

Anderson–Fabry Disease: A New Piece of the Lysosomal Puzzle in Parkinson Disease?
 ,
, 

 and
and
Abstract
1. Introduction
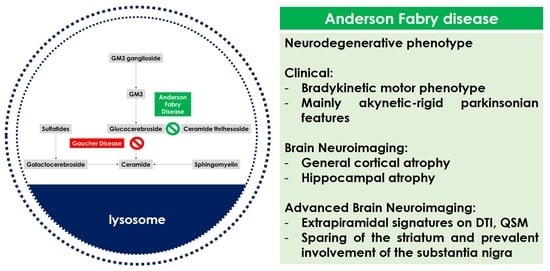
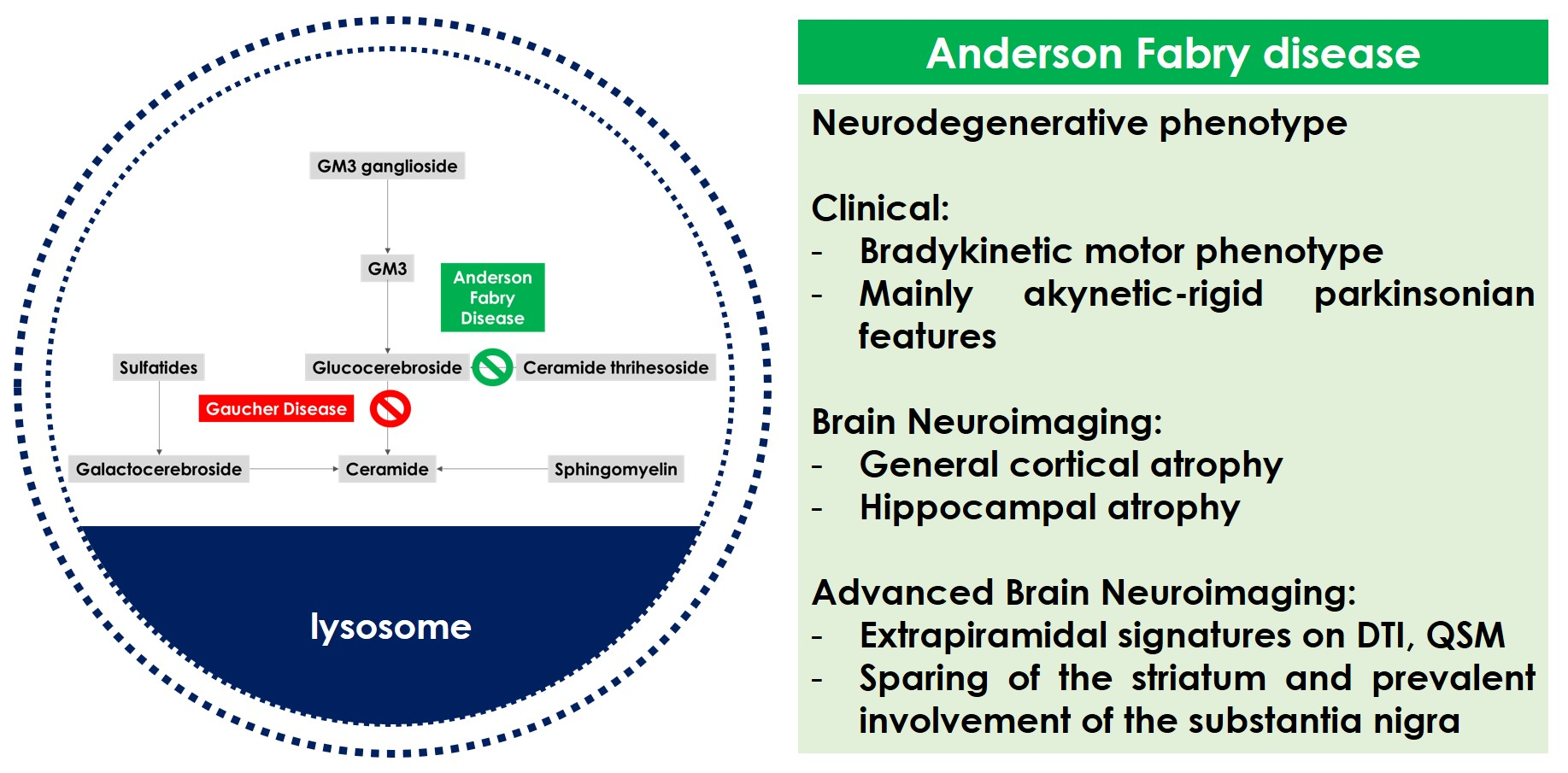
2. Anderson–Fabry Disease, Brain and Neurodegeneration
3. Anderson–Fabry Disease and Extrapyramidal Phenotype
3.1. Literature Data
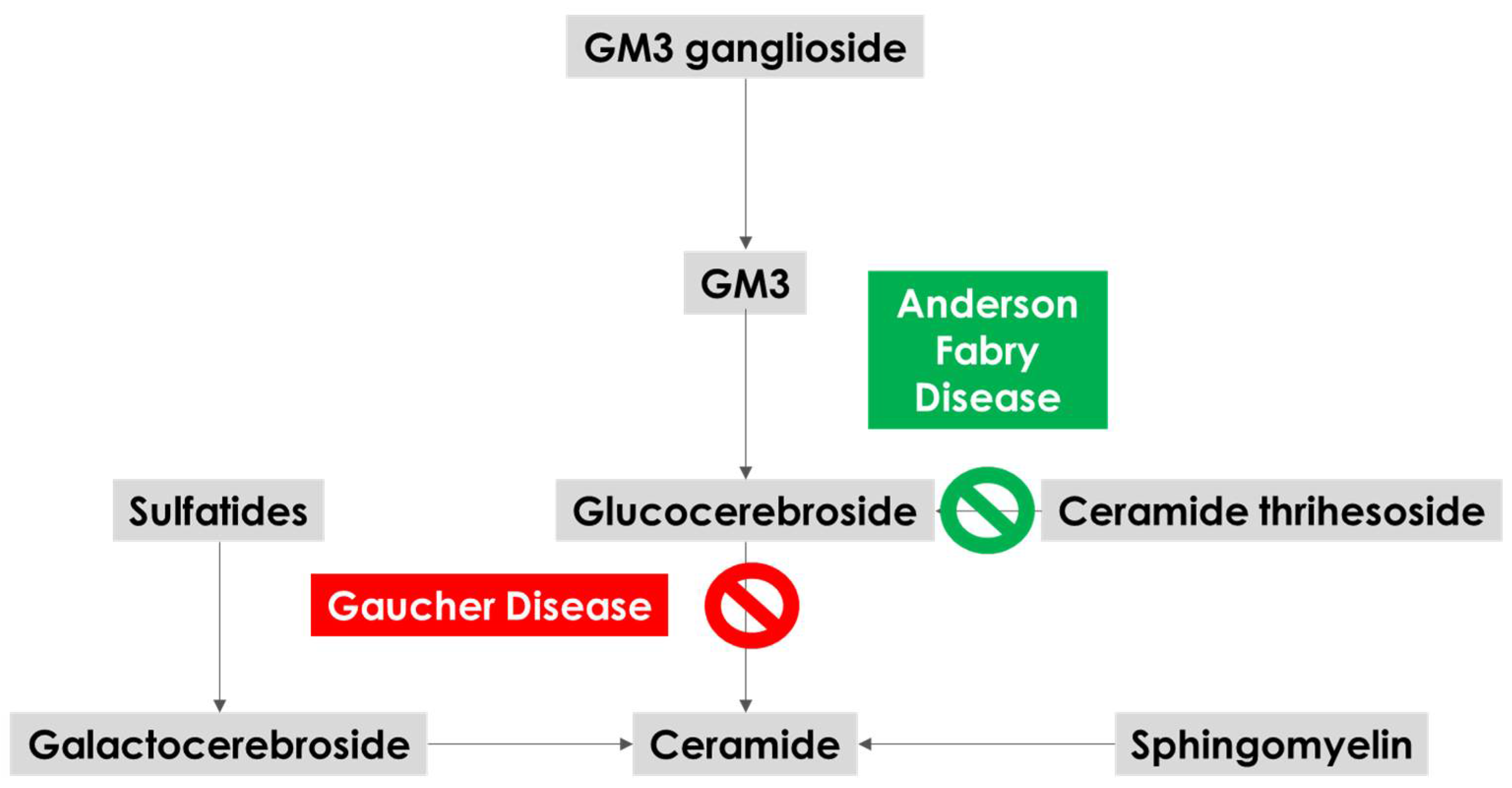
3.2. Biological Mechanisms
4. Research Perspectives and Therapeutic Implications
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Calhoun, D.H.; Bishop, D.F.; Bernstein, H.S.; Quinn, M.; Hantzopoulos, P.; Desnick, R.J. Fabry disease: Isolation of a cDNA clone encoding human α-galactosidase A. Proc. Natl. Acad. Sci. USA 1985, 82, 7364–7368. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ncbi.nlm.nih.gov/gene/2717 (accessed on 20 October 2022).
