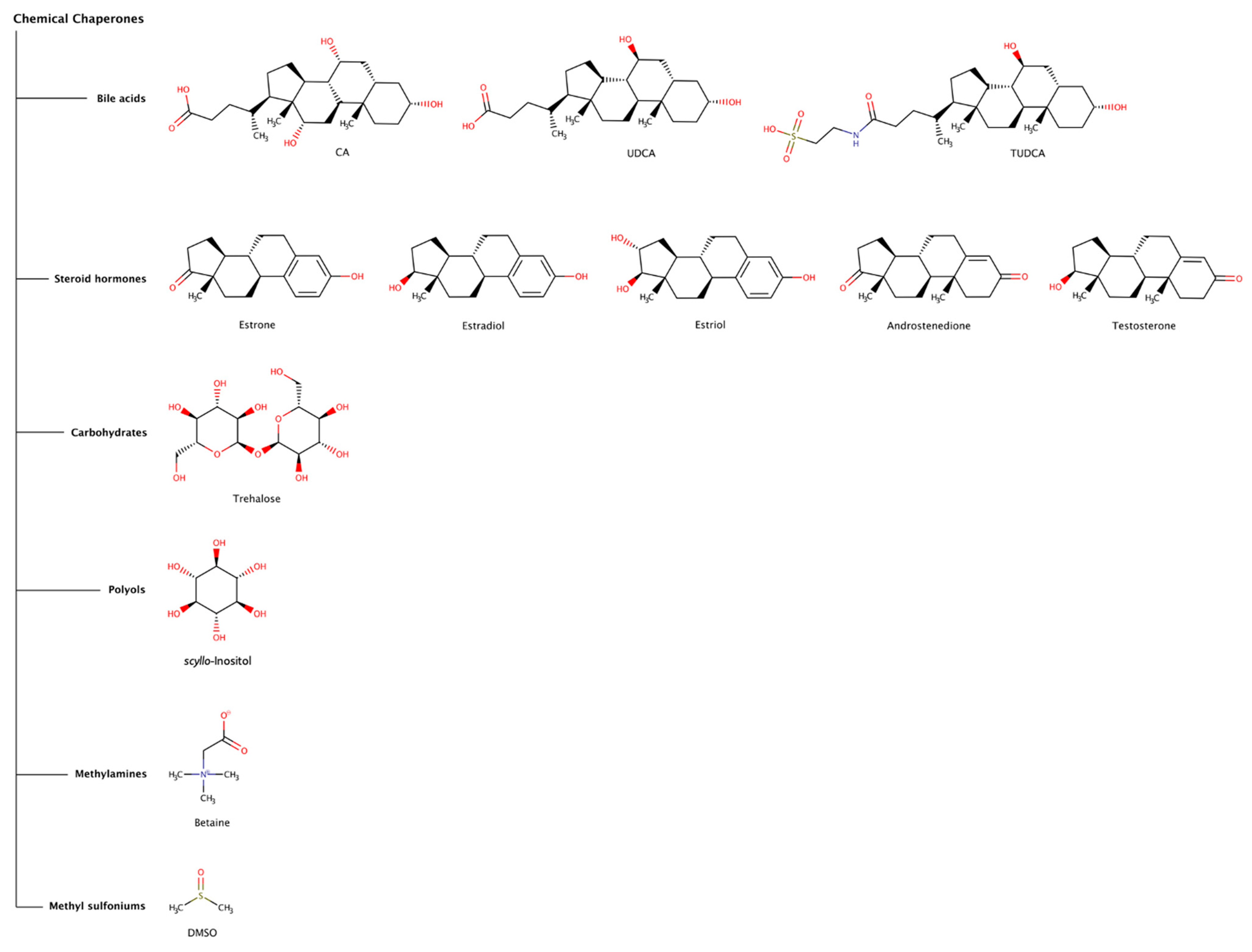
2.2. Chemical Chaperones in Amyloid Disassembly
2.2.1. Bile Acids
2.2.2. Steroid Hormones
2.2.3. Trehalose
2.2.4. Scyllo-Inositol
2.2.5. Betaine
2.2.6. Dimethyl Sulfoxide (DMSO)
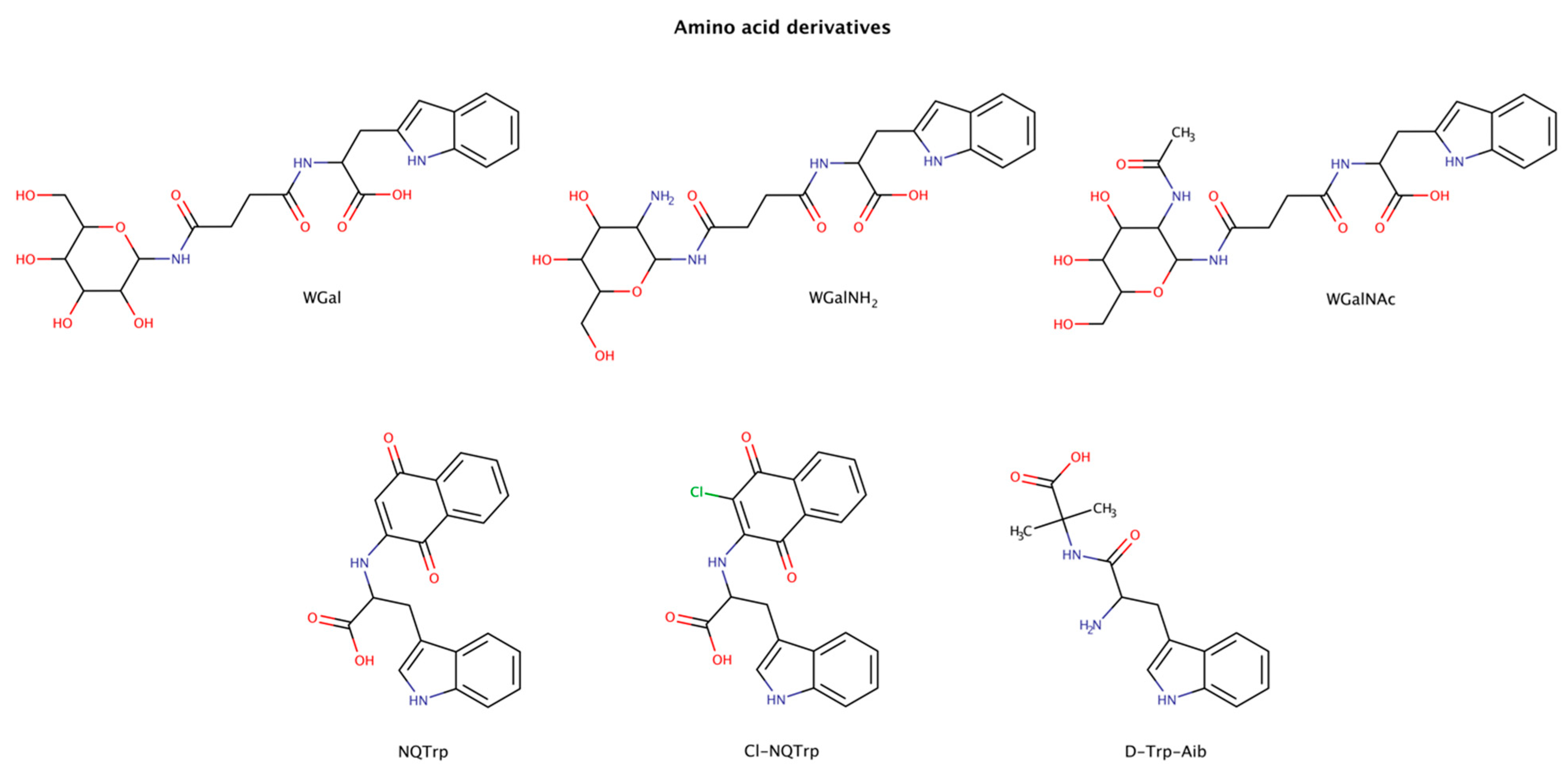
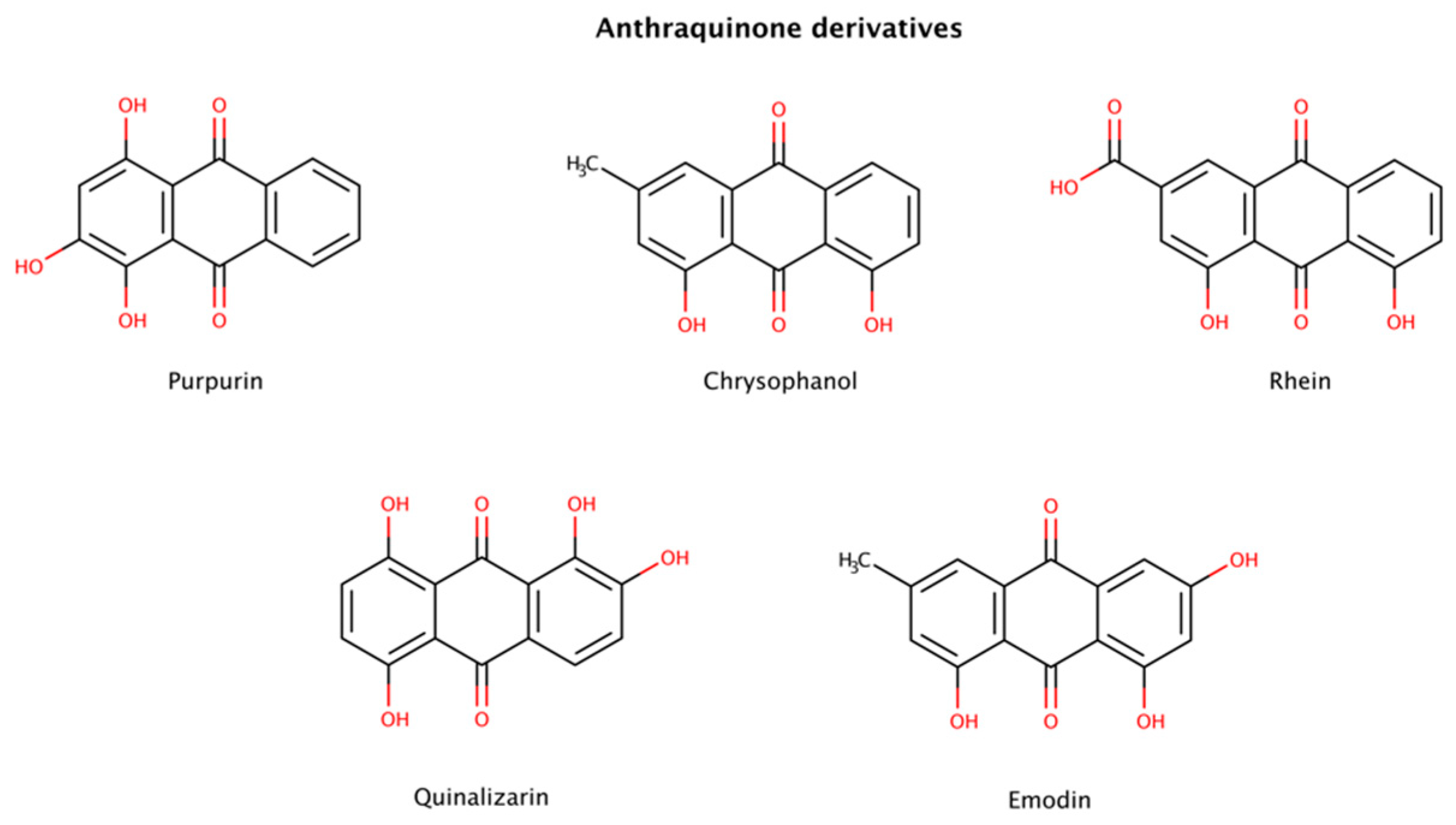
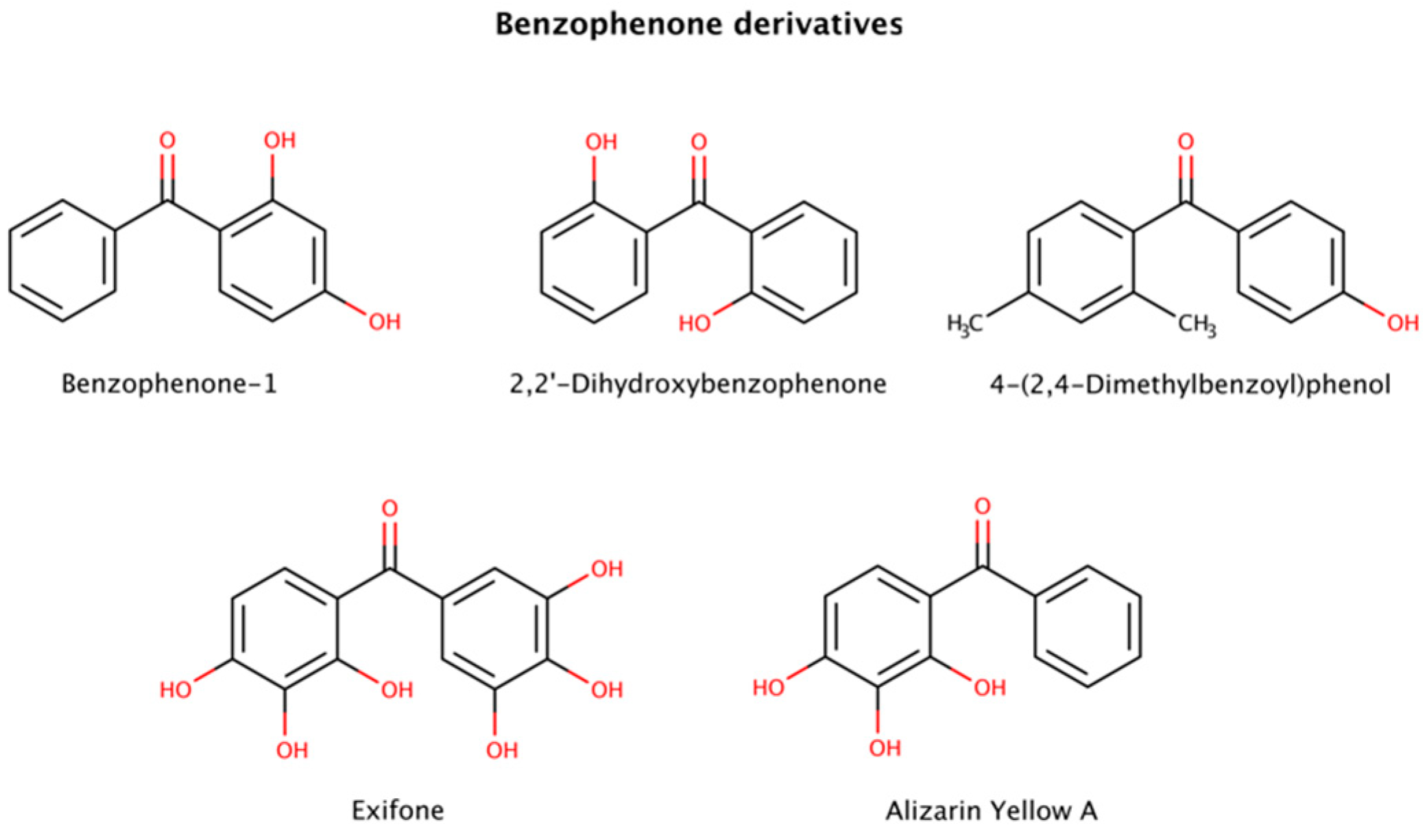
2.3. Pharmacological Chaperones in Amyloid Disassembly
2.3.1. Amino-Acid Derivatives
2.3.2. Anthraquinone Derivatives
2.3.3. Benzophenone Derivatives
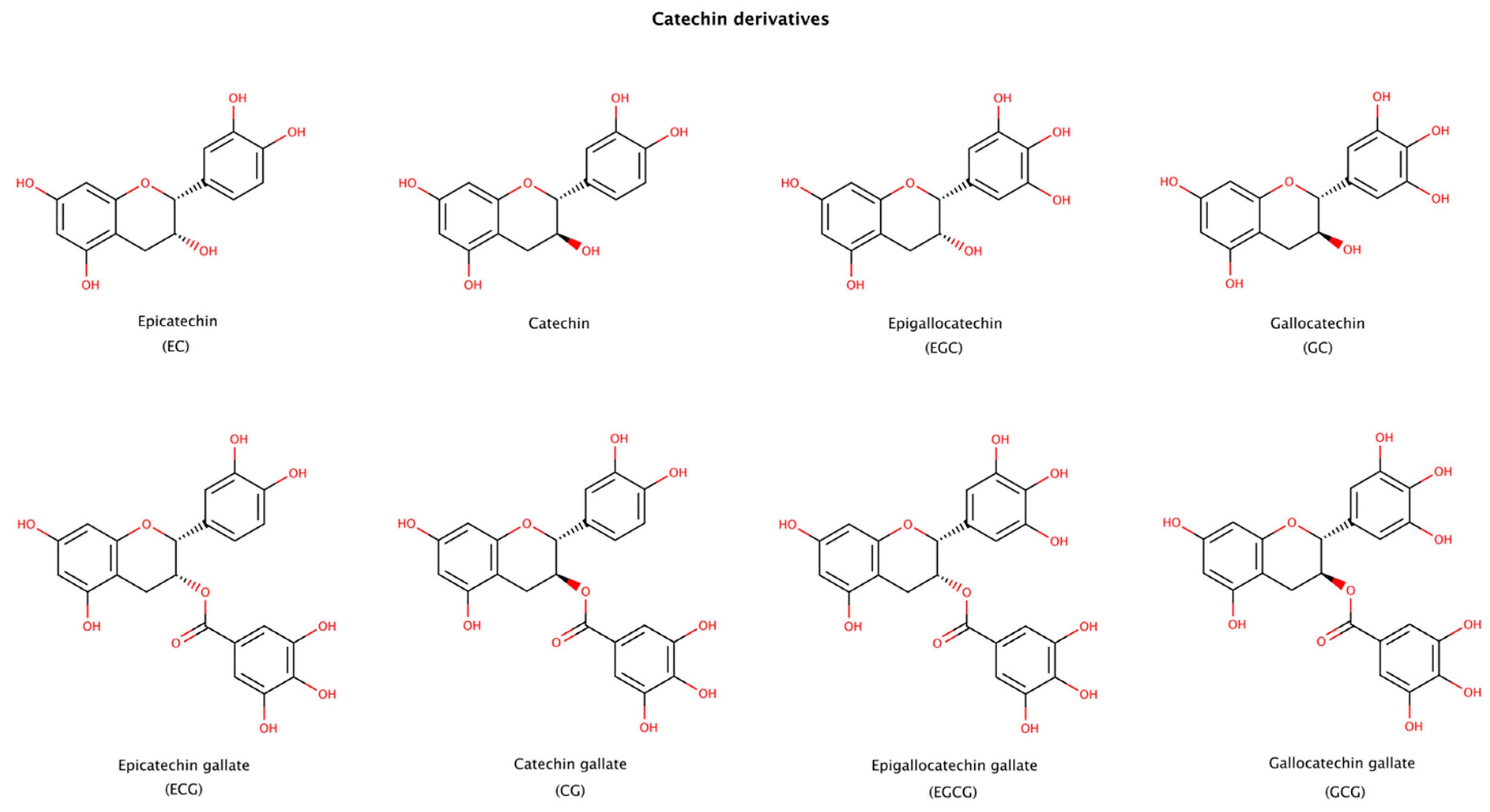
2.3.4. Catechin Derivatives
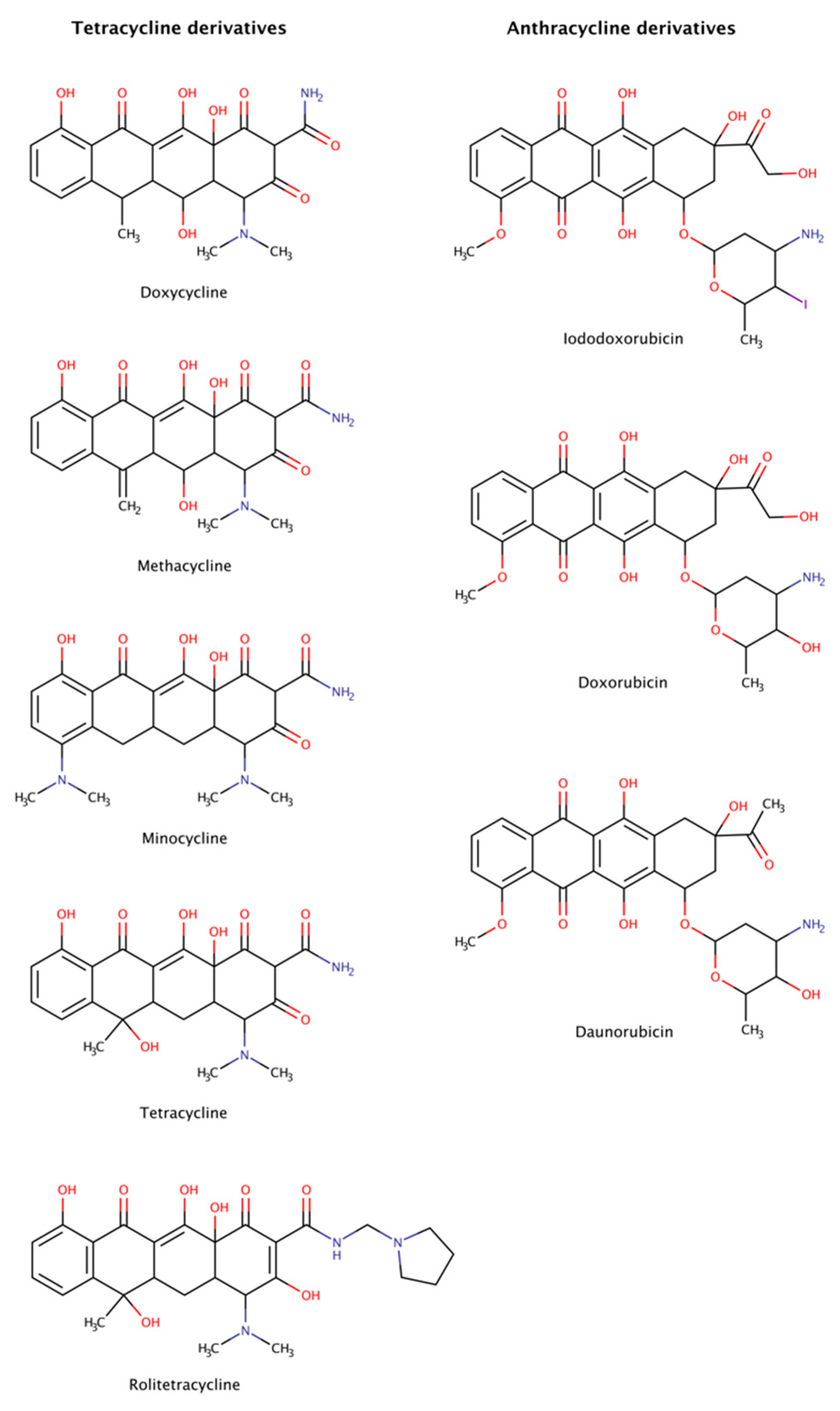
2.3.5. Anthracycline and Tetracycline Derivatives
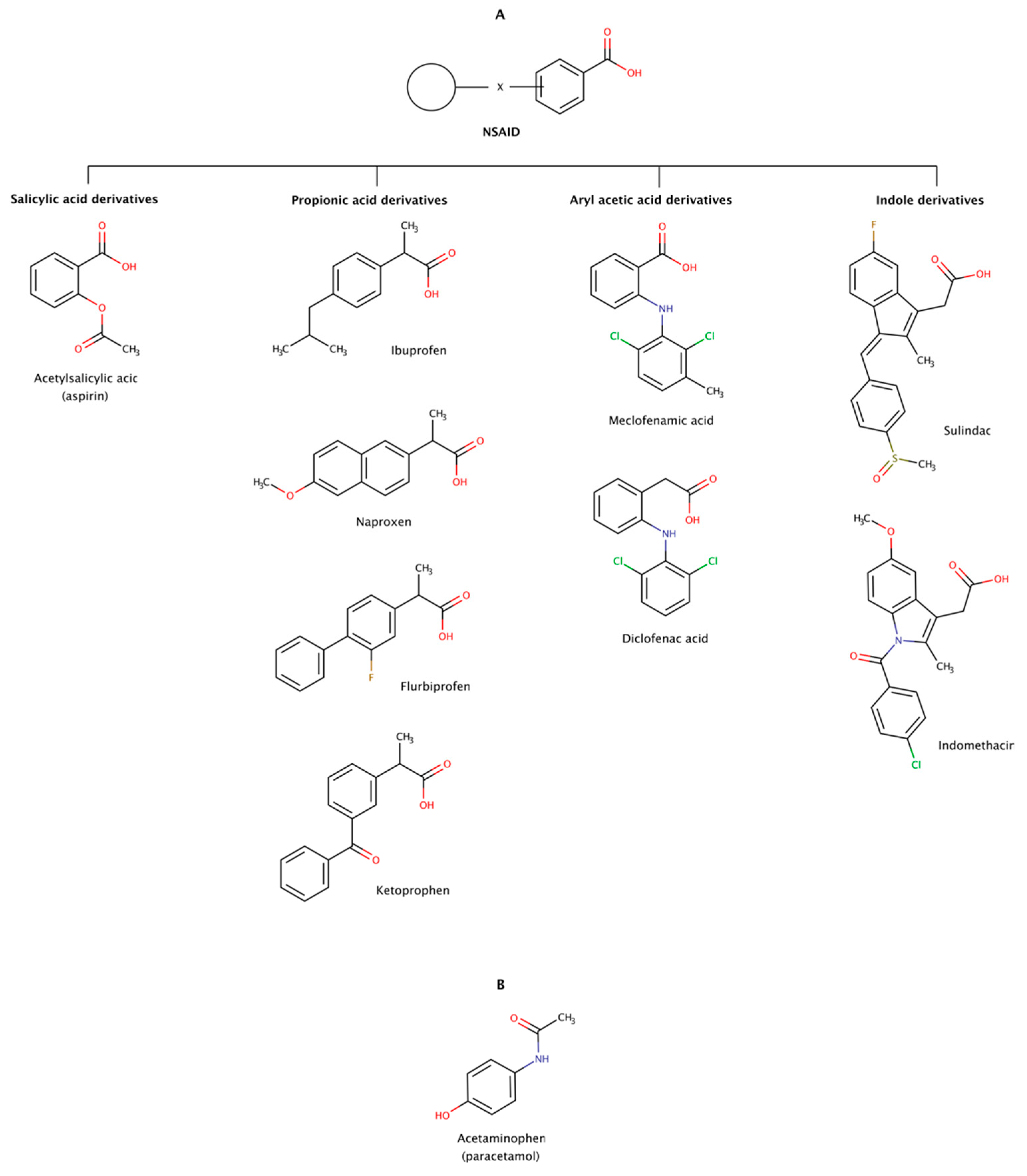
2.3.6. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
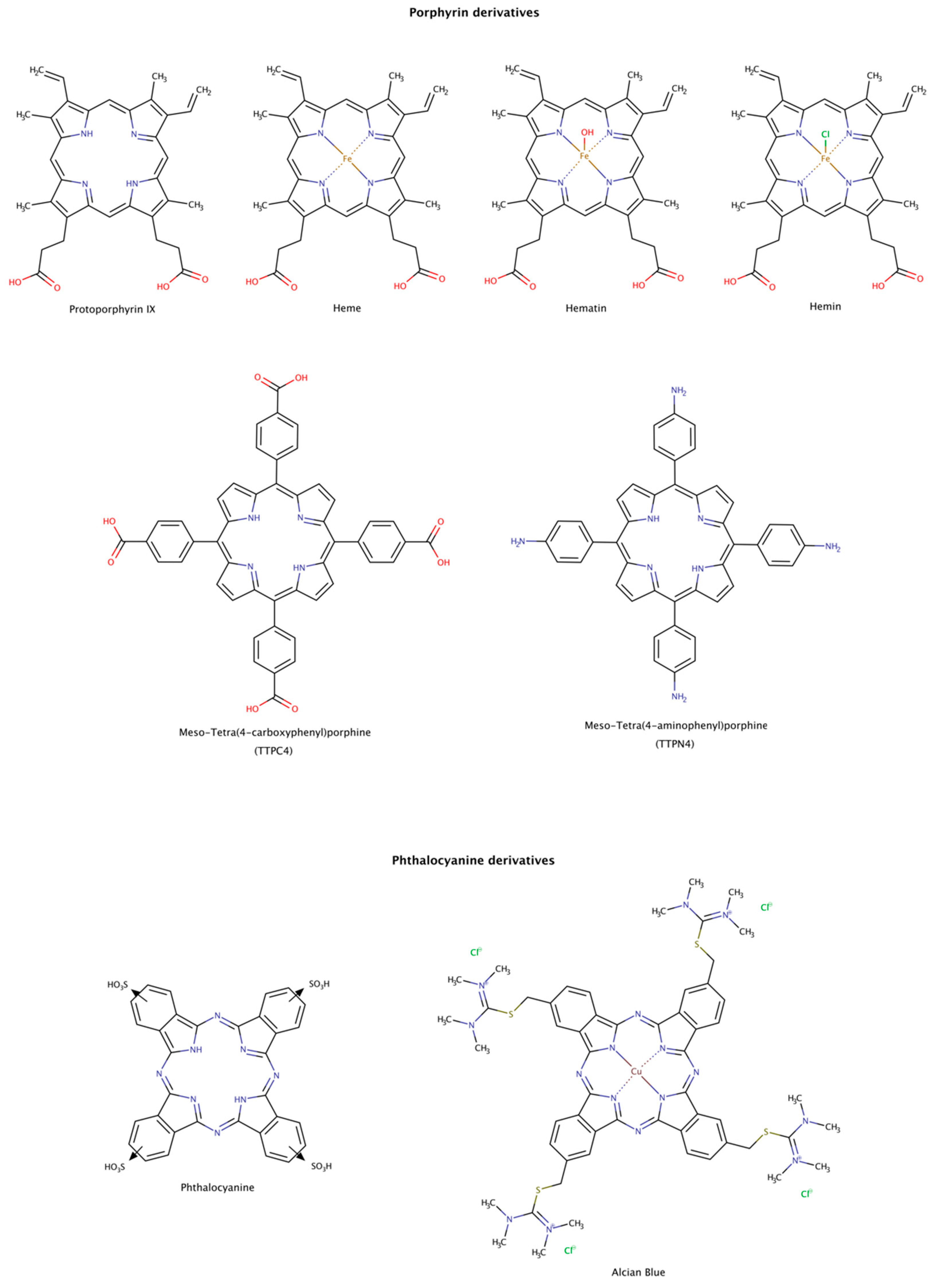
2.3.7. Porphyrin and Phthalocyanine Derivatives
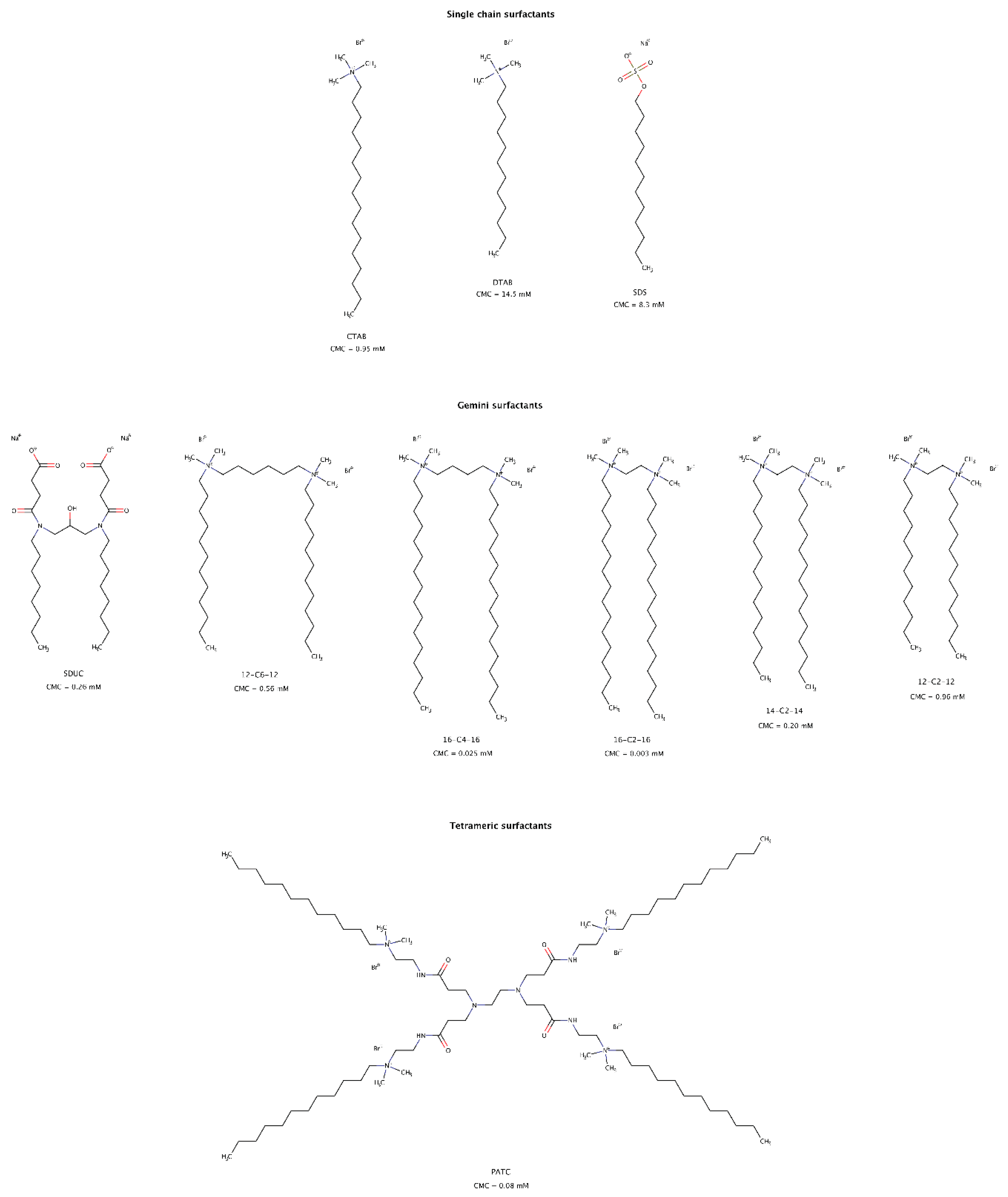
2.3.8. Surfactants
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Almeida, Z.L.; Brito, R.M.M. Structure and Aggregation Mechanisms in Amyloids. Molecules 2020, 25, 1195. [Google Scholar] [CrossRef] [PubMed]
- Adamcik, J.; Jung, J.M.; Flakowski, J.; De Los Rios, P.; Dietler, G.; Mezzenga, R. Understanding amyloid aggregation by statistical analysis of atomic force microscopy images. Nat. Nanotechnol. 2010, 5, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Arseni, D.; Zhang, W.; Huang, M.; Lövestam, S.; Schweighauser, M.; Kotecha, A.; Murzin, A.G.; Peak-Chew, S.Y.; MacDonald, J.; et al. Cryo-EM structures of amyloid-β 42 filaments from human brains. Science 2022, 375, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Wiese, S.; Adak, V.; Engler, J.; Agarwal, S.; Fritz, G.; Westermark, P.; Zacharias, M.; Fändrich, M. Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Pujols, J.; Peña-Díaz, S.; Pallarès, I.; Ventura, S. Chemical Chaperones as Novel Drugs for Parkinson’s Disease. Trends Mol. Med. 2020, 26, 408–421. [Google Scholar] [CrossRef]
- Ren, B.; Liu, Y.; Zhang, Y.; Zhang, M.; Sun, Y.; Liang, G.; Xu, J.; Zheng, J. Tanshinones inhibit hIAPP aggregation, disaggregate preformed hIAPP fibrils, and protect cultured cells. J. Mater. Chem. B 2017, 6, 56–67. [Google Scholar] [CrossRef]
- Choi, E.Y.; Kang, S.S.; Lee, S.K.; Han, B.H. Polyphenolic biflavonoids inhibit amyloid-beta fibrillation and disaggregate preformed amyloid-beta fibrils. Biomol. Ther. 2020, 28, 145–151. [Google Scholar] [CrossRef]
- Khan, A.N.; Qureshi, I.A.; Khan, U.K.; Uversky, V.N.; Khan, R.H. Inhibition and disruption of amyloid formation by the antibiotic levofloxacin: A new direction for antibiotics in an era of multi-drug resistance. Arch. Biochem. Biophys. 2021, 714, 109077. [Google Scholar] [CrossRef]
- Abioye, R.O.; Okagu, O.D.; Udenigwe, C.C. Inhibition of Islet Amyloid Polypeptide Fibrillation by Structurally Diverse Phenolic Compounds and Fibril Disaggregation Potential of Rutin and Quercetin. J. Agric. Food Chem. 2022, 70, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Mandal, H.; Basak, A.; Prabhu, T.; Kolli, V.; Sarkar, N. Inhibitory as well as Disaggregation Potential of Selected Hydroxy Benzoic Phytochemicals on Hen Egg-White Lysozyme Amyloidogenesis. Curr. Proteomics 2021, 18, 349–361. [Google Scholar] [CrossRef]
- Huang, C.; Rossi, P.; Saio, T.; Kalodimos, C.G. Structural basis for the antifolding activity of a molecular chaperone. Nature 2016, 537, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Protein quality control by molecular chaperones in neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Mannini, B.; Chiti, F. Chaperones as suppressors of protein misfolded oligomer toxicity. Front. Mol. Neurosci. 2017, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Low, K.J.Y.; Venkatraman, A.; Mehta, J.S.; Pervushin, K. Molecular mechanisms of amyloid disaggregation. J. Adv. Res. 2022, 36, 113–132. [Google Scholar] [CrossRef]
- Ohtsuka, K.; Kawashima, D.; Gu, Y.; Saito, K. Inducers and co-inducers of molecular chaperones. Int. J. Hyperth. 2005, 21, 703–711. [Google Scholar] [CrossRef]
- Shorter, J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 2011, 6, e26319. [Google Scholar] [CrossRef]
- Rüdiger, S.; Germeroth, L.; Schneider-Mergener, J.; Bukau, B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997, 16, 1501–1507. [Google Scholar] [CrossRef]
- Nillegoda, N.B.; Bukau, B. Metazoan Hsp70-based protein disaggregases: Emergence and mechanisms. Front. Mol. Biosci. 2015, 2, 57. [Google Scholar] [CrossRef]
- Nillegoda, N.B.; Stank, A.; Malinverni, D.; Alberts, N.; Szlachcic, A.; Barducci, A.; De Los Rios, P.; Wade, R.C.; Bukau, B. Evolution of an intricate J-protein network driving protein disaggregation in eukaryotes. Elife 2017, 6, e24560. [Google Scholar] [CrossRef] [PubMed]
- Duennwald, M.L.; Echeverria, A.; Shorter, J. Small Heat Shock Proteins Potentiate Amyloid Dissolution by Protein Disaggregases from Yeast and Humans. PLoS Biol. 2012, 10, e1001346. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Carroni, M.; Nussbaum-Krammer, C.; Mogk, A.; Nillegoda, N.B.; Szlachcic, A.; Guilbride, D.L.; Saibil, H.R.; Mayer, M.P.; Bukau, B. Human Hsp70 Disaggregase Reverses Parkinson’s-Linked α-Synuclein Amyloid Fibrils. Mol. Cell 2015, 59, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Scior, A.; Buntru, A.; Arnsburg, K.; Ast, A.; Iburg, M.; Juenemann, K.; Pigazzini, M.L.; Mlody, B.; Puchkov, D.; Priller, J.; et al. Complete suppression of Htt fibrilization and disaggregation of Htt fibrils by a trimeric chaperone complex. EMBO J. 2018, 37, 282–299. [Google Scholar] [CrossRef]
- Ferrari, L.; Geerts, W.J.C.; van Wezel, M.; Kos, R.; Konstantoulea, A.; van Bezouwen, L.S.; Förster, F.G.; Rüdiger, S.G.D. Human chaperones untangle fibrils of the Alzheimer protein Tau. bioRxiv 2018, 426650. [Google Scholar] [CrossRef]
- Nachman, E.; Wentink, A.S.; Madiona, K.; Bousset, L.; Katsinelos, T.; Allinson, K.; Kampinga, H.; McEwan, W.A.; Jahn, T.R.; Melki, R.; et al. Disassembly of Tau fibrils by the human Hsp70 disaggregation machinery generates small seeding-competent species. J. Biol. Chem. 2020, 295, 9676–9690. [Google Scholar] [CrossRef]
- Nillegoda, N.B.; Kirstein, J.; Szlachcic, A.; Berynskyy, M.; Stank, A.; Stengel, F.; Arnsburg, K.; Gao, X.; Scior, A.; Aebersold, R.; et al. Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature 2015, 524, 247–251. [Google Scholar] [CrossRef]
