
Sexual Dimorphism in Interstitial Lung Disease
 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Interstitial Lung Diseases
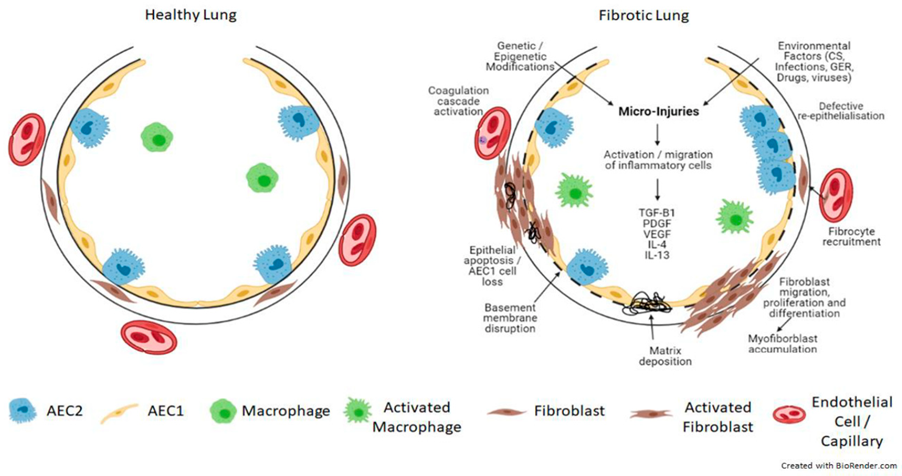
3. Pathogenesis of Pulmonary Fibrosis
4. Sex Differences in IPF
5. Connective Tissue Diseases
6. Sex Hormones and the X Chromosome in Autoimmunity
7. Gender Differences in Clinical Research
8. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Townsend, E.A.; Miller, V.M.; Prakash, Y.S. Sex Differences and Sex Steroids in Lung Health and Disease. Endocr. Rev. 2012, 33, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Torday, J.S.; Nielsen, H.C.; Fencl, M.D.M.; Avery, M.E. Sex differences in fetal lung maturation. Am. Rev. Respir. Dis. 1981, 123, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Perelman, R.H.; Palta, M.; Kirby, R.; Farrell, P.M. Discordance between male and female deaths due to the respiratory distress syndrome. Pediatrics 1986, 78, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Carey, M.A.; Card, J.W.; Voltz, J.W.; Germolec, D.R.; Korach, K.S.; Zeldin, D.C. The impact of sex and sex hormones on lung physiology and disease: Lessons from animal studies. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L272–L278. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.R.; Castile, R.G.; Fredberg, J.J.; Wohl, M.E.; Mead, J. Airway size is related to sex but not lung size in normal adults. J. Appl. Physiol. 1987, 63, 2042–2047. [Google Scholar] [CrossRef]
- Wuyts, W.A.; Agostini, C.; Antoniou, K.M.; Bouros, D.; Chambers, R.; Cottin, V.; Egan, J.J.; Lambrecht, B.N.; Lories, R.; Parfrey, H.; et al. The pathogenesis of pulmonary fibrosis: A moving target. Eur. Respir. J. 2013, 41, 1207–1218. [Google Scholar] [CrossRef]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef]
- Meyer, K.C. Pulmonary fibrosis, part I: Epidemiology, pathogenesis, and diagnosis. Expert. Rev. Respir. Med. 2017, 11, 343–359. [Google Scholar] [CrossRef]
- Marshall, R.P.; McAnulty, R.J.; Laurent, G.J. The pathogenesis of pulmonary fibrosis: Is there a fibrosis gene? Int. J. Biochem. Cell Biol. 1997, 29, 107–120. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Revealing the Pathogenic and Aging-related Mechanisms of the Enigmatic Idiopathic Pulmonary Fibrosis. An Integral Model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef]
- Wang, E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995, 55, 2284–2292. [Google Scholar]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.-S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1119–L1126. [Google Scholar] [CrossRef]
- Dove, E.P.; Olson, A.L.; Glassberg, M.K. Trends in Idiopathic Pulmonary Fibrosis–related Mortality in the United States: 2000–2017. Am. J. Respir. Crit. Care Med. 2019, 200, 929–931. [Google Scholar] [CrossRef]
- Navaratnam, V.; Hubbard, R.B. The Mortality Burden of Idiopathic Pulmonary Fibrosis in the United Kingdom. Am. J. Respir. Crit. Care Med. 2019, 200, 256–258. [Google Scholar] [CrossRef]
- Han, M.K.; Murray, S.; Fell, C.D.; Flaherty, K.R.; Toews, G.B.; Myers, J.; Colby, T.V.; Travis, W.D.; Kazerooni, E.A.; Gross, B.H.; et al. Sex differences in physiological progression of idiopathic pulmonary fibrosis. Eur. Respir. J. 2008, 31, 1183–1188. [Google Scholar] [CrossRef]
- Erbes, R.; Schaberg, T.; Loddenkemper, R. Lung Function Tests in Patients With Idiopathic Pulmonary Fibrosis. Are they helpful for predicting outcome? Chest 1997, 111, 51–57. [Google Scholar] [CrossRef]
- Ganesh Raghu, H.R.C.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; Lynch, D.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmoanry Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Crowson, C.S.; Matteson, E.L.; Myasoedova, E.; Michet, C.J.; Ernste, F.C.; Warrington, K.; Davis, J.; Hunder, G.G.; Therneau, T.M.; Gabriel, S.E. The lifetime risk of adult-onset rheumatoid arthritis and other inflammatory autoimmune rheumatic diseases. Arthritis Rheum. 2011, 63, 633–639. [Google Scholar] [CrossRef]
- Bossini-Castillo, L.; López-Isac, E.; Mayes, M.D.; Martín, J. Genetics of systemic sclerosis. Semin. Immunopathol. 2015, 37, 443–451. [Google Scholar] [CrossRef]
- Somers, E.C.; Marder, W.; Cagnoli, P.; Lewis, E.E.; Deguire, P.; Gordon, C.; Helmick, C.G.; Wang, L.; Wing, J.J.; Dhar, J.P.; et al. Population-Based Incidence and Prevalence of Systemic Lupus Erythematosus: The Michigan Lupus Epidemiology and Surveillance Program. Arthritis Rheumatol. 2014, 66, 369–378. [Google Scholar] [CrossRef]