- Favalli, V.; Disabella, E.; Molinaro, M.; Tagliani, M.; Scarabotto, A.; Serio, A.; Grasso, M.; Narula, N.; Giorgianni, C.; Caspani, C.; et al. Genetic Screening of Anderson-Fabry Disease in Probands Referred From Multispecialty Clinics. J. Am. Coll. Cardiol. 2016, 68, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Pan, J.; Tang, S.; Duan, D.; Yu, D.; Nong, H.; Wang, Z. Global Trends in the Incidence, Prevalence, and Years Lived with Disability of Parkinson’s Disease in 204 Countries/Territories from 1990 to 2019. Front. Public Health 2021, 9, 776847. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, Y.; Zhang, C.; Wang, Y.X.; Fernandez-Funez, P. Genetics of Parkinson’s disease and related disorders. J. Med. Genet. 2018, 55, 73–80. [Google Scholar] [CrossRef]
- Funayama, M.; Nishioka, K.; Li, Y.; Hattori, N. Molecular genetics of Parkinson’s disease: Contributions and global trends. J. Hum. Genet. 2022. [Google Scholar] [CrossRef]
- Skrahina, V.; Gaber, H.; Vollstedt, E.J.; Förster, T.M.; Usnich, T.; Curado, F.; Brüggemann, N.; Paul, J.; Bogdanovic, X.; Zülbahar, S.; et al. ROPAD Study Group. The Rostock International Parkinson’s Disease (ROPAD) Study: Protocol and Initial Findings. Mov. Disord. 2021, 36, 1005–1010. [Google Scholar] [CrossRef]
- Jia, F.; Fellner, A.; Kumar, K.R. Monogenic Parkinson’s Disease: Genotype, Phenotype, Pathophysiology, and Genetic Testing. Genes 2022, 13, 471. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef]
- Cabezudo, D.; Baekelandt, V.; Lobbestael, E. Multiple-Hit Hypothesis in Parkinson’s Disease: LRRK2 and Inflammation. Front. Neurosci. 2020, 14, 376. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Kandasamy, N.; Ullah, A.; Paramasivam, N.; Öztürk, M.A.; Naureen, S.; Arshad, A.; Badshah, M.; Khan, K.; Wajid, M.; et al. Putative second hit rare genetic variants in families with seemingly GBA-associated Parkinson’s disease. npj Genom. Med. 2021, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Cavallieri, F.; Fioravanti, V.; Toschi, G.; Grisanti, S.; Napoli, M.; Moratti, C.; Pascarella, R.; Versari, A.; Fraternali, A.; Casali, M.; et al. COVID-19 and Parkinson’s disease: A casual association or a possible second hit in neurodegeneration? J. Neurol. 2021, 269, 59–61. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, G.; Desouza, R.-M.; Balestrino, R.; Schapira, A.H. Glucocerebrosidase Mutations in Parkinson Disease. J. Park. Dis. 2017, 7, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Menozzi, E.; Schapira, A.H.V. Exploring the Genotype–Phenotype Correlation in GBA-Parkinson Disease: Clinical Aspects, Biomarkers, and Potential Modifiers. Front. Neurol. 2021, 12, 694764. [Google Scholar] [CrossRef]
- Malek, N.; Weil, R.S.; Bresner, C.; Lawton, M.A.; Grosset, K.A.; Tan, M.; Bajaj, N.; Barker, R.A.; Burn, D.J.; Foltynie, T.; et al. Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkin-son’s study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 702–709. [Google Scholar] [CrossRef]
- Löhle, M.; Hughes, D.; Milligan, A.; Richfield, L.; Reichmann, H.; Mehta, A.; Schapira, A.H. Clinical prodromes of neurodegeneration in Anderson-Fabry disease. Neurology 2015, 84, 1454–1464. [Google Scholar] [CrossRef]
- Kahn, P. Anderson-Fabry disease: A histopathological study of three cases with observations on the mechanism of production of pain. J. Neurol. Neurosurg. Psychiatry 1973, 36, 1053–1062. [Google Scholar] [CrossRef]
- Garzuly, F.; Maródi, L.; Erdös, M.; Grubits, J.; Varga, Z.; Gelpi, E.; Rohonyi, B.; Mázló, M.; Molnár, A.; Budka, H. Megadolichobasilar anomaly with thrombosis in a family with Fabry’s disease and a novel mutation in the α-galactosidase A gene. Brain 2005, 128, 2078–2083. [Google Scholar] [CrossRef]