- Mok, S.A.; Condello, C.; Freilich, R.; Gillies, A.; Arhar, T.; Oroz, J.; Kadavath, H.; Julien, O.; Assimon, V.A.; Rauch, J.N.; et al. Mapping interactions with the chaperone network reveals factors that protect against tau aggregation. Nat. Struct. Mol. Biol. 2018, 25, 384–393. [Google Scholar] [CrossRef]
- Tittelmeier, J.; Sandhof, C.A.; Ries, H.M.; Druffel-Augustin, S.; Mogk, A.; Bukau, B.; Nussbaum-Krammer, C. The HSP110/HSP70 disaggregation system generates spreading-competent toxic α-synuclein species. EMBO J. 2020, 39, e103954. [Google Scholar] [CrossRef]
- Carlomagno, Y.; Zhang, Y.; Davis, M.; Lin, W.L.; Cook, C.; Dunmore, J.; Tay, W.; Menkosky, K.; Cao, X.; Petrucelli, L.; et al. Casein kinase II induced polymerization of soluble TDP-43 into filaments is inhibited by heat shock proteins. PLoS ONE 2014, 9, e90452. [Google Scholar] [CrossRef]
- Ali, Y.O.; Allen, H.M.; Yu, L.; Li-Kroeger, D.; Bakhshizadehmahmoudi, D.; Hatcher, A.; McCabe, C.; Xu, J.; Bjorklund, N.; Taglialatela, G.; et al. NMNAT2:HSP90 Complex Mediates Proteostasis in Proteinopathies. PLOS Biol. 2016, 14, e1002472. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, H.; Wu, W.; Zhuo, W.; Liu, W.; Zhang, Y.; Cheng, M.; Chen, Y.G.; Gao, N.; Yu, H.; et al. Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 2014, 24, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.P.; Haubrich, K.; Perez-Borrajero, C.; Hennig, J. Emerging RNA-binding roles in the TRIM family of ubiquitin ligases. Biol. Chem. 2019, 400, 1443–1464. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Harischandra, D.S.; Ghaisas, S.; Zhang, P.; Prall, W.; Huang, L.; Maghames, C.; Guo, L.; Luna, E.; Mack, K.L.; et al. TRIM11 Prevents and Reverses Protein Aggregation and Rescues a Mouse Model of Parkinson’s Disease. Cell Rep. 2020, 33, 108418. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Giasson, B.I.; Glavis-Bloom, A.; Brewer, M.D.; Shorter, J.; Gitler, A.D.; Yang, X. A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol. Cell 2014, 55, 15–30. [Google Scholar] [CrossRef]
- Tumani, H.; Huss, A.; Bachhuber, F. The cerebrospinal fluid and barriers—Anatomic and physiologic considerations. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 146, pp. 3–20. [Google Scholar]
- Lescuyer, P.; Gandini, A.; Burkhard, P.R.; Hochstrasser, D.F.; Sanchez, J.-C. Prostaglandin D2 synthase and its post-translational modifications in neurological disorders. Electrophoresis 2005, 26, 4563–4570. [Google Scholar] [CrossRef]
- Qu, W.M.; Huang, Z.L.; Xu, X.H.; Aritake, K.; Eguchi, N.; Nambu, F.; Narumiya, S.; Urade, Y.; Hayaishi, O. Lipocalin-type prostaglandin D syntase produces prostaglandin D2 involved in regulation of physiological sleep. Proc. Natl. Acad. Sci. USA 2006, 103, 17949–17954. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Ban, T.; Aritake, K.; Huang, Z.L.; Qu, W.M.; Okazaki, I.; Mohri, I.; Murayama, S.; Ozono, K.; Taniike, M.; et al. Lipocalin-type prostaglandin D synthase/β-trace is a major amyloid β-chaperone in human cerebrospinal fluid. Proc. Natl. Acad. Sci. USA 2007, 104, 6412–6417. [Google Scholar] [CrossRef]
- Kannaian, B.; Sharma, B.; Phillips, M.; Chowdhury, A.; Manimekalai, M.S.S.; Adav, S.S.; Ng, J.T.Y.; Kumar, A.; Lim, S.; Mu, Y.; et al. Abundant neuroprotective chaperone Lipocalin-type prostaglandin D synthase (L-PGDS) disassembles the Amyloid-β fibrils. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef]
- Esler, W.P.; Stimson, E.R.; Ghilardi, J.R.; Lu, Y.-A.; Felix, A.M.; Vinters, H.V.; Mantyh, P.W.; Lee, J.P.; Maggio, J.E. Point Substitution in the Central Hydrophobic Cluster of a Human-Amyloid Congener Disrupts Peptide Folding and Abolishes Plaque Competence. Biochemistry 1996, 35, 13914–13921. [Google Scholar] [CrossRef]
- Fukuyama, R.; Mizuno, T.; Mizuno, T.; Mori, S.; Nakajima, K.; Fushiki, S.; Yanagisawa, K. Age-dependent change in the levels of Aβ40 and Aβ42 in cerebrospinal fluid from control subjects, and a decrease in the ratio of Aβ42 to Aβ40 level in cerebrospinal fluid from Alzheimer’s disease patients. Eur. Neurol. 2000, 43, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Puchades, M.; Hansson, S.F.; Nilsson, C.L.; Andreasen, N.; Blennow, K.; Davidsson, P. Proteomic studies of potential cerebrospinal fluid protein markers for Alzheimer’s disease. Mol. Brain Res. 2003, 118, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Hansson, S.F.; Andréasson, U.; Wall, M.; Skoog, I.; Andreasen, N.; Wallin, A.; Zetterberg, H.; Blennow, K. Reduced levels of amyloid-β-binding proteins in cerebrospinal fluid from Alzheimer’s disease patients. J. Alzheimer’s Dis. 2009, 16, 389–397. [Google Scholar] [CrossRef]
- Frangolho, A.; Correia, B.E.; Vaz, D.C.; Almeida, Z.L.; Brito, R.M.M. Oligomerization Profile of Human Transthyretin Variants with Distinct Amyloidogenicity. Molecules 2020, 25, 5698. [Google Scholar] [CrossRef]
- Ferreira, E.; Almeida, Z.L.; Cruz, P.F.; Silva e Sousa, M.; Veríssimo, P.; Brito, R.M.M. Searching for the Best Transthyretin Aggregation Protocol to Study Amyloid Fibril Disruption. Int. J. Mol. Sci. 2021, 23, 391. [Google Scholar] [CrossRef] [PubMed]
- Quintas, A.; Vaz, D.C.; Cardoso, I.; Saraiva, M.J.M.; Brito, R.M.M. Tetramer Dissociation and Monomer Partial Unfolding Precedes Protofibril Formation in Amyloidogenic Transthyretin Variants. J. Biol. Chem. 2001, 276, 27207–27213. [Google Scholar] [CrossRef] [PubMed]
- Hurshman, A.R.; White, J.T.; Powers, E.T.; Kelly, J.W. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 2004, 43, 7365–7381. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Murphy, R.M. Characterization of the interaction of β-Amyloid with Transthyretin monomers and tetramers. Biochemistry 2010, 49, 8276–8289. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Ladiwala, A.R.A.; Du, D.; Yadav, J.K.; Tessier, P.M.; Wright, P.E.; Kelly, J.W.; Buxbaum, J.N. Mechanisms of transthyretin inhibition of β-amyloid aggregation in vitro. J. Neurosci. 2013, 33, 19423–19433. [Google Scholar] [CrossRef]
- Cotrina, E.Y.; Gimeno, A.; Llop, J.; Jiménez-Barbero, J.; Quintana, J.; Valencia, G.; Cardoso, I.; Prohens, R.; Arsequell, G. Calorimetric Studies of Binary and Ternary Molecular Interactions between Transthyretin, Aβ Peptides, and Small-Molecule Chaperones toward an Alternative Strategy for Alzheimer’s Disease Drug Discovery. J. Med. Chem. 2020, 63, 3205–3214. [Google Scholar] [CrossRef]