- Qin, B.; Wang, J.; Yang, Z.; Yang, M.; Ma, N.; Huang, F.; Zhong, R. Epidemiology of primary Sjogren’s syndrome: A systematic review and meta-analysis. Ann. Rheum. Dis. 2015, 74, 1983–1989. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, J.; Cobo, T.; Balsa, A.; Descalzo, M.A.; Carmona, L.; SERAP Study Group. The incidence of rheumatoid arthritis in Spain: Results from a nationwide primary care registry. Rheumatology 2008, 47, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Mayes, M.D.; Lacey, J.V., Jr.; Beebe-Dimmer, J.; Gillespie, B.W.; Cooper, B.; Laing, T.J.; Schottenfeld, D. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003, 48, 2246–2255. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.I. Sjogren’s syndrome. Lancet 2005, 366, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-K.; Kim, D.S.; Yoo, B.; Seo, J.B.; Rho, J.-Y.; Colby, T.V.; Kitaichi, M. Histopathologic Pattern and Clinical Features of Rheumatoid Arthritis-Associated Interstitial Lung Disease. Chest 2005, 127, 2019–2027. [Google Scholar] [CrossRef]
- Bouros, D.; Wells, A.U.; Nicholson, A.G.; Colby, T.V.; Polychronopoulos, V.; Pantelidis, P.; Haslam, P.L.; Vassilakis, D.A.; Black, C.M.; du Bois, R.M. Histopathologic Subsets of Fibrosing Alveolitis in Patients with Systemic Sclerosis and Their Relationship to Outcome. Am. J. Respir. Crit. Care Med. 2002, 165, 1581–1586. [Google Scholar] [CrossRef]
- Enomoto, N.; Egashira, R.; Tabata, K.; Hashisako, M.; Kitani, M.; Waseda, Y.; Ishizuka, T.; Watanabe, S.; Kasahara, K.; Izumi, S.; et al. Analysis of systemic lupus erythematosus-related interstitial pneumonia: A retrospective multicentre study. Sci. Rep. 2019, 9, 7355. [Google Scholar] [CrossRef]
- Manuel, R.C.; Brito-Zerón, P.; Seror, R.; Bootsma, H.; Bowman, S.J.; Dörner, T.; Gottenberg, J.-E.; Mariette, X.; Theander, E.; Bombardieri, S.; et al. Characterization of systemic disease in primary Sjogren’s syndrome: EULAR-SS Task Force recommendations for articular, cutaneous, pulmonary and renal involvements. Rheumatology 2017, 56, 1245. [Google Scholar] [CrossRef]
- Restrepo, J.F.; Del Rincón, I.; Battafarano, D.; Haas, R.W.; Doria, M.; Escalante, A. Clinical and laboratory factors associated with interstitial lung disease in rheumatoid arthritis. Clin. Rheumatol. 2015, 34, 1529–1536. [Google Scholar] [CrossRef]
- Steen, V.D.; Conte, C.; Owens, G.R.; Medsger, T.A. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum. 1994, 37, 1283–1289. [Google Scholar] [CrossRef]
- Schurawitzki, H.; Stiglbauer, R.; Graninger, W.; Herold, C.; Pölzleitner, D.; Burghuber, O.; Tscholakoff, D. Interstitial lung disease in progressive systemic sclerosis: High-resolution CT versus radiography. Radiology 1990, 176, 755–759. [Google Scholar] [CrossRef]
- Demoruelle, M.K.; Mittoo, S.; Solomon, J.J. Connective tissue disease-related interstitial lung disease. Best Pract. Res. Clin. Rheumatol. 2016, 30, 39–52. [Google Scholar] [CrossRef]
- Fell, C.D.; Mittoo, S. Pulmonary Manifestations of Systemic Lupus Erythematosus. Semin. Respir. Crit. Care Med. 2014, 35, 249–254. [Google Scholar] [CrossRef]
- Cervera, R.; Khamashta, M.A.; Font, J.; Sebastiani, G.D.; Gil, A.; Lavilla, P.; Doménech, I.; Aydintug, A.O.; Jedryka-Góral, A.; De Ramón, E. Systemic lupus erythematosus: Clinical and immunologic patterns of disease expression in a cohort of 1000 patients. The European Working Party on Systemic Lupus Erythematosus. Medicine 1993, 72, 113–124. [Google Scholar] [CrossRef]
- Fairfax, A.J.; Bigot, A.; Chaigne, B.; Henique, H.; Diot, E.; Marchand-Adam, S. Pulmonary disorders associated with Sjogren’s syndrome. Q. J. Med. 1981, 50, 279–295. [Google Scholar]
- Dong, X.; Zheng, Y.; Wang, L.; Chen, W.-H.; Zhang, Y.-G.; Fu, Q. Clinical characteristics of autoimmune rheumatic disease-related organizing pneumonia. Clin. Rheumatol. 2018, 37, 1027–1035. [Google Scholar] [CrossRef]
- Nannini, C.; Jebakumar, A.J.; Crowson, C.S.; Ryu, J.H.; Matteson, E.L. Primary Sjogren’s syndrome 1976–2005 and associated interstitial lung disease: A population-based study of incidence and mortality. BMJ Open 2013, 3, e003569. [Google Scholar] [CrossRef]
- Kelly, C.A.; Saravanan, V.; Nisar, M.; Arthanari, S.; Woodhead, F.A.; Price-Forbes, A.N.; Dawson, J.; Sathi, N.; Ahmad, Y.; Koduri, G.; et al. Rheumatoid arthritis-related interstitial lung disease: Associations, prognostic factors and physiological and radiological characteristics—A large multicentre UK study. Rheumatology 2014, 53, 1676–1682. [Google Scholar] [CrossRef]
- Zhu, J.; Zhou, Y.; Chen, X.; Li, J. A Metaanalysis of the Increased Risk of Rheumatoid Arthritis-related Pulmonary Disease as a Result of Serum Anticitrullinated Protein Antibody Positivity. J. Rheumatol. 2014, 41, 1282–1289. [Google Scholar] [CrossRef]