- Schiffmann, R.; Rapkiewicz, A.; Abu-Asab, M.; Ries, M.; Askari, H.; Tsokos, M.; Quezado, M. Pathological findings in a patient with Fabry disease who died after 2.5 years of enzyme replacement. Virchows Arch. 2005, 448, 337–343. [Google Scholar] [CrossRef]
- Lelieveld, I.M.; Böttcher, A.; Hennermann, J.B.; Beck, M.; Fellgiebel, A. Eight-Year Follow-Up of Neuropsychiatric Symptoms and Brain Structural Changes in Fabry Disease. PLoS ONE 2015, 10, e0137603. [Google Scholar] [CrossRef] [PubMed]
- Fellgiebel, A.; Wolf, D.O.; Kolodny, E.; Müller, M.J. Hippocampal atrophy as a surrogate of neuronal involvement in Fabry disease. J. Inherit. Metab. Dis. 2011, 35, 363–367. [Google Scholar] [CrossRef] [PubMed]
- DeVeber, G.A.; Schwarting, G.A.; Kolodny, E.H. Fabry disease: Immunocytochemical characterization of neuronal involvement. Ann. Neurol. 1992, 31, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Kaye, E.M.; Kolodny, E.H.; Logigian, E.L.; Ullman, M.D. Nervous system involvement in Fabry’s disease: Clinicopathological and biochemical correlation. Ann. Neurol. 1988, 23, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Okeda, R.; Nisihara, M. An autopsy case of Fabry disease with neuropathological investigation of the pathogenesis of associated dementia. Neuropathology 2008, 28, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Cocozza, S.; Russo, C.; Pontillo, G.; Pisani, A.; Brunetti, A. Neuroimaging in Fabry disease: Current knowledge and future directions. Insights Imaging 2018, 9, 1077–1088. [Google Scholar] [CrossRef]
- Buechner, S.; Moretti, M.; Burlina, A.P.; Cei, G.; Manara, R.; Ricci, R.; Mignani, R.; Parini, R.; Di Vito, R.; Giordano, G.P.; et al. Central nervous system involvement in Anderson-Fabry disease: A clinical and MRI retrospective study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1249–1254. [Google Scholar] [CrossRef]
- Paavilainen, T.; Lepomäki, V.; Saunavaara, J.; Borra, R.; Nuutila, P.; Kantola, I.; Parkkola, R. Diffusion tensor imaging and brain volumetry in Fabry disease patients. Neuroradiology 2013, 55, 551–558. [Google Scholar] [CrossRef]
- Cocozza, S.; Pisani, A.; Olivo, G.; Saccà, F.; Ugga, L.; Riccio, E.; Migliaccio, S.; Brescia Morra, V.; Brunetti, A.; Quarantelli, M.; et al. Alterations of functional connectivity of the motor cortex in Fabry disease: An RS-fMRI study. Neurology 2017, 88, 1822–1829. [Google Scholar] [CrossRef]
- Pontillo, G.; Cocozza, S.; Brunetti, A.; Brescia Morra, V.; Riccio, E.; Russo, C.; Saccà, F.; Tedeschi, E.; Pisani, A.; Quarantelli, M. Reduced Intracranial Volume in Fabry Disease: Evidence of Abnormal Neurodevelopment? Front. Neurol. 2018, 9, 672. [Google Scholar] [CrossRef]
- Fellgiebel, A.; Albrecht, J.; Dellani, P.R.; Schermuly, I.; Stoeter, P.; Müller, M.J. Quantification of brain tissue alterations in Fabry disease using diffusion-tensor imaging. Acta Paediatr. 2007, 96, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.F.; Schiffmann, R.; Ulug, A.M. Elevated CNS average diffusion constant in Fabry disease. Acta Paediatr. 2002, 91, 67–68. [Google Scholar] [CrossRef] [PubMed]
- Fellgiebel, A.; Mazanek, M.; Whybra, C.; Beck, M.; Hartung, R.; Müller, K.M.; Scheurich, A.; Dellani, P.R.; Stoeter, P.; Müller, M.J. Pattern of microstructural brain tissue alterations in Fabry disease: A diffusion-tensor imaging study. J. Neurol. 2006, 253, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, J.; Dellani, P.R.; Müller, M.J.; Schermuly, I.; Beck, M.; Stoeter, P.; Gerhard, A.; Fellgiebel, A. Voxel based analyses of diffusion tensor imaging in Fabry disease. J. Neurol. Neurosurg Psychiatry 2007, 78, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Jenkinson, M.; Johansen-Berg, H.; Rueckert, D.; Nichols, T.E.; Mackay, C.E.; Watkins, K.E.; Ciccarelli, O.; Cader, M.Z.; Matthews, P.M.; et al. Tract-based spatial statistics: Voxelwise analysis of multi-subject diffusion data. Neuroimage 2006, 31, 1487–1505. [Google Scholar] [CrossRef]
