
The Long-Term Effects of Prenatal Hypoxia on Coronary Artery Function of the Male and Female Offspring

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Model of Prenatal Hypoxia
2.2. Coronary Artery Vascular Function by Wire Myography
2.3. Molecular Assessment of Plasma ET-1 Levels with ELISA
2.4. Immunofluorescent Staining of the Main Left Coronary Artery for ET-1, ETA, ETB, eNOS, PGHS-1, and PGHS-2
2.5. Image Analysis of Immunofluorescence Staining
2.6. Statistical Analysis
3. Results
3.1. Coronary Artery Endothelium-Dependent and Endothelium-Independent Vasodilation Responses
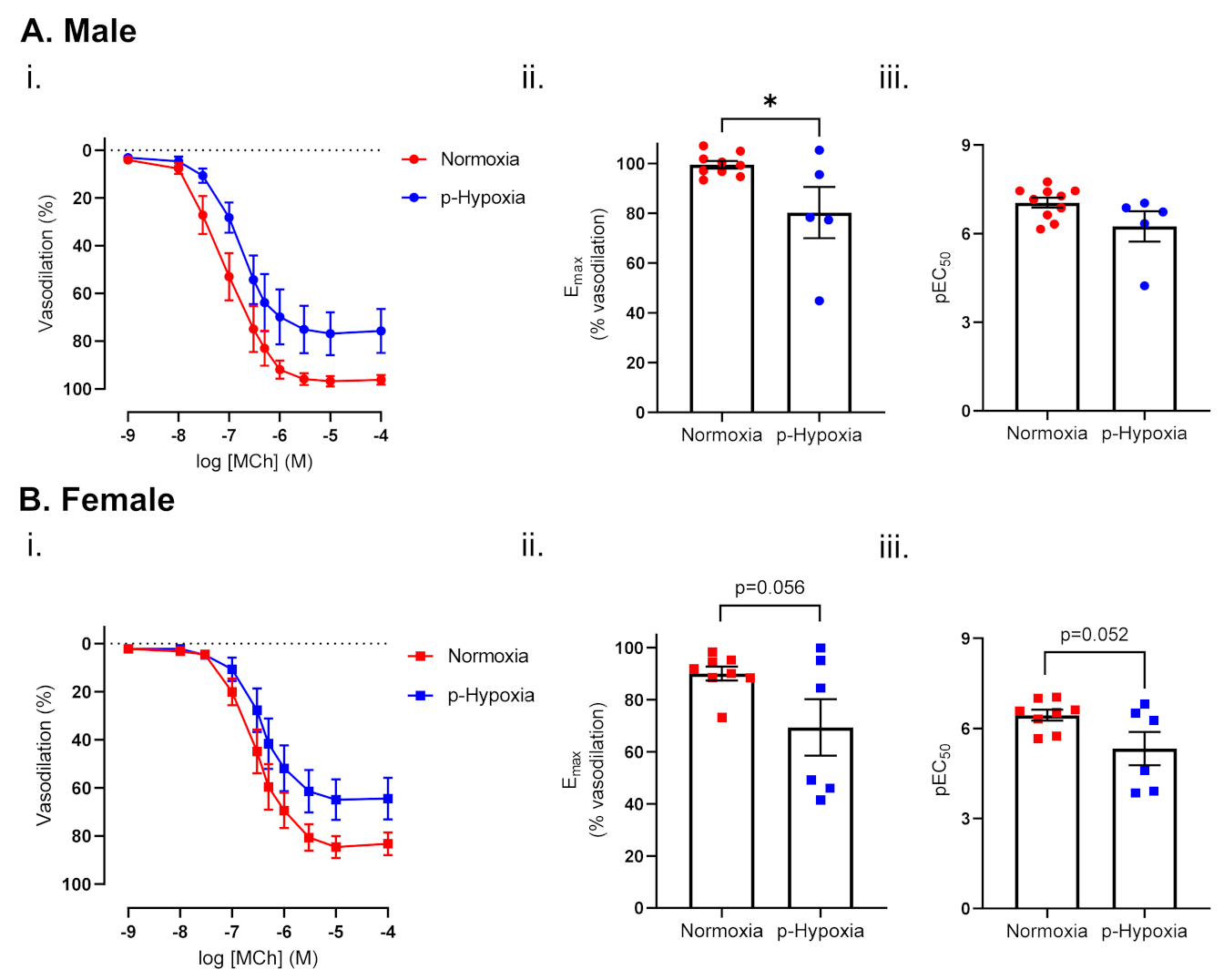