- Nilsson, L.; Pamrén, A.; Islam, T.; Brännström, K.; Golchin, S.A.; Pettersson, N.; Iakovleva, I.; Sandblad, L.; Gharibyan, A.L.; Olofsson, A. Transthyretin Interferes with Aβ Amyloid Formation by Redirecting Oligomeric Nuclei into Non-Amyloid Aggregates. J. Mol. Biol. 2018, 430, 2722–2733. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Gonçalves, A.; Saraiva, M.J.; Cardoso, I. Transthyretin binding to A-Beta peptide - Impact on A-Beta fibrillogenesis and toxicity. FEBS Lett. 2008, 582, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Buxbaum, J.N. Transthyretin and the brain re-visited: Is neuronal synthesis of transthyretin protective in Alzheimer’s disease? Mol. Neurodegener. 2011, 6, 79. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Conti, S.; Mannini, B.; Li, X.; Buxbaum, J.N.; Tiribilli, B.; Chiti, F.; Cecchi, C. Transthyretin suppresses the toxicity of oligomers formed by misfolded proteins in vitro. Biochim. Biophys. Acta—Mol. Basis Dis. 2013, 1832, 2302–2314. [Google Scholar] [CrossRef]
- Garai, K.; Posey, A.E.; Li, X.; Buxbaum, J.N.; Pappu, R.V. Inhibition of amyloid beta fibril formation by monomeric human transthyretin. Protein Sci. 2018, 27, 1252–1261. [Google Scholar] [CrossRef]
- Vatassery, G.T.; Quach, H.T.; Smith, W.E.; Benson, B.A.; Eckfeldt, J.H. A sensitive assay of transthyretin (prealbumin) in human cerebrospinal fluid in nanogram amounts by ELISA. Clin. Chim. Acta 1991, 197, 19–25. [Google Scholar] [CrossRef]
- Smith, F.R.; Goodman, D.S. The effects of diseases of the liver, thyroid, and kidneys on the transport of vitamin A in human plasma. J. Clin. Invest. 1971, 50, 2426–2436. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Serot, J.M.; Christmann, D.; Dubost, T.; Couturier, M. Cerebrospinal fluid transthyretin: Aging and late onset Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 63, 506–508. [Google Scholar] [CrossRef]
- Han, S.H.; Jung, E.S.; Sohn, J.H.; Hong, H.J.; Hong, H.S.; Kim, J.W.; Na, D.L.; Kim, M.; Kim, H.; Ha, H.J.; et al. Human serum transthyretin levels correlate inversely with Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 25, 77–84. [Google Scholar] [CrossRef]
- Gimeno, A.; Santos, L.M.; Alemi, M.; Rivas, J.; Blasi, D.; Cotrina, E.Y.; Llop, J.; Valencia, G.; Cardoso, I.; Quintana, J.; et al. Insights on the Interaction between Transthyretin and Aβ in Solution. A Saturation Transfer Difference (STD) NMR Analysis of the Role of Iododiflunisal. J. Med. Chem. 2017, 60, 5749–5758. [Google Scholar] [CrossRef] [PubMed]
- Schwarzman, A.L.; Goldgaber, D. Interaction of transthyretin with amyloid β-protein: Binding and inhibition of amyloid formation. CIBA Found. Symp. 1996, 199, 146–164. [Google Scholar] [PubMed]
- Du, J.; Cho, P.Y.; Yang, D.T.; Murphy, R.M. Identification of beta-amyloid-binding sites on transthyretin. Protein Eng. Des. Sel. 2012, 25, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.D.; Shelton, L.B.; Zheng, D.; Favretto, F.; Nordhues, B.A.; Darling, A.; Sullivan, L.E.; Sun, Z.; Solanki, P.K.; Martin, M.D.; et al. Human cyclophilin 40 unravels neurotoxic amyloids. PLOS Biol. 2017, 15, e2001336. [Google Scholar] [CrossRef]
- Gur, M.; Blackburn, E.A.; Ning, J.; Narayan, V.; Ball, K.L.; Walkinshaw, M.D.; Erman, B. Molecular dynamics simulations of site point mutations in the TPR domain of cyclophilin 40 identify conformational states with distinct dynamic and enzymatic properties. J. Chem. Phys. 2018, 148, 145101. [Google Scholar] [CrossRef]
- Papp, E.; Csermely, P. Chemical chaperones: Mechanisms of action and potential use. Handb. Exp. Pharmacol. 2006, 172, 405–416. [Google Scholar]
- Kushwah, N.; Jain, V.; Yadav, D. Osmolytes: A possible therapeutic molecule for ameliorating the neurodegeneration caused by protein misfolding and aggregation. Biomolecules 2020, 10, 132. [Google Scholar] [CrossRef]
- Schonewille, M.; de Boer, J.F.; Groen, A.K. Bile salts in control of lipid metabolism. Curr. Opin. Lipidol. 2016, 27, 295–301. [Google Scholar] [CrossRef]
- Ackerman, H.D.; Gerhard, G.S. Bile acids in neurodegenerative disorders. Front. Aging Neurosci. 2016, 8, 263. [Google Scholar] [CrossRef]
- Majid, N.; Siddiqi, M.K.; Khan, A.N.; Shabnam, S.; Malik, S.; Alam, A.; Uversky, V.N.; Khan, R.H. Biophysical Elucidation of Amyloid Fibrillation Inhibition and Prevention of Secondary Nucleation by Cholic Acid: An Unexplored Function of Cholic Acid. ACS Chem. Neurosci. 2019, 10, 4704–4715. [Google Scholar] [CrossRef]
- Nunes, A.F.; Amaral, J.D.; Lo, A.C.; Fonseca, M.B.; Viana, R.J.S.; Callaerts-Vegh, Z.; D’Hooge, R.; Rodrigues, C.M.P. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-β deposition in APP/PS1 mice. Mol. Neurobiol. 2012, 45, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.C.; Callaerts-Vegh, Z.; Nunes, A.F.; Rodrigues, C.M.P.; D’Hooge, R. Tauroursodeoxycholic acid (TUDCA) supplementation prevents cognitive impairment and amyloid deposition in APP/PS1 mice. Neurobiol. Dis. 2013, 50, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Macedo, B.; Batista, A.R.; Ferreira, N.; Almeida, M.R.; Saraiva, M.J. Anti-apoptotic treatment reduces transthyretin deposition in a transgenic mouse model of Familial Amyloidotic Polyneuropathy. Biochim. Biophys. Acta—Mol. Basis Dis. 2008, 1782, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, I.; Martins, D.; Ribeiro, T.; Merlini, G.; Saraiva, M.J. Synergy of combined Doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: Studies in FAP mouse models. J. Transl. Med. 2010, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Obici, L.; Cortese, A.; Lozza, A.; Lucchetti, J.; Gobbi, M.; Palladini, G.; Perlini, S.; Saraiva, M.J.; Merlini, G. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: A phase II study. Amyloid 2012, 19, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Wixner, J.; Pilebro, B.; Lundgren, H.-E.; Olsson, M.; Anan, I. Effect of doxycycline and ursodeoxycholic acid on transthyretin amyloidosis. Amyloid 2017, 24, 78–79. [Google Scholar] [CrossRef]
- Karlstedt, E.; Jimenez-Zepeda, V.; Howlett, J.G.; White, J.A.; Fine, N.M. Clinical Experience With the Use of Doxycycline and Ursodeoxycholic Acid for the Treatment of Transthyretin Cardiac Amyloidosis. J. Card. Fail. 2019, 25, 147–153. [Google Scholar] [CrossRef]