- Cocozza, S.; Pontillo, G.; Quarantelli, M.; Saccà, F.; Riccio, E.; Costabile, T.; Olivo, G.; Brescia Morra, V.; Pisani, A.; Brunetti, A.; et al. Default mode network modifications in Fabry disease: A resting-state fMRI study with structural correlations. Hum Brain Mapp. 2018, 39, 1755–1764. [Google Scholar] [CrossRef]
- Gavazzi, C.; Borsini, W.; Guerrini, L.; Della Nave, R.; Rocca, M.A.; Tessa, C.; Buchner, S.; Belli, G.; Filippi, M.; Villari, N.; et al. Subcortical damage and cortical functional changes in men and women with Fabry disease: A multifaceted MR study. Radiology 2006, 241, 492–500. [Google Scholar] [CrossRef]
- Russo, C.; Pontillo, G.; Pisani, A.; Saccà, F.; Riccio, E.; Macera, A.; Rusconi, G.; Stanzione, A.; Borrelli, P.; Morra, V.B.; et al. Striatonigral involvement in Fabry Disease: A quantitative and volumetric Magnetic Resonance Imaging study. Park. Relat. Disord. 2018, 57, 27–32. [Google Scholar] [CrossRef]
- Ravanfar, P.; Loi, S.M.; Syeda, W.T.; Van Rheenen, T.E.; Bush, A.I.; Desmond, P.; Cropley, V.L.; Lane, D.J.R.; Opazo, C.M.; Moffat, B.A.; et al. Systematic Review: Quantitative Susceptibility Mapping (QSM) of Brain Iron Profile in Neurodegenerative Diseases. Front. Neurosci. 2021, 15, 618435. [Google Scholar] [CrossRef]
- Murakami, Y.; Kakeda, S.; Watanabe, K.; Ueda, I.; Ogasawara, A.; Moriya, J.; Ide, S.; Futatsuya, K.; Sato, T.; Okada, K.; et al. Usefulness of Quantitative Susceptibility Mapping for the Diagnosis of Parkinson Disease. Am. J. Neuroradiol. 2015, 36, 1102–1108. [Google Scholar] [CrossRef]
- Sjöström, H.; Granberg, T.; Westman, E.; Svenningsson, P. Quantitative susceptibility mapping differentiates between parkinsonian disorders. Park. Relat. Disord. 2017, 44, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Dagan, E.; Schlesinger, I.; Ayoub, M.; Mory, A.; Nassar, M.; Kurolap, A.; Peretz-Aharon, J.; Gershoni-Baruch, R. The contribution of Niemann-Pick SMPD1 mutations to Parkinson disease in Ashkenazi Jews. Parkinsonism Relat Disord. 2015, 21, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.N.; Liany, H.; Bei, J.X.; Yu, X.Q.; Liu, J.; Au, W.L.; Prakash, K.M.; Tan, L.C.; Tan, E.K. Rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson’s disease. Neurobiol. Aging 2013, 34, 2890.e13–2890.e15. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Ozelius, L.J.; Bar-Shira, A.; Saunders-Pullman, R.; Mirelman, A.; Kornreich, R.; Gana-Weisz, M.; Raymond, D.; Rozenkrantz, L.; Deik, A.; et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 2013, 80, 1606–1610. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Levy, O.A.; Wolf, P.; Oliva, P.; Zhang, X.K.; Waters, C.H.; Fahn, S.; Kang, U.; Liong, C.; Ford, B. SCARB2 variants and glucocerebrosidase activity in Parkinson’s disease. npj Park. Dis. 2016, 2, 16004. [Google Scholar] [CrossRef]
- Van der Lienden, M.; Aten, J.; Marques, A.; Waas, I.; Larsen, P.; Claessen, N.; van der Wel, N.; Ottenhoff, R.; van Eijk, M.; Aerts, J. GCase and LIMP2 Abnormalities in the Liver of Niemann Pick Type C Mice. Int. J. Mol. Sci. 2021, 22, 2532. [Google Scholar] [CrossRef]
- Wu, G.; Yan, B.; Wang, X.; Feng, X.; Zhang, A.; Xu, X.; Dong, H. Decreased activities of lysosomal acid alpha-D-galactosidase A in the leukocytes of sporadic Parkinson’s disease. J. Neurol. Sci. 2008, 271, 168–173. [Google Scholar] [CrossRef]
- Nelson, M.P.; Tse, T.E.; O’Quinn, D.B.; Percival, S.M.; Jaimes, E.A.; Warnock, D.G.; Shacka, J.J. Autophagy-lysosome pathway associated neuropathology and axonal degeneration in the brains of alpha-galactosidase A-deficient mice. Acta Neuropathol Commun. 2014, 2, 20. [Google Scholar] [CrossRef]