- Juge, P.-A.; Lee, J.S.; Ebstein, E.; Furukawa, H.; Dobrinskikh, E.; Gazal, S.; Kannengiesser, C.; Ottaviani, S.; Oka, S.; Tohma, S.; et al. MUC5B Promoter Variant and Rheumatoid Arthritis with Interstitial Lung Disease. N. Engl. J. Med. 2018, 379, 2209–2219. [Google Scholar] [CrossRef]
- Bongartz, T.; Nannini, C.; Medina-Velasquez, Y.F.; Achenbach, S.J.; Crowson, C.S.; Ryu, J.; Vassallo, R.; Gabriel, S.E.; Matteson, E.L. Incidence and mortality of interstitial lung disease in rheumatoid arthritis: A population-based study. Arthritis Rheum. 2010, 62, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Saag, K.G.; Kolluri, S.; Koehnke, R.K.; Georgou, T.A.; Rachow, J.W.; Hunninghake, G.W.; Schwartz, D.A. Rheumatoid arthritis lung disease. Determinants of radiographic and physiologic abnormalities. Arthritis Rheum. 1996, 39, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Steen, V.D. Autoantibodies in Systemic Sclerosis. Semin. Arthritis Rheum. 2005, 35, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Al-Sheikh, H.; Ahmad, Z.; Johnson, S.R. Ethnic Variations in Systemic Sclerosis Disease Manifestations, Internal Organ Involvement, and Mortality. J. Rheumatol. 2019, 46, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Peoples, C.; Medsger, T.A.; Lucas, M.; Rosario, B.L.; Feghali-Bostwick, C.A.; Medsger, J.T.A. Gender differences in systemic sclerosis: Relationship to clinical features, serologic status and outcomes. J. Scleroderma Relat. Disord. 2016, 1, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, H.; Dubois, E.L.; Sherwin, R.P.; Balchum, O.J. Diffuse Interstitial Lung Disease in Systemic Lupus Erythematosus. Ann. Intern. Med. 1973, 79, 37–45. [Google Scholar] [CrossRef]
- Ward, M.M.; Polisson, R.P. A meta-analysis of the clinical manifestations of older-onset systemic lupus erythematosus. Arthritis Rheum. 1989, 32, 1226–1232. [Google Scholar] [CrossRef]
- ter Borg, E.J.; Groen, H.; Horst, G.; Limburg, P.C.; Wouda, A.A.; Kallenberg, C.G. Clinical associations of antiribonucleoprotein antibodies in patients with systemic lupus erythematosus. Semin. Arthritis Rheum. 1990, 20, 164–173. [Google Scholar] [CrossRef]
- Li, X.; Xu, B.; Ma, Y.; Li, X.; Cheng, Q.; Wang, X.; Wang, G.; Qian, L.; Wei, L. Clinical and laboratory profiles of primary Sjogren’s syndrome in a Chinese population: A retrospective analysis of 315 patients. Int. J. Rheum. Dis. 2015, 18, 439–446. [Google Scholar] [CrossRef]
- He, C.; Chen, Z.; Liu, S.; Chen, H.; Zhang, F. Prevalence and risk factors of interstitial lung disease in patients with primary Sjogren’s syndrome: A systematic review and meta-analysis. Int. J. Rheum. Dis. 2020, 23, 1009–1018. [Google Scholar] [CrossRef]
- Marshall, D.C.; Salciccioli, J.D.; Shea, B.S.; Akuthota, P. Trends in mortality from idiopathic pulmonary fibrosis in the European Union: An observational study of the WHO mortality database from 2001–2013. Eur. Respir. J. 2018, 51, 1701603. [Google Scholar] [CrossRef]
- Solomon, J.J.; Chung, J.H.; Cosgrove, G.P.; Demoruelle, M.K.; Pérez, E.F.; Fischer, A.; Frankel, S.K.; Hobbs, S.B.; Huie, T.J.; Ketzer, J.; et al. Predictors of mortality in rheumatoid arthritis-associated interstitial lung disease. Eur. Respir. J. 2016, 47, 588–596. [Google Scholar] [CrossRef]
- Ippolito, A.; Petri, M. An update on mortality in systemic lupus erythematosus. Clin. Exp. Rheumatol. 2008, 26, S72–S79. [Google Scholar]
- Palm, O.; Garen, T.; Enger, T.B.; Jensen, J.L.; Lund, M.-B.; Aaløkken, T.M.; Gran, J.T. Clinical pulmonary involvement in primary Sjogren’s syndrome: Prevalence, quality of life and mortality—A retrospective study based on registry data. Rheumatology 2013, 52, 173–179. [Google Scholar] [CrossRef]
- Borie, R.; Kannengiesser, C.; Nathan, N.; Tabèze, L.; Pradère, P.; Crestani, B. Familial pulmonary fibrosis. Rev. Mal. Respir. 2015, 32, 413–434. [Google Scholar] [CrossRef]
- Silbiger, S.R.; Neugarten, J. The impact of gender on the progression of chronic renal disease. Am. J. Kidney Dis. 1995, 25, 515–533. [Google Scholar] [CrossRef]
- Dai, W.J.; Jiang, H.C. Advances in gene therapy of liver cirrhosis: A review. World J. Gastroenterol. 2001, 7, 1–8. [Google Scholar] [CrossRef]
- Lekgabe, E.D.; Royce, S.G.; Hewitson, T.D.; Tang, M.L.K.; Zhao, C.; Moore, X.L.; Tregear, G.W.; Bathgate, R.A.D.; Du, X.-J.; Samuel, C.S. The Effects of Relaxin and Estrogen Deficiency on Collagen Deposition and Hypertrophy of Nonreproductive Organs. Endocrinology 2006, 147, 5575–5583. [Google Scholar] [CrossRef]