- Wehling, M. Specific, Nongenomic Actions of Steroid Hormones. Annu. Rev. Physiol. 1997, 59, 365–393. [Google Scholar] [CrossRef]
- Campbell, C.M.; LoRusso, S.; Dispenzieri, A.; Kristen, A.V.; Maurer, M.S.; Rapezzi, C.; Lairez, O.; Drachman, B.; Garcia-Pavia, P.; Grogan, M.; et al. Sex Differences in Wild-Type Transthyretin Amyloidosis: An Analysis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Cardiol. Ther. 2022, 11, 393–405. [Google Scholar] [CrossRef]
- Caponetti, A.G.; Rapezzi, C.; Gagliardi, C.; Milandri, A.; Dispenzieri, A.; Kristen, A.V.; Wixner, J.; Maurer, M.S.; Garcia-Pavia, P.; Tournev, I.; et al. Sex-Related Risk of Cardiac Involvement in Hereditary Transthyretin Amyloidosis: Insights From THAOS. JACC Hear. Fail. 2021, 9, 736–746. [Google Scholar] [CrossRef]
- Van Den Eeden, S.K. Incidence of Parkinson’s Disease: Variation by Age, Gender, and Race/Ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Vest, R.S.; Pike, C.J. Gender, sex steroid hormones, and Alzheimer’s disease. Horm. Behav. 2013, 63, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, A.; Hirohata, M.; Ono, K.; Yamada, M. Estrogen has anti-amyloidogenic effects on Alzheimer’s β-amyloid fibrils in vitro. Biochem. Biophys. Res. Commun. 2007, 359, 697–702. [Google Scholar] [CrossRef]
- Hirohata, M.; Ono, K.; Morinaga, A.; Ikeda, T.; Yamada, M. Anti-aggregation and fibril-destabilizing effects of sex hormones on α-synuclein fibrils in vitro. Exp. Neurol. 2009, 217, 434–439. [Google Scholar] [CrossRef]
- Song, Y.; Li, S.; Li, X.; Chen, X.; Wei, Z.; Liu, Q.; Cheng, Y. The Effect of Estrogen Replacement Therapy on Alzheimer’s Disease and Parkinson’s Disease in Postmenopausal Women: A Meta-Analysis. Front. Neurosci. 2020, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Goodman, Y.; Bruce, A.J.; Cheng, B.; Mattson, M.P. Estrogens Attenuate and Corticosterone Exacerbates Excitotoxicity, Oxidative Injury, and Amyloid β-Peptide Toxicity in Hippocampal Neurons. J. Neurochem. 1996, 66, 1836–1844. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Shen, Y.; Yang, L.-B.; Lue, L.-F.; Finch, C.; Rogers, J. Estrogen Enhances Uptake of Amyloid β-Protein by Microglia Derived from the Human Cortex. J. Neurochem. 2002, 75, 1447–1454. [Google Scholar] [CrossRef]
- Ragonese, P.; D’Amelio, M.; Savettieri, G. Implications for Estrogens in Parkinson’s Disease: An Epidemiological Approach. Ann. N. Y. Acad. Sci. 2006, 1089, 373–382. [Google Scholar] [CrossRef]
- Pinkerton, J.A.V.; Henderson, V.W. Estrogen and cognition, with a focus on Alzheimer’s disease. Semin. Reprod. Med. 2005, 23, 172–179. [Google Scholar] [CrossRef]
- Yu, W.B.; Jiang, T.; Lan, D.M.; Lu, J.H.; Yue, Z.Y.; Wang, J.; Zhou, P. Trehalose inhibits fibrillation of A53T mutant alpha-synuclein and disaggregates existing fibrils. Arch. Biochem. Biophys. 2012, 523, 144–150. [Google Scholar] [CrossRef]
- Liu, R.; Barkhordarian, H.; Emadi, S.; Chan, B.P.; Sierks, M.R. Trehalose differentially inhibits aggregation and neurotoxicity of beta-amyloid 40 and 42. Neurobiol. Dis. 2005, 20, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, R.; Kolli, V.; Sarkar, N. Trehalose and Magnesium Chloride Exert a Common Anti-amyloidogenic Effect Towards Hen Egg White Lysozyme. Protein J. 2017, 36, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Lan, C.P.; Hasan, S.; Brown, M.E.; McLaurin, J.A. Scyllo-Inositol promotes Robust Mutant Huntingtin Protein degradation. J. Biol. Chem. 2014, 289, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.; McLaurin, J.A. α-Synuclein aggregation, seeding and inhibition by scyllo-inositol. Biochem. Biophys. Res. Commun. 2016, 469, 529–534. [Google Scholar] [CrossRef]
- McLaurin, J.A.; Kierstead, M.E.; Brown, M.E.; Hawkes, C.A.; Lambermon, M.H.L.; Phinney, A.L.; Darabie, A.A.; Cousins, J.E.; French, J.E.; Lan, M.F.; et al. Cyclohexanehexol inhibitors of Aβ aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat. Med. 2006, 12, 801–808. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Keren, R.; Porsteinsson, A.P.; Van Dyck, C.H.; Tariot, P.N.; Gilman, S.; Arnold, D.; Abushakra, S.; Hernandez, C.; et al. A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology 2011, 77, 1253–1262. [Google Scholar] [CrossRef]
- Fenili, D.; Brown, M.; Rappaport, R.; McLaurin, J.A. Properties of scyllo-inositol as a therapeutic treatment of AD-like pathology. J. Mol. Med. 2007, 85, 603–611. [Google Scholar] [CrossRef]
- Townsend, M.; Cleary, J.P.; Mehta, T.; Hofmeister, J.; Lesne, S.; O’Hare, E.; Walsh, D.M.; Selkoe, D.J. Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-β oligomers. Ann. Neurol. 2006, 60, 668–676. [Google Scholar] [CrossRef]
- Ma, K.; Thomason, L.A.M.; McLaurin, J.A. Scyllo-Inositol, Preclinical, and Clinical Data for Alzheimer’s Disease. In Advances in Pharmacology; Academic Press Inc.: Cambridge, MA, USA, 2012; Volume 64, pp. 177–212. [Google Scholar]
- Natalello, A.; Liu, J.; Ami, D.; Doglia, S.M.; de Marco, A. The osmolyte betaine promotes protein misfolding and disruption of protein aggregates. Proteins Struct. Funct. Bioinforma. 2009, 75, 509–517. [Google Scholar] [CrossRef]
- De Marco, A.; Vigh, L.; Diamant, S.; Goloubinoff, P. Native folding of aggregation-prone recombinant proteins in Escherichia coli by osmolytes, plasmid- or benzyl alcohol-overexpressed molecular chaperones. Cell Stress Chaperones 2005, 10, 329–339. [Google Scholar] [CrossRef]
- Santos, N.C.; Figueira-Coelho, J.; Martins-Silva, J.; Saldanha, C. Multidisciplinary utilization of dimethyl sulfoxide: Pharmacological, cellular, and molecular aspects. Biochem. Pharmacol. 2003, 65, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Hanslick, J.L.; Lau, K.; Noguchi, K.K.; Olney, J.W.; Zorumski, C.F.; Mennerick, S.; Farber, N.B. Dimethyl sulfoxide (DMSO) produces widespread apoptosis in the developing central nervous system. Neurobiol. Dis. 2009, 34, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nasrallah, F.A.; Garner, B.; Ball, G.E.; Rae, C. Modulation of brain metabolism by very low concentrations of the commonly used drug delivery vehicle dimethyl sulfoxide (DMSO). J. Neurosci. Res. 2008, 86, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Institóris, Á.; Domoki, F.; Mihály, A.; Luiten, P.G.M.; Bari, F. Diazoxide and dimethyl sulphoxide prevent cerebral hypoperfusion-related learning dysfunction and brain damage after carotid artery occlusion. Brain Res. 2004, 1008, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Hirota-Nakaoka, N. Dissolution of 2-Microglobulin Amyloid Fibrils by Dimethylsulfoxide. J. Biochem. 2003, 134, 159–164. [Google Scholar] [CrossRef]