- Orimo, S.; Iwasaki, T.; Yoshino, H.; Arai, M.; Hiyamuta, E. An autopsied case of Fabry’s disease presenting with parkinsonism and cardiomegaly as a cardinal clinical manifestation. Rinsho Shinkeigaku 1994, 34, 1003–1007. [Google Scholar]
- Buechner, S.; De Cristofaro, M.T.R.; Ramat, S.; Borsini, W. Parkinsonism and Anderson Fabry’s disease: A case report. Mov. Disord. 2006, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Duran, R.; Proukakis, C.; Bras, J.; Hughes, D.; Mehta, A.; Hardy, J.; Wood, N.W.; Schapira, A.H. Hyposmia and cognitive impairment in Gaucher disease patients and carriers. Mov. Disord. 2012, 27, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Wise, A.H.; Yang, A.; Naik, H.; Stauffer, C.; Zeid, N.; Liong, C.; Balwani, M.; Desnick, R.J.; Alcalay, R.N. Parkinson’s disease prevalence in Fabry disease: A survey study. Mol. Genet. Metab. Rep. 2017, 14, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Marder, K.; Levy, G.; Louis, E.D.; Mejia-Santana, H.; Cote, L.; Andrews, H.; Harris, J.; Waters, C.; Ford, B.; Frucht, S.; et al. Accuracy of family history data on Parkinson’s disease. Neurology 2003, 61, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Wang, Y.; Alcalay, R.N.; Mejia-Santana, H.; Saunders-Pullman, R.; Bressman, S.; Corvol, J.-C.; Brice, A.; Lesage, S.; Mangone, G.; et al. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov. Disord. 2017, 32, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Dinur, T.; Quinn, T.; Sakanaka, K.; Levy, O.; Waters, C.; Fahn, S.; Dorovski, T.; Chung, W.K.; Pauciulo, M.; et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 2014, 71, 752–757. [Google Scholar] [CrossRef]
- Glass, R.B.; Astrin, K.H.; Norton, K.I.; Parsons, R.; Eng, C.M.; Banikazemi, M.; Desnick, R.J. Fabry disease: Renal sonographic and magnetic resonance imaging findings in affected males and carrier females with the classic and cardiac variant phenotypes. J. Comput. Assist. Tomogr. 2004, 28, 158–168. [Google Scholar] [CrossRef]
- Eng, C.M.; Niehaus, D.J.; Enriquez, A.L.; Burgert, T.S.; Ludman, M.D.; Desnick, R.J. Fabry disease: Twenty-three mutations including sense and antisense CpG alterations and identification of a deletional hot-spot in the alpha-galactosidase a gene. Hum. Mol. Genet. 1994, 3, 1795–1799. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Gago, M.F.; Azevedo, O.; Guimarães, A.; Teresa Vide, A.; Lamas, N.J.; Oliveira, T.G.; Gaspar, P.; Bicho, E.; Miltenberger-Miltenyi, G.; Ferreira, J.; et al. Parkinson’s Disease and Fabry Disease: Clinical, Biochemical and Neuroimaging Analysis of Three Pedigrees. J. Parkinsons Dis. 2020, 10, 141–152. [Google Scholar] [CrossRef]
- Ferreira, J.J.; Goncalves, N.; Valadas, A.; Januario, C.; Silva, M.R.; Nogueira, L.; Vieira, J.L.M.; Lima, A.B. Prevalence of Parkinson’s disease: A population-based study in Portugal. Eur. J. Neurol. 2017, 24, 748–750. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, P.; Azevedo, O.; Pinto, R.; Marino, J.; Cardoso, C.; Sousa, N.; Cunha, D.; Hughes, D.; Ducla Soares, J.L. Biomarkers of myocardial fibrosis: Revealing the natural history of fibrogenesis in fabry disease cardiomyopathy. J. Am. Heart Assoc. 2018, 7, e007124. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, O.; Gal, A.; Faria, R.; Gaspar, P.; Miltenberger-Milteny, G.; Gago, M.F.; Dias, F.; Martins, A.; Rodrigues, J.; Reimão, P.; et al. Founder effect of Fabry disease due to p.F113L mutation: Clinical profile of a late-onset phenotype. Mol. Genet. Metab. 2019, 129, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Moccia, M.; Tedeschi, E.; Ugga, L.; Erro, R.; Picillo, M.; Caranci, F.; Barone, P.; Brunetti, A. White matter changes and the development of motor phenotypes in de novo Parkinson’s Disease. J. Neurol. Sci. 2016, 367, 215–219. [Google Scholar] [CrossRef]