- Voltz, J.W.; Card, J.W.; Carey, M.A.; DeGraff, L.M.; Ferguson, C.D.; Flake, G.P.; Bonner, J.C.; Korach, K.S.; Zeldin, D.C. Male Sex Hormones Exacerbate Lung Function Impairment after Bleomycin-Induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 45–52. [Google Scholar] [CrossRef]
- Redente, E.F.; Jacobsen, K.M.; Solomon, J.J.; Lara, A.R.; Faubel, S.; Keith, R.C.; Henson, P.M.; Downey, G.P.; Riches, D.W.H. Age and sex dimorphisms contribute to the severity of bleomycin-induced lung injury and fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2011, 301, L510–L518. [Google Scholar] [CrossRef]
- Gharaee-Kermani, M.; Hatano, K.; Nozaki, Y.; Phan, S. Gender-Based Differences in Bleomycin-Induced Pulmonary Fibrosis. Am. J. Pathol. 2005, 166, 1593–1606. [Google Scholar] [CrossRef] [PubMed]
- Elliot, S.; Periera-Simon, S.; Xia, X.; Catanuto, P.; Rubio, G.; Shahzeidi, S.; El Salem, F.; Shapiro, J.; Briegel, K.; Korach, K.S.; et al. MicroRNA let-7 Downregulates Ligand-Independent Estrogen Receptor–mediated Male-Predominant Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 1246–1257. [Google Scholar] [CrossRef] [PubMed]
- Tatler, A.L.; Habgood, A.; Porte, J.; John, A.E.; Stavrou, A.; Hodge, E.; Kerama-Likoko, C.; Violette, S.M.; Weinreb, P.H.; Knox, A.J. Reduced Ets Domain-containing Protein Elk1 Promotes Pulmonary Fibrosis via Increased Integrin alphavbeta6 Expression. J. Biol. Chem. 2016, 291, 9540–9553. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Strek, M.E.; Cottin, V.; Dellaripa, P.F.; Bernstein, E.J.; Brown, K.K.; Danoff, S.K.; Distler, O.; Hirani, N.; Jones, K.D.; et al. Proceedings of the American College of Rheumatology/Association of Physicians of Great Britain and Ireland Connective Tissue Disease–Associated Interstitial Lung Disease Summit: A Multidisciplinary Approach to Address Challenges and Opportunities. Arthritis Rheumatol. 2019, 71, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Ortona, E.; Pierdominici, M.; Maselli, A.; Veroni, C.; Aloisi, F.; Shoenfeld, Y. Sex-based differences in autoimmune diseases. Ann. Ist. Super Sanita 2016, 52, 205–212. [Google Scholar] [CrossRef]
- Proudman, S.; Lake, F. Rheumatoid Arthritis and Lung Disease: From Mechanisms to a Practical Approach. Semin. Respir. Crit. Care Med. 2014, 35, 222–238. [Google Scholar] [CrossRef]
- Khanna, D.; Tashkin, D.P.; Denton, C.P.; Renzoni, E.A.; Desai, S.R.; Varga, J. Etiology, Risk Factors, and Biomarkers in Systemic Sclerosis with Interstitial Lung Disease. Am. J. Respir. Crit. Care Med. 2020, 201, 650–660. [Google Scholar] [CrossRef]
- Jawaheer, D.; Lum, R.F.; Gregersen, P.K.; Criswell, L.A. Influence of male sex on disease phenotype in familial rheumatoid arthritis. Arthritis Rheum. 2006, 54, 3087–3094. [Google Scholar] [CrossRef]
- Weyand, C.M.; Schmidt, D.; Wagner, U. The influence of sex on the phenotype of rheumatoid arthritis. Arthritis Rheum. 1998, 41, 817–822. [Google Scholar] [CrossRef]
- Kiely, P.; Busby, A.D.; Nikiphorou, E.; Sullivan, K.; Walsh, D.A.; Creamer, P.; Dixey, J.; Young, A. Is incident rheumatoid arthritis interstitial lung disease associated with methotrexate treatment? Results from a multivariate analysis in the ERAS and ERAN inception cohorts. BMJ Open 2019, 9, e028466. [Google Scholar] [CrossRef]
- Yin, Y.; Liang, D.; Zhao, L.; Li, Y.; Liu, W.; Ren, Y.; Li, Y.; Zeng, X.; Zhang, F.; Tang, F.; et al. Anti-Cyclic Citrullinated Peptide Antibody Is Associated with Interstitial Lung Disease in Patients with Rheumatoid Arthritis. PLoS ONE 2014, 9, e92449. [Google Scholar] [CrossRef]
- Palomäki, A.; Palotie, A.; Koskela, J.; Eklund, K.K.; Pirinen, M.; Ripatti, S.; Laitinen, T.; Mars, N.; FinnGen Rheumatology Clinical Expert Group; Gen, F. Lifetime risk of rheumatoid arthritis-associated interstitial lung disease in MUC5B mutation carriers. Ann. Rheum. Dis. 2021, 80, 1530–1536. [Google Scholar] [CrossRef]
- Chifflot, H.; Fautrel, B.; Sordet, C.; Chatelus, E.; Sibilia, J. Incidence and Prevalence of Systemic Sclerosis: A Systematic Literature Review. Semin. Arthritis Rheum. 2008, 37, 223–235. [Google Scholar] [CrossRef]
- Tyndall, A.J.; Bannert, B.; Vonk, M.; Airò, P.; Cozzi, F.; Carreira, P.E.; Bancel, D.F.; Allanore, Y.; Müller-Ladner, U.; Distler, O. Causes and risk factors for death in systemic sclerosis: A study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann. Rheum. Dis. 2010, 69, 1809–1815. [Google Scholar] [CrossRef]
- Steen, V.D. The lung in systemic sclerosis. J. Clin. Rheumatol. 2005, 11, 40–46. [Google Scholar] [CrossRef]
- Weinrib, L.; Sharma, O.P.; Quismorio, F.P., Jr. A long-term study of interstitial lung disease in systemic lupus erythematosus. Semin. Arthritis Rheum. 1990, 20, 48–56. [Google Scholar] [CrossRef]