- Loksztejn, A.; Dzwolak, W. Noncooperative dimethyl sulfoxide-induced dissection of insulin fibrils: Toward soluble building blocks of amyloid. Biochemistry 2009, 48, 4846–4851. [Google Scholar] [CrossRef]
- Roy, S.; Bagchi, B. Comparative study of protein unfolding in aqueous urea and dimethyl sulfoxide solutions: Surface polarity, solvent specificity, and sequence of secondary structure melting. J. Phys. Chem. B 2014, 118, 5691–5697. [Google Scholar] [CrossRef]
- Coelho, T.; Merlini, G.; Bulawa, C.E.; Fleming, J.A.; Judge, D.P.; Kelly, J.W.; Maurer, M.S.; Planté-Bordeneuve, V.; Labaudinière, R.; Mundayat, R.; et al. Mechanism of Action and Clinical Application of Tafamidis in Hereditary Transthyretin Amyloidosis. Neurol. Ther. 2016, 5, 1–25. [Google Scholar] [CrossRef]
- Faria, T.Q.; Almeida, Z.L.; Cruz, P.F.; Jesus, C.S.H.; Castanheira, P.; Brito, R.M.M. A look into amyloid formation by transthyretin: Aggregation pathway and a novel kinetic model. Phys. Chem. Chem. Phys. 2015, 17, 7255–7263. [Google Scholar] [CrossRef]
- Jesus, C.S.H.; Almeida, Z.L.; Vaz, D.C.; Faria, T.Q.; Brito, R.M.M. A new folding kinetic mechanism for human transthyretin and the influence of the amyloidogenic V30M mutation. Int. J. Mol. Sci. 2016, 17, 1428. [Google Scholar] [CrossRef]
- KrishnaKumar, V.G.; Paul, A.; Gazit, E.; Segal, D. Mechanistic insights into remodeled Tau-derived PHF6 peptide fibrils by Naphthoquinone-Tryptophan hybrids. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Viswanathan, G.K.; Mahapatra, S.; Balboni, G.; Pacifico, S.; Gazit, E.; Segal, D. Antagonistic Activity of Naphthoquinone-Based Hybrids toward Amyloids Associated with Alzheimer’s Disease and Type-2 Diabetes. ACS Chem. Neurosci. 2019, 10, 3510–3520. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, G.K.; Paul, A.; Gazit, E.; Segal, D. Naphthoquinone Tryptophan Hybrids: A Promising Small Molecule Scaffold for Mitigating Aggregation of Amyloidogenic Proteins and Peptides. Front. Cell Dev. Biol. 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.P.; DuBay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of “aggregation-prone” and “aggregation- susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Scherzer-Attali, R.; Pellarin, R.; Convertino, M.; Frydman-Marom, A.; Egoz-Matia, N.; Peled, S.; Levy-Sakin, M.; Shalev, D.E.; Caflisch, A.; Gazit, E.; et al. Complete phenotypic recovery of an Alzheimer’s disease model by a quinone-tryptophan hybrid aggregation inhibitor. PLoS ONE 2010, 5, e11101. [Google Scholar] [CrossRef]
- Paul, A.; Zhang, B.-D.; Mohapatra, S.; Li, G.; Li, Y.-M.; Gazit, E.; Segal, D. Novel Mannitol-Based Small Molecules for Inhibiting Aggregation of α-Synuclein Amyloids in Parkinson’s Disease. Front. Mol. Biosci. 2019, 6, 16. [Google Scholar] [CrossRef]
- Scherzer-Attali, R.; Shaltiel-Karyo, R.; Adalist, Y.; Segal, D.; Gazit, E. Generic inhibition of amyloidogenic proteins by two naphthoquinone-tryptophan hybrid molecules. Proteins Struct. Funct. Bioinforma. 2012, 80, 1962–1973. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 1–10. [Google Scholar] [CrossRef]
- Hudak, J.E.; Bertozzi, C.R. Glycotherapy: New advances inspire a reemergence of glycans in medicine. Chem. Biol. 2014, 21, 16–37. [Google Scholar] [CrossRef]
- Paul, A.; Frenkel-Pinter, M.; Escobar Alvarez, D.; Milordini, G.; Gazit, E.; Zacco, E.; Segal, D. Tryptophan-galactosylamine conjugates inhibit and disaggregate amyloid fibrils of Aβ42 and hIAPP peptides while reducing their toxicity. Commun. Biol. 2020, 3, 1–12. [Google Scholar] [CrossRef]
- Frydman-Marom, A.; Shaltiel-Karyo, R.; Moshe, S.; Gazit, E. The generic amyloid formation inhibition effect of a designed small aromatic β-breaking peptide. Amyloid 2011, 18, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Gazit, E. A possible role for π-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, J.G. Health functions and structure-activity relationships of natural anthraquinones from plants. Food Funct. 2018, 9, 6063–6080. [Google Scholar] [CrossRef] [PubMed]
- Nam, W.; Kim, S.; Nam, S.; Friedman, M. Structure-Antioxidative and Anti-Inflammatory Activity Relationships of Purpurin and Related Anthraquinones in Chemical and Cell Assays. Molecules 2017, 22, 265. [Google Scholar] [CrossRef]
- Viswanathan, G.K.; Shwartz, D.; Losev, Y.; Arad, E.; Shemesh, C.; Pichinuk, E.; Engel, H.; Raveh, A.; Jelinek, R.; Cooper, I.; et al. Purpurin modulates Tau-derived VQIVYK fibrillization and ameliorates Alzheimer’s disease-like symptoms in animal model. Cell. Mol. Life Sci. 2020, 77, 2795–2813. [Google Scholar] [CrossRef]
- Pickhardt, M.; Gazova, Z.; Von Bergen, M.; Khlistunova, I.; Wang, Y.; Hascher, A.; Mandelkow, E.M.; Biernat, J.; Mandelkow, E. Anthraquinones inhibit tau aggregation and dissolve Alzheimer’s paired helical filaments in vitro and in cells. J. Biol. Chem. 2005, 280, 3628–3635. [Google Scholar] [CrossRef]
- Gong, H.; He, Z.; Peng, A.; Zhang, X.; Cheng, B.; Sun, Y.; Zheng, L.; Huang, K. Effects of several quinones on insulin aggregation. Sci. Rep. 2014, 4, 5648. [Google Scholar] [CrossRef]
- Wu, S.B.; Long, C.; Kennelly, E.J. Structural diversity and bioactivities of natural benzophenones. Nat. Prod. Rep. 2014, 31, 1158–1174. [Google Scholar] [CrossRef]
- Ladiwala, A.R.A.; Mora-Pale, M.; Lin, J.C.; Bale, S.S.; Fishman, Z.S.; Dordick, J.S.; Tessier, P.M. Polyphenolic Glycosides and Aglycones Utilize Opposing Pathways To Selectively Remodel and Inactivate Toxic Oligomers of Amyloid β. ChemBioChem 2011, 12, 1749–1758. [Google Scholar] [CrossRef]
- Castillo, G.M.; Choi, P.Y.; Snow, A.D. Polyhydroxylated Aromatic Compounds for The Treatment of Amyloidosis and Α-Synuclein Fibril Diseases. WO2001049281A2, 28 December 2000. [Google Scholar]
- Bae, J.; Kim, N.; Shin, Y.; Kim, S.-Y.; Kim, Y.-J. Activity of catechins and their applications. Biomed. Dermatology 2020, 4, 1–10. [Google Scholar] [CrossRef]
- Andrich, K.; Bieschke, J. The effect of (-)-epigallo-catechin-(3)-gallate on amyloidogenic proteins suggests a common mechanism. Adv. Exp. Med. Biol. 2015, 863, 139–161. [Google Scholar] [PubMed]