- Cocozza, S.; Schiavi, S.; Pontillo, G.; Battocchio, M.; Riccio, E.; Caccavallo, S.; Russo, C.; Di Risi, T.; Pisani, A.; Daducci, A.; et al. Microstructural damage of the cortico-striatal and thalamo-cortical fibers in Fabry disease: A diffusion MRI tractometry study. Neuroradiology 2020, 62, 1459–1466. [Google Scholar] [CrossRef]
- He, N.; Huang, P.; Ling, H.; Langley, J.; Liu, C.; Ding, B.; Huang, J.; Xu, H.; Zhang, Y.; Zhang, Z.; et al. Dentate nucleus iron deposition is a potential biomarker for tremor-dominant Parkinson’s disease. NMR Biomed. 2017, 30, e3554. [Google Scholar] [CrossRef]
- Menke, R.A.; Jbabdi, S.; Miller, K.L.; Matthews, P.M.; Zarei, M. Connectivity-based segmentation of the substantia nigra in human and its implications in Parkinson’s disease. Neuroimage 2010, 52, 1175–1180. [Google Scholar] [CrossRef]
- Camlidag, I.; Kocabicak, E.; Sahin, B.; Jahanshahi, A.; Incesu, L.; Aygun, D.; Yildiz, O.; Temel, Y.; Belet, U. Volumetric analysis of the subthalamic and red nuclei based on magnetic resonance imaging in patients with Parkinson’s disease. Int. J. Neurosci. 2014, 124, 291–295. [Google Scholar] [CrossRef]
- Menke, R.A.; Scholz, J.; Miller, K.L.; Deoni, S.; Jbabdi, S.; Matthews, P.M.; Zarei, M. MRI characteristics of the substantia nigra in Parkinson’s disease: A combined quantitative T1 and DTI study. Neuroimage 2009, 47, 435–441. [Google Scholar] [CrossRef]
- Del Tredici, K.; Ludolph, A.C.; Feldengut, S.; Jacob, C.; Reichmann, H.; Bohl, J.R.; Braak, H. Fabry Disease with Concomitant Lewy Body Disease. J. Neuropathol. Exp. Neurol. 2020, 79, 378–392. [Google Scholar] [CrossRef]
- Russo, C.; Pontillo, G.; Saccà, F.; Riccio, E.; Cocozza, S.; Pane, C.; Tedeschi, E.; Pisani, A.; Pappatà, S. Nonvascular Parkinsonism in Fabry Disease: Results From Magnetic Resonance and Dopamine Transporter Imaging. J. Neuropathol. Exp. Neurol. 2021, 80, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Cociasu, I.; Sorbera, C.; Tuttolomondo, A.; Morgante, F. Anderson-Fabry Disease: A Rare Cause of Levodopa-Responsive Early-Onset Parkinsonism. Mov. Disord. Clin. Pr. 2021, 8 (Suppl. S1), S32–S34. [Google Scholar] [CrossRef] [PubMed]
- Wallom, K.-L.; Fernández-Suárez, M.E.; Priestman, D.A.; Vruchte, D.T.; Huebecker, M.; Hallett, P.J.; Isacson, O.; Platt, F.M. Glycosphingolipid metabolism and its role in ageing and Parkinson’s disease. Glycoconj. J. 2021, 39, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, G.; Fratila, O.; Buhas, C.; Judea-Pusta, C.T.; Negrut, N.; Bustea, C.; Bungau, S. Cross-talks among GBA mutations, glucocerebrosidase, and α-synuclein in GBA-associated Parkinson’s disease and their targeted therapeutic approaches: A comprehensive review. Transl. Neurodegener. 2021, 10, 4. [Google Scholar] [CrossRef]
- Pchelina, S.; Emelyanov, A.; Baydakova, G.; Andoskin, P.; Senkevich, K.; Nikolaev, M.; Miliukhina, I.; Yakimovskii, A.; Timofeeva, A.; Fedotova, E.; et al. Oligomeric alpha-synuclein and glucocerebrosidase activity levels in GBA-associated Parkinson’s disease. Neurosci. Lett. 2017, 636, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.; Wolf, P.; Levy, O.; Kang, U.; Waters, C.; Fahn, S.; Ford, B.; Kuo, S.; Vanegas, N.; Shah, H.; et al. Alpha galactosidase A activity in Parkinson’s disease. Neurobiol. Dis. 2018, 112, 85–90. [Google Scholar] [CrossRef]
- Duran, R.; Mencacci, N.E.; Angeli, A.V.; Shoai, M.; Deas, E.; Houlden, H.; Mehta, A.; Hughes, D.; Cox, T.M.; Deegan, P.; et al. The glucocerobrosidase E326K variant predisposes to Parkinson’s disease, but does not cause Gaucher’s disease. Mov. Disord. 2013, 28, 232–236. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Giladi, N.; Rozovski, U.; Shifrin, C.; Rosner, S.; Gurevich, T.; Bar-Shira, A.; Orr-Urtreger, A. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008, 70, 2277–2283. [Google Scholar] [CrossRef]