3.1.1. Endothelium-Dependent Vasodilation Was Impaired in Prenatally Hypoxic Male and Female 4-Month-Old Offspring
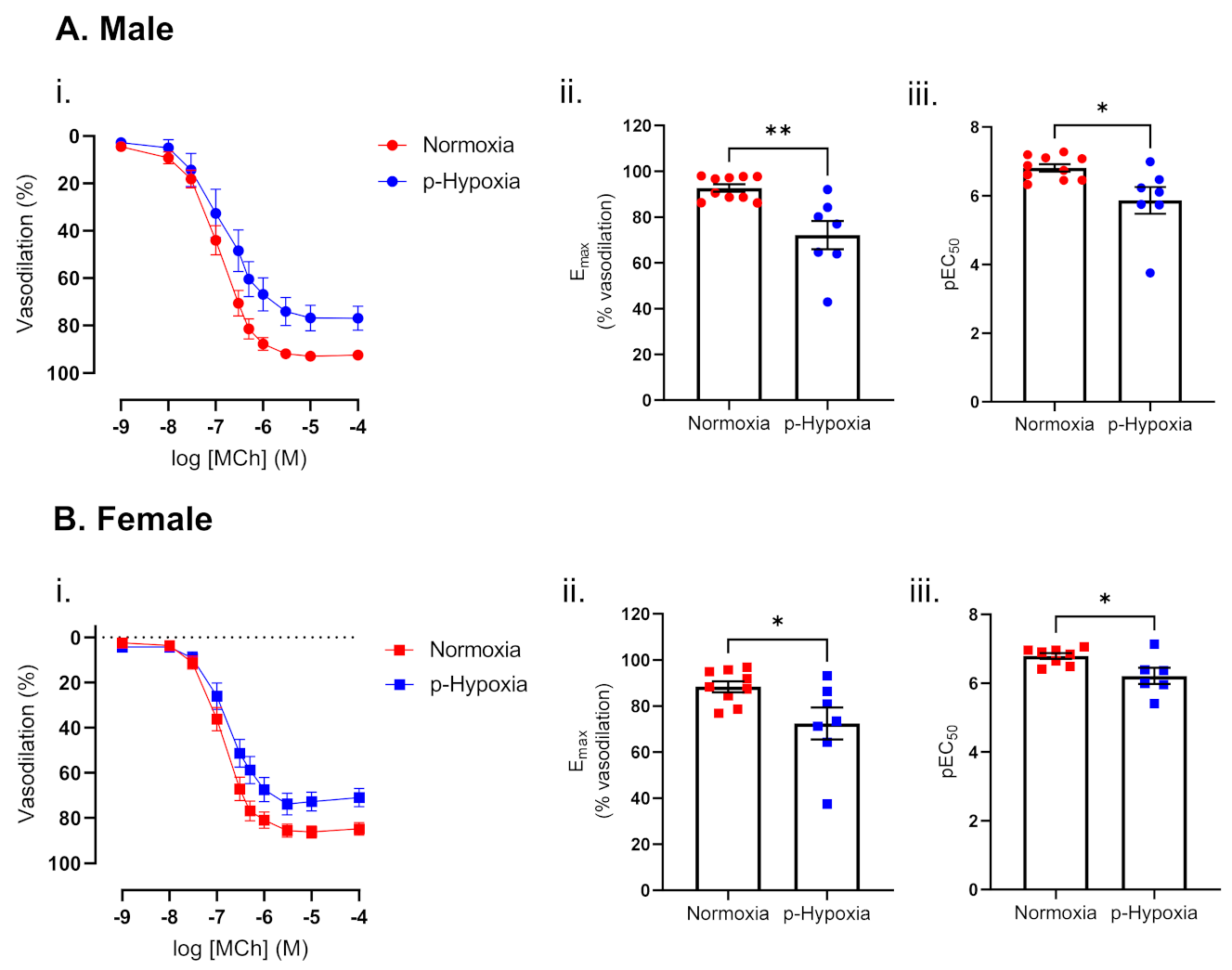
3.1.2. Impaired Endothelium-Dependent Vasodilation in Prenatally Hypoxic Male and Female 9.5-Month-Old Offspring
3.2. Mechanisms of Endothelium-Dependent Vasodilation in Male and Female 9.5-Moth-Old Offspring
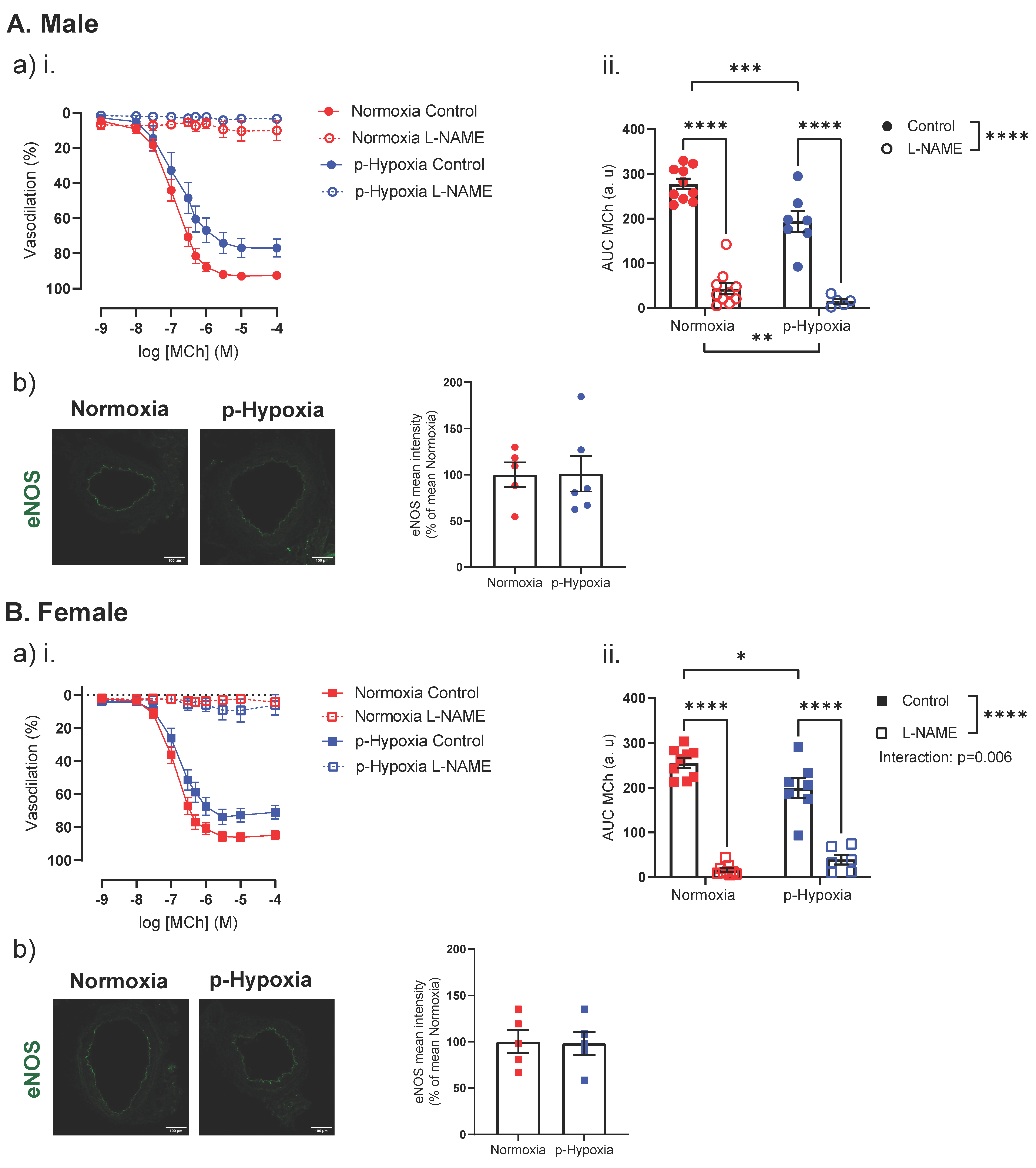