- Wiedemann, H.P.; Matthay, R.A. Pulmonary manifestations of systemic lupus erythematosus. J. Thorac. Imaging 1992, 7, 1–18. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Chen, X.; Liang, H.; Yang, X. Association of Interstitial Lung Disease with Clinical Characteristics of Chinese Patients With Systemic Lupus Erythematosus. Arch. Rheumatol. 2020, 35, 239–246. [Google Scholar] [CrossRef]
- Frankel, S.K.; Cosgrove, G.P.; Fischer, A.; Meehan, R.T.; Brown, K.K. Update in the Diagnosis and Management of Pulmonary Vasculitis. Chest 2006, 129, 452–465. [Google Scholar] [CrossRef]
- Amarnani, R.; Yeoh, S.-A.; Denneny, E.K.; Wincup, C. Lupus and the Lungs: The Assessment and Management of Pulmonary Manifestations of Systemic Lupus Erythematosus. Front. Med. 2021, 7, 610257. [Google Scholar] [CrossRef]
- Parambil, J.G.; Myers, J.L.; Lindell, R.M.; Matteson, E.L.; Ryu, J.H. Interstitial Lung Disease in Primary Sjögren Syndrome. Chest 2006, 130, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Solans, R.; Rosas, J.; Camps, M.T.; Gil, A.; Del Pino-Montes, J.; Calvo-Alen, J.; Jiménez-Alonso, J.; Micó, M.-L.; Beltrán, J.; et al. Primary Sjogren syndrome in Spain: Clinical and immunologic expression in 1010 patients. Medicine 2008, 87, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Hansell, A.L. Chapter and verse on why lung disease is under-recognised and underestimated. Int. J. Tuberc. Lung Dis. 2008, 12, 358. [Google Scholar] [PubMed]
- Seror, R.; Bootsma, H.; Saraux, A.; Bowman, S.J.; Theander, E.; Brun, J.G.; Baron, G.; Le Guern, V.; Devauchelle-Pensec, V.; Ramos-Casals, M.; et al. Defining disease activity states and clinically meaningful improvement in primary Sjogren’s syndrome with EULAR primary Sjogren’s syndrome disease activity (ESSDAI) and patient-reported indexes (ESSPRI). Ann. Rheum. Dis. 2016, 75, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.E.; Cox, D.; Shiboski, C.H.; Schiødt, M.; Wu, A.; Lanfranchi, H.; Umehara, H.; Zhao, Y.; Challacombe, S.; Lam, M.Y.; et al. Associations between salivary gland histopathologic diagnoses and phenotypic features of Sjogren’s syndrome among 1726 registry participants. Arthritis Rheum. 2011, 63, 2021–2030. [Google Scholar] [CrossRef]
- Hewagama, A.; Patel, D.; Yarlagadda, S.; Strickland, F.M.; Richardson, B.C. Stronger inflammatory/cytotoxic T-cell response in women identified by microarray analysis. Genes Immun. 2009, 10, 509–516. [Google Scholar] [CrossRef]
- Moulton, V.R. Sex Hormones in Acquired Immunity and Autoimmune Disease. Front. Immunol. 2018, 9, 2279. [Google Scholar] [CrossRef]
- Ngo, S.T.; Steyn, F.J.; McCombe, P.A. Gender differences in autoimmune disease. Front. Neuroendocrinol. 2014, 35, 347–369. [Google Scholar] [CrossRef]
- Rubtsova, K.; Marrack, P.; Rubtsov, A.V. Sexual dimorphism in autoimmunity. J. Clin. Investig. 2015, 125, 2187–2193. [Google Scholar] [CrossRef]
- Wasef, S.Z.Y. Gender differences in systemic lupus erythematosus. Gend. Med. 2004, 1, 12–17. [Google Scholar] [CrossRef]
- Gourdy, P.; Araujo, L.A.; Zhu, R.; Garmy-Susini, R.; Diem, S.; Laurell, H.; Leite-de-Moraes, M.; Dy, M.; Arnal, J.F.; Bayard, F.; et al. Relevance of sexual dimorphism to regulatory T cells: Estradiol promotes IFN-gamma production by invariant natural killer T cells. Blood 2005, 105, 2415–2420. [Google Scholar] [CrossRef]
- Young, N.A.; Wu, L.-C.; Burd, C.J.; Friedman, A.K.; Kaffenberger, B.H.; Rajaram, M.V.S.; Schlesinger, L.S.; James, H.; Shupnik, M.A.; Jarjour, W.N. Estrogen modulation of endosome-associated toll-like receptor 8: An IFNalpha-independent mechanism of sex-bias in systemic lupus erythematosus. Clin. Immunol. 2014, 151, 66–77. [Google Scholar] [CrossRef]
- Singh, R.P.; Bischoff, D.S. Sex Hormones and Gender Influence the Expression of Markers of Regulatory T Cells in SLE Patients. Front. Immunol. 2021, 12, 619268. [Google Scholar] [CrossRef]
- Lahita, R.G.; Bradlow, H.L. Klinefelter’s syndrome: Hormone metabolism in hypogonadal males with systemic lupus erythematosus. J. Rheumatol. Suppl. 1987, 14, 154–157. [Google Scholar]
- Lahita, R.G.; Bradlow, H.L.; Ginzler, E.; Pang, S.; New, M. Low plasma androgens in women with systemic lupus erythematosus. Arthritis Rheum. 1987, 30, 241–248. [Google Scholar] [CrossRef]
- Sequeira, J.F.; Keser, G.; Greenstein, B.; Wheeler, M.J.; Duarte, P.C.; Khamashta, M.A.; Hughes, R.R. Systemic Lupus Erythematosus: Sex Hormones in Male Patients. Lupus 1993, 2, 315–317. [Google Scholar] [CrossRef]