- Nelson, M.P.; Boutin, M.; Tse, T.E.; Lu, H.; Haley, E.D.; Ouyang, X.; Zhang, J.; Auray-Blais, C.; Shacka, J.J. The lysosomal enzyme alpha-Galactosidase A is deficient in Parkinson’s disease brain in association with the pathologic accumulation of alpha-synuclein. Neurobiol Dis. 2018, 110, 68–81. [Google Scholar] [CrossRef]
- Huebecker, M.; Moloney, E.B.; van der Spoel, A.C.; Priestman, D.A.; Isacson, O.; Hallett, P.J.; Platt, F.M. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 40. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Pierguidi, L.; Persichetti, E.; Parnetti, L.; Sbaragli, M.; Tassi, C.; Orlacchio, A.; Calabresi, P.; Beccari, T.; Rossi, A. Lysosomal hydrolases in cerebrospinal fluid from subjects with Parkinson’s disease. Mov. Disord. 2007, 22, 1481–1484. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.M.; Dardis, A.; Deganuto, M.; De Carlo, C.; Castrioto, A.; et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. 2014, 29, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Paciotti, S.; Eusebi, P.; Dardis, A.; Zampieri, S.; Chiasserini, D.; Tasegian, A.; Tambasco, N.; Bembi, B.; Calabresi, P.; et al. Cerebrospinal fluid β-glucocerebrosidase activity is reduced in parkinson’s disease patients. Mov. Disord. 2017, 32, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, K.D.; Persichetti, E.; Chiasserini, D.; Eusebi, P.; Beccari, T.; Calabresi, P.; Berendse, H.W.; Parnetti, L.; van de Berg, W.D. Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov. Disord. 2013, 28, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, L.; Jette, N.; Frolkis, A.; Steeves, T.; Pringsheim, T. The incidence of Parkinson’s disease: A systematic review and meta-analysis. Neuroepidemiology 2016, 46, 292–300. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Level | Mechanism |
|---|---|
| Biological | Reduced a-GAL A activity in brain samples of PD patients. |
| Histopathological | Neurodegenerative hallmarks in few brain autopsies of AFD patients with concomitant extrapyramidal manifestations during life. Signs of substrate accumulation in brain areas crucial for extrapyramidal pathways in brain autopsies of AFD patients. |
| Clinical | Neurodegenerative prodromal phenotype in AFD patients. |
| Neuroradiological | MRI markers of neurodegenerative disease and extrapyramidal dysfunction in selected AFD patients’ subgroups. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zedde, M.; Pascarella, R.; Cavallieri, F.; Pezzella, F.R.; Grisanti, S.; Di Fonzo, A.; Valzania, F. Anderson–Fabry Disease: A New Piece of the Lysosomal Puzzle in Parkinson Disease? Biomedicines 2022, 10, 3132. https://doi.org/10.3390/biomedicines10123132
Zedde M, Pascarella R, Cavallieri F, Pezzella FR, Grisanti S, Di Fonzo A, Valzania F. Anderson–Fabry Disease: A New Piece of the Lysosomal Puzzle in Parkinson Disease? Biomedicines. 2022; 10(12):3132. https://doi.org/10.3390/biomedicines10123132
Chicago/Turabian StyleZedde, Marialuisa, Rosario Pascarella, Francesco Cavallieri, Francesca Romana Pezzella, Sara Grisanti, Alessio Di Fonzo, and Franco Valzania. 2022. "Anderson–Fabry Disease: A New Piece of the Lysosomal Puzzle in Parkinson Disease?" Biomedicines 10, no. 12: 3132. https://doi.org/10.3390/biomedicines10123132
APA StyleZedde, M., Pascarella, R., Cavallieri, F., Pezzella, F. R., Grisanti, S., Di Fonzo, A., & Valzania, F. (2022). Anderson–Fabry Disease: A New Piece of the Lysosomal Puzzle in Parkinson Disease? Biomedicines, 10(12), 3132. https://doi.org/10.3390/biomedicines10123132