- Liu, Y.; Liu, Y.; Wang, S.; Dong, S.; Chang, P.; Jiang, Z. Structural characteristics of (-)-epigallocatechin-3-gallate inhibiting amyloid Aβ42 aggregation and remodeling amyloid fibers. RSC Adv. 2015, 5, 62402–62413. [Google Scholar] [CrossRef]
- Cao, P.; Raleigh, D.P. Analysis of the inhibition and remodeling of islet amyloid polypeptide amyloid fibers by flavanols. Biochemistry 2012, 51, 2670–2683. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.E.; Xing, Y.F.; Huang, B.O.; Yi-Zheng Zhang, A.; Zeng, C.M. Tea catechins induce the conversion of preformed lysozyme amyloid fibrils to amorphous aggregates. J. Agric. Food Chem. 2009, 57, 11391–11396. [Google Scholar]
- Zhan, C.; Chen, Y.; Tang, Y.; Wei, G. Green Tea Extracts EGCG and EGC Display Distinct Mechanisms in Disrupting Aβ42Protofibril. ACS Chem. Neurosci. 2020, 11, 1841–1851. [Google Scholar] [CrossRef]
- Acharya, A.; Stockmann, J.; Beyer, L.; Rudack, T.; Nabers, A.; Gumbart, J.C.; Gerwert, K.; Batista, V.S. The Effect of (−)-Epigallocatechin-3-Gallate on the Amyloid-β Secondary Structure. Biophys. J. 2020, 119, 349–359. [Google Scholar] [CrossRef]
- Li, F.; Zhan, C.; Dong, X.; Wei, G. Molecular mechanisms of resveratrol and EGCG in the inhibition of Aβ42aggregation and disruption of Aβ42protofibril: Similarities and differences. Phys. Chem. Chem. Phys. 2021, 23, 18843–18854. [Google Scholar] [CrossRef]
- Yao, Y.; Tang, Y.; Wei, G. Epigallocatechin Gallate Destabilizes α-Synuclein Fibril by Disrupting the E46-K80 Salt-Bridge and Inter-protofibril Interface. ACS Chem. Neurosci. 2020, 11, 4351–4361. [Google Scholar] [CrossRef]
- Seidler, P.; Boyer, D.; Sawaya, M.; Ge, P.; Shin, W.; DeTure, M.; Dickson, D.; Jiang, L.; Eisenberg, D. CryoEM reveals how the small molecule EGCG binds to Alzheimer’s brain-derived tau fibrils and initiates fibril disaggregation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Roy, S.; Bhat, R. Suppression, disaggregation, and modulation of γ-Synuclein fibrillation pathway by green tea polyphenol EGCG. Protein Sci. 2019, 28, 382–402. [Google Scholar] [CrossRef]
- Kristen, A.V.; Lehrke, S.; Buss, S.; Mereles, D.; Steen, H.; Ehlermann, P.; Hardt, S.; Giannitsis, E.; Schreiner, R.; Haberkorn, U.; et al. Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clin. Res. Cardiol. 2012, 101, 805–813. [Google Scholar] [CrossRef] [PubMed]
- aus dem Siepen, F.; Buss, S.J.; Andre, F.; Seitz, S.; Giannitsis, E.; Steen, H.; Katus, H.A.; Kristen, A.V. Extracellular remodeling in patients with wild-type amyloidosis consuming epigallocatechin-3-gallate: Preliminary results of T1 mapping by cardiac magnetic resonance imaging in a small single center study. Clin. Res. Cardiol. 2015, 104, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, F.; Martone, R.; Taborchi, G.; Morini, S.; Bartolini, S.; Angelotti, P.; Farsetti, S.; Di Mario, C.; Perfetto, F. Epigallocatechin-3-gallate tolerability and impact on survival in a cohort of patients with transthyretin-related cardiac amyloidosis. A single-center retrospective study. Intern. Emerg. Med. 2018, 13, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Lange, K.W.; Lange, K.M.; Nakamura, Y. Green tea, epigallocatechin gallate and the prevention of Alzheimer’s disease: Clinical evidence. Food Sci. Hum. Wellness 2022, 11, 765–770. [Google Scholar] [CrossRef]
- Narayan, M.; Henríquez, G.; Gomez, A.; Guerrero, E. Potential role of natural polyphenols against protein aggregation toxicity: In vitro, in vivo, and clinical studies. ACS Chem. Neurosci. 2020, 11, 2915–2934. [Google Scholar]
- Baell, J.B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef]
- DI MARCO, A. Adriamycin (NSC-123,127): A new antibiotic with antitumor activity. Cancer Chemother. Rep. 1969, 53, 33–37. [Google Scholar]
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-hydroxydaimomycin, a new antitumor antibiotic fromS. Peucetius var.caesius. Biotechnol. Bioeng. 1969, 11, 1101–1110. [Google Scholar] [CrossRef]
- Post, C.; Tagliavini, F.; McArthur, R.A.; Della Vedova, F.; Gerna, M.; Bandiera, T.; Varasi, M.; Molinari, A.; Lansen, J. Anthracyclines and Amyloidosis. In Advances in Behavioral Biology; Springer: Boston, MA, USA, 1998; pp. 197–204. [Google Scholar]
- Stoilova, T.; Colombo, L.; Forloni, G.; Tagliavini, F.; Salmona, M. A new face for old antibiotics: Tetracyclines in treatment of amyloidoses. J. Med. Chem. 2013, 56, 5987–6006. [Google Scholar] [CrossRef]
- Forloni, G.; Colombo, L.; Girola, L.; Tagliavini, F.; Salmona, M. Anti-amyloidogenic activity of tetracyclines: Studies in vitro. FEBS Lett. 2001, 487, 404–407. [Google Scholar] [CrossRef]
- Ono, K.; Yamada, M. Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. J. Neurochem. 2006, 97, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hirohata, M.; Yamada, M. Anti-fibrillogenic and fibril-destabilizing activities of anti-Parkinsonian agents for α-synuclein fibrils in vitro. J. Neurosci. Res. 2007, 85, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, I.; Merlini, G.; Saraiva, M.J. 4 ′-iodo-4′-Deoxydoxorubicin and tetracyclines disrupt transthyretin amyloid fibrils in vitro producing noncytotoxic species: Screening for TTR fibril disrupters. FASEB J. 2003, 17, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, I.; Saraiva, M.J. Doxycycline disrupts transthyretin amyloid: Evidence from studies in a FAP transgenic mice model. FASEB J. 2006, 20, 234–239. [Google Scholar] [CrossRef]
- Giorgetti, S.; Raimondi, S.; Pagano, K.; Relini, A.; Bucciantini, M.; Corazza, A.; Fogolari, F.; Codutti, L.; Salmona, M.; Mangione, P.; et al. Effect of tetracyclines on the dynamics of formation and destructuration of β2-microglobulin amyloid fibrils. J. Biol. Chem. 2011, 286, 2121–2131. [Google Scholar] [CrossRef]
- Aitken, J.F.; Loomes, K.M.; Konarkowska, B.; Cooper, G.J.S. Suppression by polycyclic compounds of the conversion of human amylin into insoluble amyloid. Biochem. J. 2003, 374, 779–784. [Google Scholar] [CrossRef]
- Xu, J.; Zhao, C.; Huang, X.; Du, W. Tetracycline derivatives resist the assembly behavior of human islet amyloid polypeptide. Biochimie 2020, 174, 95–106. [Google Scholar] [CrossRef]
- Ward, J.E.; Ren, R.; Toraldo, G.; SooHoo, P.; Guan, J.; O’Hara, C.; Jasuja, R.; Trinkaus-Randall, V.; Liao, R.; Connors, L.H.; et al. Doxycycline reduces fibril formation in a transgenic mouse model of AL amyloidosis. Blood 2011, 118, 6610–6617. [Google Scholar] [CrossRef]