- Verheul, H.A.; Verveld, M.; Hoefakker, S.; Schuurs, A.H. Effects of Ethinylestradiol on the Course of Spontaneous Autoimmune Disease in NZB/W and Nod Mice. Immunopharmacol. Immunotoxicol. 1995, 17, 163–180. [Google Scholar] [CrossRef]
- Spector, T.D.; Perry, L.A.; Tubb, G.; Silman, A.J.; Huskisson, E.C. Low free testosterone levels in rheumatoid arthritis. Ann. Rheum. Dis. 1988, 47, 65–68. [Google Scholar] [CrossRef]
- Castagnetta, L.A.; Carruba, G.; Granata, O.M.; Stefano, R.; Miele, M.; Schmidt, M.; Cutolo, M.; Straub, R.H. Increased estrogen formation and estrogen to androgen ratio in the synovial fluid of patients with rheumatoid arthritis. J. Rheumatol. 2003, 30, 2597–2605. [Google Scholar]
- Frost, D.B.; Wolf, B.; Peoples, C.; Fike, J.; Silver, K.; Laffoon, M.; Medsger, T.A., Jr.; Feghali-Bostwick, C. Estradiol levels are elevated in older men with diffuse cutaneous SSc and are associated with decreased survival. Arthritis Res. Ther. 2019, 21, 85. [Google Scholar] [CrossRef]
- Shi-Wen, X.; Panesar, M.; Msc, R.V.; Mason, J.; Haskard, D.; Black, C.; Olsen, I.; Abraham, D. Expression and shedding of intercellular adhesion molecule 1 and lymphocyte function–associated antigen 3 by normal and scleroderma fibroblasts. Effects of interferon-gamma, tumor necrosis factor alpha, and estrogen. Arthritis Rheum. 1994, 37, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.K.; Brinton, R.D. Autoimmune Disease in Women: Endocrine Transition and Risk Across the Lifespan. Front. Endocrinol. 2019, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Quintero, O.L.; Amador-Patarroyo, M.J.; Montoya-Ortiz, G.; Rojas-Villarraga, A.; Anaya, J.-M. Autoimmune disease and gender: Plausible mechanisms for the female predominance of autoimmunity. J. Autoimmun. 2012, 38, J109–J119. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.L.; Furst, D.E.; Maloney, S.; Gooley, T.; Evans, P.C.; Smith, A.; Bean, M.A.; Ober, C.; Bianchi, D.W. Microchimerism and HLA-compatible relationships of pregnancy in scleroderma. Lancet 1998, 351, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Artlett, C.M.; Smith, J.B.; Jimenez, S.A. Identification of Fetal DNA and Cells in Skin Lesions from Women with Systemic Sclerosis. N. Engl. J. Med. 1998, 338, 1186–1191. [Google Scholar] [CrossRef]
- Ichikawa, N.; Nakauchi, H.; Sumida, T. Microchimerism in Japanese patients with systemic sclerosis. Arthritis Rheum. 2001, 44, 1226–1228. [Google Scholar] [CrossRef]
- Murata, H.; Nakauchi, H.; Sumida, T. Microchimerism in Japanese women patients with systemic sclerosis. Lancet 1999, 354, 220. [Google Scholar] [CrossRef]
- Ross, M.T.; Grafham, D.V.; Coffey, A.J.; Scherer, S.; McLay, K.; Muzny, D.; Platzer, M.; Howell, G.R.; Burrows, C.; Bird, C.P.; et al. The DNA sequence of the human X chromosome. Nature 2005, 434, 325–337. [Google Scholar] [CrossRef]
- Scofield, R.H.; Bruner, G.R.; Namjou, B.; Kimberly, R.; Ramsey-Goldman, R.; Petri, M.; Reveille, J.D.; Alarcón, G.S.; Vilá, L.M.; Reid, J.; et al. Klinefelter’s syndrome (47,XXY) in male systemic lupus erythematosus patients: Support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum. 2008, 58, 2511–2517. [Google Scholar] [CrossRef]
- Smith-Bouvier, D.L.; Divekar, A.A.; Sasidhar, M.; Du, S.; Tiwari-Woodruff, S.K.; King, J.K.; Arnold, A.P.; Singh, R.R.; Voskuhl, R.R. A role for sex chromosome complement in the female bias in autoimmune disease. J. Exp. Med. 2008, 205, 1099–1108. [Google Scholar] [CrossRef]
- Libert, C.; Dejager, L.; Pinheiro, I. The X chromosome in immune functions: When a chromosome makes the difference. Nat. Rev. Immunol. 2010, 10, 594–604. [Google Scholar] [CrossRef]
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef]
- Lu, Q.; Wu, A.; Tesmer, L.; Ray, N.; Yousif, D.; Richardson, B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J. Immunol. 2007, 179, 6352–6358. [Google Scholar] [CrossRef]
- Liao, J.; Liang, G.; Xie, S.; Zhao, H.; Zuo, X.; Li, F.; Chen, J.; Zhao, M.; Chan, T.; Lu, Q. CD40L demethylation in CD4+ T cells from women with rheumatoid arthritis. Clin. Immunol. 2012, 145, 13–18. [Google Scholar] [CrossRef]
- Lian, X.; Xiao, R.; Hu, X.; Kanekura, T.; Jiang, H.; Li, Y.; Wang, Y.; Yang, Y.; Zhao, M.; Lu, Q. DNA demethylation of CD40L in CD4+ T cells from women with systemic sclerosis: A possible explanation for female susceptibility. Arthritis Rheum. 2012, 64, 2338–2345. [Google Scholar] [CrossRef]
- Di Palo, A.; Siniscalchi, C.; Salerno, M.; Russo, A.; Gravholt, C.H.; Potenza, N. What microRNAs could tell us about the human X chromosome. Cell. Mol. Life Sci. 2020, 77, 4069–4080. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Gibson, A.; Edberg, J.; Kimberly, R.P.; Absher, D.M. Skewed allelic expression on X chromosome associated with aberrant expression of XIST on systemic lupus erythematosus lymphocytes. Hum. Mol. Genet. 2020, 29, 2523–2534. [Google Scholar] [CrossRef]