- Tagliavini, F.; Forloni, G.; Colombo, L.; Rossi, G.; Girola, L.; Canciani, B.; Angeretti, N.; Giampaolo, L.; Peressini, E.; Awan, T.; et al. Tetracycline affects abnormal properties of synthetic PrP peptides and PrP(Sc) in vitro. J. Mol. Biol. 2000, 300, 1309–1322. [Google Scholar] [CrossRef]
- Smith, D.L.; Woodman, B.; Mahal, A.; Sathasivam, K.; Ghazi-Noori, S.; Lowden, P.A.S.; Bates, G.P.; Hockly, E. Minocycline and doxycycline are not beneficial in a model of Huntington’s disease. Ann. Neurol. 2003, 54, 186–196. [Google Scholar] [CrossRef]
- Gertz, M.A.; Zeldenrust, S.R. Treatment of immunoglobulin light chain amyloidosis. Curr. Hematol. Malig. Rep. 2009, 4, 91–98. [Google Scholar] [CrossRef] [PubMed]
- SEBASTIÃO, M.P.; MERLINI, G.; SARAIVA, M.J.; DAMAS, A.M. The molecular interaction of 4′-iodo-4′-deoxydoxorubicin with Leu-55Pro transthyretin ‘amyloid-like’ oligomer leading to disaggregation. Biochem. J. 2000, 351, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Bellotti, V.; Gianni, A.M.; Merlini, G. New drug therapy of amyloidoses: Resorption of AL-type deposits with 4’- iodo-4’-deoxydoxorubicin. Blood 1995, 86, 855–861. [Google Scholar] [CrossRef]
- Merlini, G.; Ascari, E.; Amboldi, N.; Bellotti, V.; Arbustini, E.; Perfetti, V.; Ferrari, M.; Zorzoli, I.; Marinone, M.G.; Garini, P.; et al. Interaction of the anthracycline 4’-iodo-4’-deoxydoxorubicin with amyloid fibrils: Inhibition of amyloidogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 2959–2963. [Google Scholar] [CrossRef]
- Vivenza, D.; Feola, M.; Garrone, O.; Monteverde, M.; Merlano, M.; Lo Nigro, C. Role of the renin-angiotensin-aldosterone system and the glutathione S-transferase Mu, Pi and Theta gene polymorphisms in cardiotoxicity after anthracycline chemotherapy for breast carcinoma. Int. J. Biol. Markers 2013, 28, e336–e347. [Google Scholar] [CrossRef]
- Narezkina, A.; Narayan, H.K.; Zemljic-Harpf, A.E. Molecular mechanisms of anthracycline cardiovascular toxicity. Clin. Sci. 2021, 135, 1311–1332. [Google Scholar] [CrossRef]
- Gautieri, A.; Beeg, M.; Gobbi, M.; Rigoldi, F.; Colombo, L.; Salmona, M. The Anti-Amyloidogenic Action of Doxycycline: A Molecular Dynamics Study on the Interaction with Aβ42. Int. J. Mol. Sci. 2019, 20, 4641. [Google Scholar] [CrossRef]
- Andes, D.; Craig, W.A. Pharmacokinetics and Pharmacodynamics of Tetracyclines. In Antimicrobial Pharmacodynamics in Theory and Clinical Practice, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 279–290. [Google Scholar]
- Danesi, R.; Fogli, S.; Gennari, A.; Conte, P.; Del Tacca, M. Pharmacokinetic-pharmacodynamic relationships of the anthracycline anticancer drugs. Clin. Pharmacokinet. 2002, 41, 431–444. [Google Scholar] [CrossRef]
- Lees, P.; Landoni, M.F.; Giraudel, J.; Toutain, P.L. Pharmacodynamics and pharmacokinetics of nonsteroidal anti-inflammatory drugs in species of veterinary interest. J. Vet. Pharmacol. Ther. 2004, 27, 479–490. [Google Scholar] [CrossRef]
- Miller, S.R.; Sekijima, Y.; Kelly, J.W. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab. Investig. 2004, 84, 545–552. [Google Scholar] [CrossRef]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA - J. Am. Med. Assoc. 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y.; Tojo, K.; Morita, H.; Koyama, J.; Ikeda, S.I. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid 2015, 22, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Ikram, A.; Donnelly, J.P.; Sperry, B.W.; Samaras, C.; Valent, J.; Hanna, M. Diflunisal tolerability in transthyretin cardiac amyloidosis: A single center’s experience. Amyloid 2018, 25, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Ono, K.; Shibata, S.; Nakamura, K.; Komatsu, J.; Ikeda, Y.; Ikeda, T.; Samuraki, M.; Sakai, K.; Iwasa, K.; et al. Efficacy of diflunisal on autonomic dysfunction of late-onset familial amyloid polyneuropathy (TTR Val30Met) in a Japanese endemic area. J. Neurol. Sci. 2014, 345, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Hirohata, M.; Ono, K.; Naiki, H.; Yamada, M. Non-steroidal anti-inflammatory drugs have anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. Neuropharmacology 2005, 49, 1088–1099. [Google Scholar] [CrossRef]
- Hirohata, M.; Ono, K.; Morinaga, A.; Yamada, M. Non-steroidal anti-inflammatory drugs have potent anti-fibrillogenic and fibril-destabilizing effects for α-synuclein fibrils in vitro. Neuropharmacology 2008, 54, 620–627. [Google Scholar] [CrossRef]
- Azam, F.; Alabdullah, N.H.; Ehmedat, H.M.; Abulifa, A.R.; Taban, I.; Upadhyayula, S. NSAIDs as potential treatment option for preventing amyloid β toxicity in Alzheimer’s disease: An investigation by docking, molecular dynamics, and DFT studies. J. Biomol. Struct. Dyn. 2018, 36, 2099–2117. [Google Scholar] [CrossRef]
- Wichmann, M.A.; Cruickshanks, K.J.; Carlsson, C.M.; Chappell, R.; Fischer, M.E.; Klein, B.E.K.; Klein, R.; Schubert, C.R. NSAID Use and Incident Cognitive Impairment in a Population-based Cohort. Alzheimer Dis. Assoc. Disord. 2016, 30, 105–112. [Google Scholar] [CrossRef]
- Breitner, J.C.S.; Haneuse, S.J.P.A.; Walker, R.; Dublin, S.; Crane, P.K.; Gray, S.L.; Larson, E.B. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology 2009, 72, 1899–1905. [Google Scholar] [CrossRef]
- Thal, L.J.; Ferris, S.H.; Kirby, L.; Block, G.A.; Lines, C.R.; Yuen, E.; Assaid, C.; Nessly, M.L.; Norman, B.A.; Baranak, C.C.; et al. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 2005, 30, 1204–1215. [Google Scholar] [CrossRef]
- Cunningham, C.; Skelly, D.T. Non-steroidal anti-inflammatory drugs and cognitive function: Are prostaglandins at the heart of cognitive impairment in dementia and delirium? J. Neuroimmune Pharmacol. 2012, 7, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Valiente-Gabioud, A.A.; Miotto, M.C.; Chesta, M.E.; Lombardo, V.; Binolfi, A.; Fernández, C.O. Phthalocyanines as Molecular Scaffolds to Block Disease-Associated Protein Aggregation. Acc. Chem. Res. 2016, 49, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Ghosh, K.S. Inhibition of Amyloid Fibrillation by Small Molecules and Nanomaterials: Strategic Development of Pharmaceuticals Against Amyloidosis. Protein Pept. Lett. 2019, 26, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Gour, N.; Koshti, B. A Chemical Perspective to the Anti-Amyloid Action of Compounds and a Nanoparticle Based Assay for Screening Amyloid Inhibitors. ChemRxiv 2019. [Google Scholar] [CrossRef]