- Hewagama, A.; Gorelik, G.; Patel, D.; Liyanarachchi, P.; McCune, W.J.; Somers, E.; Gonzalez-Rivera, T.; The Michigan Lupus Cohort; Strickland, F.; Richardson, B. Overexpression of X-Linked genes in T cells from women with lupus. J. Autoimmun. 2013, 41, 60–71. [Google Scholar] [CrossRef]
- Satterthwaite, A.B. Bruton’s Tyrosine Kinase, a Component of B Cell Signaling Pathways, Has Multiple Roles in the Pathogenesis of Lupus. Front Immunol. 2017, 8, 1986. [Google Scholar] [CrossRef]
- Kanaan, S.B.; Onat, O.E.; Balandraud, N.; Martin, G.V.; Nelson, J.L.; Azzouz, D.F.; Auger, I.; Arnoux, F.; Martin, M.; Roudier, J.; et al. Evaluation of X Chromosome Inactivation with Respect to HLA Genetic Susceptibility in Rheumatoid Arthritis and Systemic Sclerosis. PLoS ONE 2016, 11, e0158550. [Google Scholar] [CrossRef]
- Chabchoub, G.; Uz, E.; Maalej, A.; Mustafa, C.A.; Rebai, A.; Mnif, M.; Bahloul, Z.; Farid, N.R.; Ozcelik, T.; Ayadi, H. Analysis of skewed X-chromosome inactivation in females with rheumatoid arthritis and autoimmune thyroid diseases. Arthritis Res. Ther. 2009, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Zito, A.; Davies, M.N.; Tsai, P.-C.; Roberts, S.; Andres-Ejarque, R.; Nardone, S.; Bell, J.T.; Wong, C.C.Y.; Small, K.S. Heritability of skewed X-inactivation in female twins is tissue-specific and associated with age. Nat. Commun. 2019, 10, 5339. [Google Scholar] [CrossRef] [PubMed]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejía, J.E.; Guéry, J.-C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018, 3, eaap8855. [Google Scholar] [CrossRef] [PubMed]
- Broen, J.; Wolvers-Tettero, I.; Bon, L.G.-V.; Vonk, M.C.; Coenen, M.; Lafyatis, R.; Radstake, T.; Langerak, A.W. Skewed X-chromosomal inactivation impacts T regulatory cell function in systemic sclerosis. Ann. Rheum. Dis. 2010, 8, 2213–2216. [Google Scholar] [CrossRef] [PubMed]
- Uz, E.; Loubiere, L.S.; Gadi, V.K.; Ozbalkan, Z.; Stewart, J.; Nelson, J.L.; Ozcelik, T. Skewed X-Chromosome Inactivation in Scleroderma. Clin. Rev. Allergy Immunol. 2008, 34, 352–355. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Özbalkan, Z.; Baǧışlar, S.; Kiraz, S.; Akyerli, C.B.; Özer, H.T.E.; Yavuz, S.; Birlik, A.M.; Çalgüneri, M.; Özçelik, T. Skewed X chromosome inactivation in blood cells of women with scleroderma. Arthritis Rheum. 2005, 52, 1564–1570. [Google Scholar] [CrossRef]
- Franks, P.; Clancy, C.M. Naumburg, Sex, access, and excess. Ann. Intern. Med. 1995, 123, 548–550. [Google Scholar] [CrossRef]
- Assayag, D.; Morisset, J.; Johannson, K.A.; Wells, A.U.; Walsh, S.L.F. Patient gender bias on the diagnosis of idiopathic pulmonary fibrosis. Thorax 2020, 75, 407–412. [Google Scholar] [CrossRef]
- Garate-Carrillo, A.; Gonzalez, J.; Ceballos, G.; Ramirez-Sanchez, I.; Villarreal, F. Sex related differences in the pathogenesis of organ fibrosis. Transl. Res. 2020, 222, 41–55. [Google Scholar] [CrossRef]
- Hirokawa, K.; Utsuyama, M.; Hayashi, Y.; Kitagawa, M.; Makinodan, T.; Fulop, T. Slower immune system aging in women versus men in the Japanese population. Immun. Ageing 2013, 10, 19. [Google Scholar] [CrossRef]
- Rubtsov, A.V.; Rubtsova, K.; Fischer, A.; Meehan, R.T.; Gillis, J.Z.; Kappler, J.W.; Marrack, P. Toll-like receptor 7 (TLR7)–driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood 2011, 118, 1305–1315. [Google Scholar] [CrossRef]
- Chagnon, P.; Provost, S.; Belisle, C.; Bolduc, V.; Gingras, M.; Busque, L. Age-associated skewing of X-inactivation ratios of blood cells in normal females: A candidate-gene analysis approach. Exp. Hematol. 2005, 33, 1209–1214. [Google Scholar] [CrossRef]
- Shah, K.; McCormack, C.E.; Bradbury, N.A. Do you know the sex of your cells? Am. J. Physiol. Cell Physiol. 2014, 306, C3–C18. [Google Scholar] [CrossRef]
- Sodhi, A.; Pisani, M.; Glassberg, M.K.; Bourjeily, G.; D’Ambrosio, C. Sex and Gender in lung diseases and sleep disorders: A state of the art review. Part 2. Chest 2022, 162, 647–658. [Google Scholar] [CrossRef]


{kind=link}
| Idiopathic Pulmonary Fibrosis | Rheumatoid Arthritis | Systemic Sclerosis | Systemic Lupus Erythematosus | Primary Sjögren’s Syndrome | |
|---|---|---|---|---|---|
| Gender ratio (F:M) | 1:1.5 to 2 [8,17] | 2:1 [18] | 3:1 [19] | 9:1 [20] | 9:1 [21] |
| Affected age group | >50 y | F: 50–60 y M: >70 y [22] | 30–60 y [23] Males peak later | Late teens to early 40s [20] | Peaks in Females ~30 y and >55 y [24] |
| Radiographic features | UIP | UIP > NSIP [25] | NSIP > UIP [26] | NSIP, OP, UIP [27] | NSIP > UIP, OP, LIP [28] |
| Lifetime Risk of ILD development | ND | 10% 9 times ↑ILD risk vs. people without RA [29] | Up to 90%, 40% have clinically significant ILD [30,31] | 1–15% [32,33,34] | 9–22% (F > M) [35,36,37] |
| Potential risks for development of ILD | Genetic predisposition, aging, male, smoking, GER, viruses [6,7] | RF [38], ACPA [39], MUC5B mutation [40], older age [41], male [41], smoking [42] | Anti-SCL70 and anti-Anti-U3RNP antibodies [43], Afro-Caribbean [44], male [45] | Anti-U1RNP [33], longstanding disease [46], older age [47], overlapping features with scleroderma [48] | Anti-SSA [49], older age, male, high CRP [50] |
| Risk of mortality | Survival worse in males [15], 3.75:1.5 per 100,000 M:F [51]. | RA-ILD median survival 3-7y [41]. RA-UIP worse survival vs. RA-NSIP 10.18 vs. 13.62 y [52] | ILD associated mortality ↑ in males. Female increased mortality associated with PAH [45] | Higher in males [53] | 4 fold after 10 y of disease compared with those without lung involvement [54] |
| Hormone | Alteration or Mechanism | Ref. |
|---|---|---|
| SLE | ||
| Estrogen | Allows autoreactive B cells to escape tolerance mechanisms | [90] |
| Promotes IFN-γ production | [91] | |
| Upregulates expression of intracellular (but not surface) Toll-like receptors | [92] | |
| Increased plasma estradiol in women with SLE versus healthy women; estradiol reduces FOXP3 expression in regulatory T cells | [93] | |
| Androgens | Testosterone suppresses anti-dsDNA antibody production | [94] |
| Women with SLE have lower plasma androgen levels than healthy women | [95] | |
| Men with SLE have low testosterone (and elevated 16-hydroxyestrone, estrone and luteinizing hormone) | [96] | |
| Sjögren’s Syndrome | ||
| Androgens | Protective against disease development—gonadectomy of non-obese diabetic (NOD) mice exacerbated the disease | [97] |
| Rheumatoid Arthritis | ||
| Estrogens | Males with RA have increased circulating levels of estradiol and decreased estrone | [98] |
| Males and females have increased synovial fluid estrogens relative to androgens which may contribute to synovial cell proliferation, e.g., macrophages and fibroblasts | [99] | |
| Androgens | Males with RA have decreased testosterone and dehydroepiandrosterone | [98] |
| Systemic Sclerosis | ||
| Estrogens | Increased estradiol in older men with dcSSc, associated with decreased survival | [100] |
| Estradiol may augment fibroblast dysfunction | [101] | |
| Disease | Mechanism | Refs. |
|---|---|---|
| SLE | X inactivation specific transcript (XIST), the non-coding RNA which orchestrates XCI, was significantly upregulated in SLE patients, together with skewed XCI in lymphocytes | [117] |
| T cells from women with SLE showed overexpression of 18 X-linked miRNAs; males with SLE did not show overexpression of any | [118] | |
| BTK (an X-linked gene) promotes activation, plasma cell differentiation, and class switching of autoreactive B cells | [119] | |
| CD40L reactivation on the inactive X chromosome in T cells of females with SLE | [113] | |
| RA | Higher incidence of skewed XCI in RA patients | [120,121,122] |
| TLR7 can escape XCI, resulting in increased gene dosage | [123] | |
| CD40L reactivation on the inactive X chromosome in T cells of females with RA | [114] | |
| SSc | Higher incidence of skewed XCI in SSc women v healthy women; association with reduced FOXP3 expression in Tregs | [120,124,125,126] |
| CD40L reactivation on the inactive X chromosome in T cells of females with SSc | [115] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozaki, M.; Glasgow, A.; Oglesby, I.K.; Ng, W.L.; Kelly, S.; Greene, C.M.; Durcan, L.; Hurley, K. Sexual Dimorphism in Interstitial Lung Disease. Biomedicines 2022, 10, 3030. https://doi.org/10.3390/biomedicines10123030
Ozaki M, Glasgow A, Oglesby IK, Ng WL, Kelly S, Greene CM, Durcan L, Hurley K. Sexual Dimorphism in Interstitial Lung Disease. Biomedicines. 2022; 10(12):3030. https://doi.org/10.3390/biomedicines10123030
Chicago/Turabian StyleOzaki, Mari, Arlene Glasgow, Irene K. Oglesby, Wan Lin Ng, Sile Kelly, Catherine M. Greene, Laura Durcan, and Killian Hurley. 2022. "Sexual Dimorphism in Interstitial Lung Disease" Biomedicines 10, no. 12: 3030. https://doi.org/10.3390/biomedicines10123030
APA StyleOzaki, M., Glasgow, A., Oglesby, I. K., Ng, W. L., Kelly, S., Greene, C. M., Durcan, L., & Hurley, K. (2022). Sexual Dimorphism in Interstitial Lung Disease. Biomedicines, 10(12), 3030. https://doi.org/10.3390/biomedicines10123030