- Liu, Y.; Carver, J.A.; Ho, L.H.; Elias, A.K.; Musgrave, I.F.; Pukala, T.L. Hemin as a generic and potent protein misfolding inhibitor. Biochem. Biophys. Res. Commun. 2014, 454, 295–300. [Google Scholar] [CrossRef]
- Sonavane, S.; Haider, S.Z.; Kumar, A.; Ahmad, B. Hemin is able to disaggregate lysozyme amyloid fibrils into monomers. Biochim. Biophys. Acta—Proteins Proteomics 2017, 1865, 1315–1325. [Google Scholar] [CrossRef]
- Yuan, C.; Gao, Z. Aβ interacts with both the iron center and the porphyrin ring of heme: Mechanism of heme’s action on Aβ aggregation and disaggregation. Chem. Res. Toxicol. 2013, 26, 262–269. [Google Scholar] [CrossRef]
- Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.I.; Iwatsubo, T.; Goedert, M.; Hasegawa, M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005, 280, 7614–7623. [Google Scholar] [CrossRef]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.K.; Roth, M.; Harrington, C.R. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef]
- Chernii, S.; Losytskyy, M.; Kelm, A.; Gorski, A.; Tretyakova, I.; Yarmoluk, S.; Chernii, V.; Kovalska, V. Study of tetraphenylporphyrins as modifiers of insulin amyloid aggregation. J. Mol. Recognit. 2020, 33, e2811. [Google Scholar] [CrossRef]
- Abelein, A.; Kaspersen, J.D.; Nielsen, S.B.; Jensen, G.V.; Christiansen, G.; Pedersen, J.S.; Danielsson, J.; Otzen, D.E.; Gräslund, A. Formation of dynamic soluble surfactant-induced amyloid β peptide aggregation intermediates. J. Biol. Chem. 2013, 288, 23518–23528. [Google Scholar] [CrossRef] [PubMed]
- Sabaté, R.; Estelrich, J. Stimulatory and inhibitory effects of alkyl bromide surfactants on β-amyloid fibrillogenesis. Langmuir 2005, 21, 6944–6949. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.S.; Chen, Y.T.; Chou, S.W. Inhibition of amyloid fibril formation of β-amyloid peptides via the amphiphilic surfactants. Biochim. Biophys. Acta—Mol. Basis Dis. 2005, 1741, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.K.; Ghosh, S.; Dasgupta, S. Effect of surfactants on preformed fibrils of human serum albumin. Int. J. Biol. Macromol. 2013, 59, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Yaseen, Z.; Rehman, S.U.; Tabish, M.; Shalla, A.H.; Kabir-ud-Din, K.-D. Modulation of bovine serum albumin fibrillation by ester bonded and conventional gemini surfactants. RSC Adv. 2015, 5, 58616–58624. [Google Scholar] [CrossRef]
- Bhat, W.F.; Bhat, I.A.; Bhat, S.A.; Bano, B. In vitro disintegration of goat brain cystatin fibrils using conventional and gemini surfactants: Putative therapeutic intervention in amyloidoses. Int. J. Biol. Macromol. 2016, 93, 493–500. [Google Scholar] [CrossRef]
- Han, Y.; He, C.; Cao, M.; Huango, X.; Wang, Y.; Li, Z. Facile disassembly of amyloid fibrils using gemini surfactant micelles. Langmuir 2010, 26, 1583–1587. [Google Scholar] [CrossRef]
- He, C.; Hou, Y.; Han, Y.; Wang, Y. Disassembly of amyloid fibrils by premicellar and micellar aggregates of a tetrameric cationic surfactant in aqueous solution. Langmuir 2011, 27, 4551–4556. [Google Scholar] [CrossRef]
- Zhu, L.; Han, Y.; He, C.; Huang, X.; Wang, Y. Disaggregation ability of different chelating molecules on copper ion-triggered amyloid fibers. J. Phys. Chem. B 2014, 118, 9298–9305. [Google Scholar] [CrossRef]
- Menger, F.M.; Keiper, J.S. Gemini Surfactants. Angew. Chemie Int. Ed. 2000, 39, 1906–1920. [Google Scholar] [CrossRef]
- Miura, M.; Kodama, M. The Second CMC of the Aqueous Solution of Sodium Dodecyl Sulfate. I. Conductivity. Bull. Chem. Soc. Jpn. 1972, 45, 428–431. [Google Scholar] [CrossRef]
- Hou, Y.; Han, Y.; Deng, M.; Xiang, J.; Wang, Y. Aggregation behavior of a tetrameric cationic surfactant in aqueous solution. Langmuir 2010, 26, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.K.; Chatterjee, S.K.; Bhattarai, A. Micellization of cationic surfactants in alcohol — water mixed solvent media. J. Mol. Liq. 2016, 222, 906–914. [Google Scholar] [CrossRef]
- Bombelli, C.; Giansanti, L.; Luciani, P.; Mancini, G. Gemini Surfactant Based Carriers in Gene and Drug Delivery. Curr. Med. Chem. 2008, 16, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A.J.; Camilleri, P.; Engberts, J.B.F.N.; Feiters, M.C.; Nolte, R.J.M.; Söderman, O.; Bergsma, M.; Bell, P.C.; Fielden, M.L.; García Rodríguez, C.L.; et al. Gemini Surfactants: New Synthetic Vectors for Gene Transfection. Angew. Chemie Int. Ed. 2003, 42, 1448–1457. [Google Scholar] [CrossRef]
- Chatani, E.; Lee, Y.H.; Yagi, H.; Yoshimura, Y.; Naiki, H.; Goto, Y. Ultrasonication-dependent production and breakdown lead to minimum-sized amyloid fibrils. Proc. Natl. Acad. Sci. USA 2009, 106, 11119–11124. [Google Scholar] [CrossRef]
- Ikenoue, T.; Lee, Y.-H.; Kardos, J.; Saiki, M.; Yagi, H.; Kawata, Y.; Goto, Y. Cold Denaturation of α-Synuclein Amyloid Fibrils. Angew. Chemie Int. Ed. 2014, 53, 7799–7804. [Google Scholar] [CrossRef]
- Arora, A.; Ha, C.; Park, C.B. Insulin amyloid fibrillation at above 100 °C: New insights into protein folding under extreme temperatures. Protein Sci. 2004, 13, 2429–2436. [Google Scholar] [CrossRef]
- Dubois, J.; Ismail, A.A.; Chan, S.L.; Ali-Khan, Z. Fourier Transform Infrared Spectroscopic Investigation of Temperature- and Pressure-Induced Disaggregation of Amyloid A. Scand. J. Immunol. 1999, 49, 376–380. [Google Scholar] [CrossRef]
- Kim, Y.S.; Randolph, T.W.; Seefeldt, M.B.; Carpenter, J.F. High-Pressure Studies on Protein Aggregates and Amyloid Fibrils. Methods Enzymol. 2006, 413, 237–253. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, Z.L.; Brito, R.M.M. Amyloid Disassembly: What Can We Learn from Chaperones? Biomedicines 2022, 10, 3276. https://doi.org/10.3390/biomedicines10123276
Almeida ZL, Brito RMM. Amyloid Disassembly: What Can We Learn from Chaperones? Biomedicines. 2022; 10(12):3276. https://doi.org/10.3390/biomedicines10123276
Chicago/Turabian StyleAlmeida, Zaida L., and Rui M. M. Brito. 2022. "Amyloid Disassembly: What Can We Learn from Chaperones?" Biomedicines 10, no. 12: 3276. https://doi.org/10.3390/biomedicines10123276
APA StyleAlmeida, Z. L., & Brito, R. M. M. (2022). Amyloid Disassembly: What Can We Learn from Chaperones? Biomedicines, 10(12), 3276. https://doi.org/10.3390/biomedicines10123276