3.2.1. The Nitric Oxide Synthase (NOS) Pathway Is a Major Contributor to Coronary Artery Endothelium-Dependent Vasodilation in Male and Female Offspring
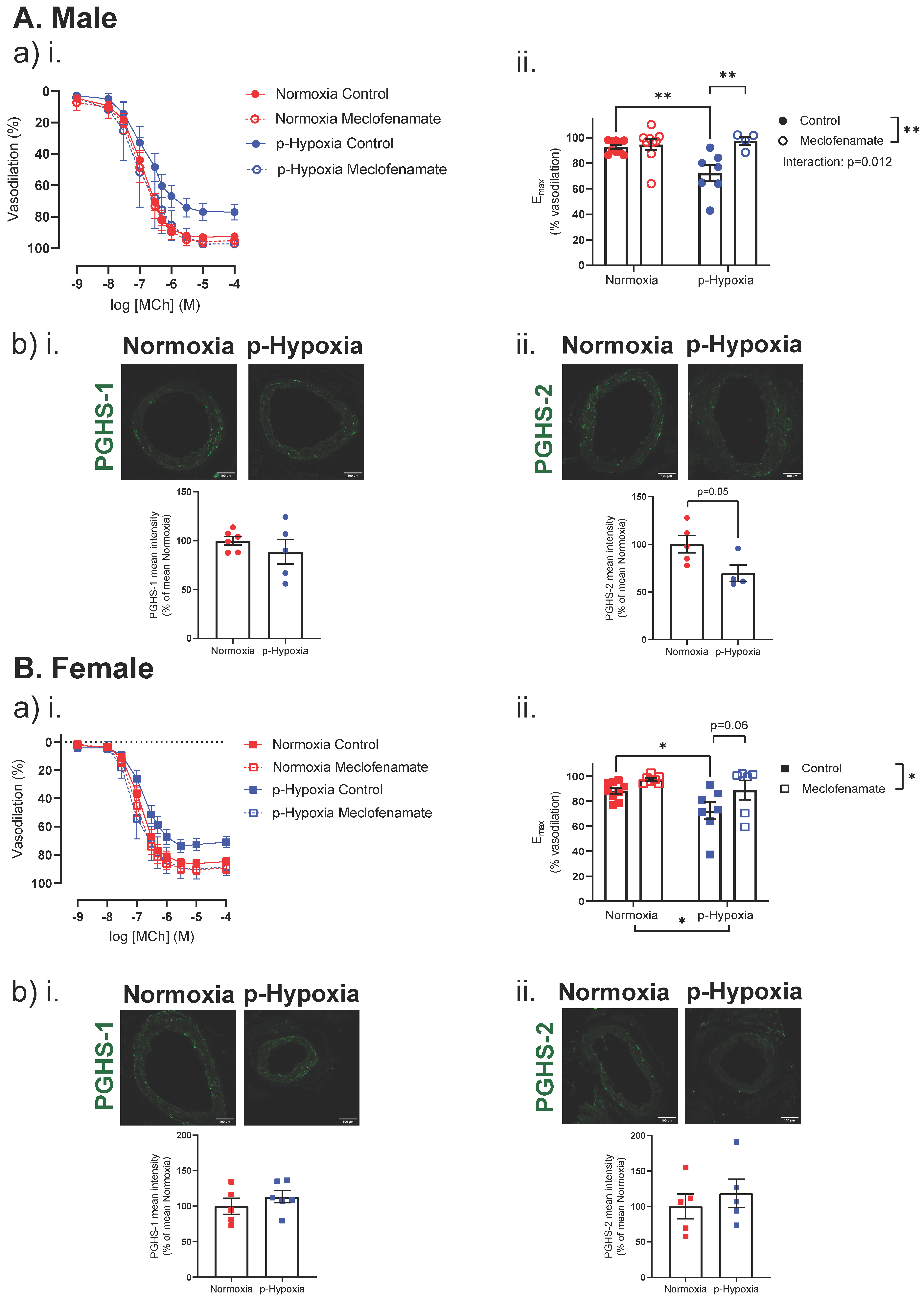
3.2.2. Enhanced Contribution of the PGHS Pathway to Coronary Artery Endothelium-Dependent Vasodilation in Prenatally Hypoxic Offspring
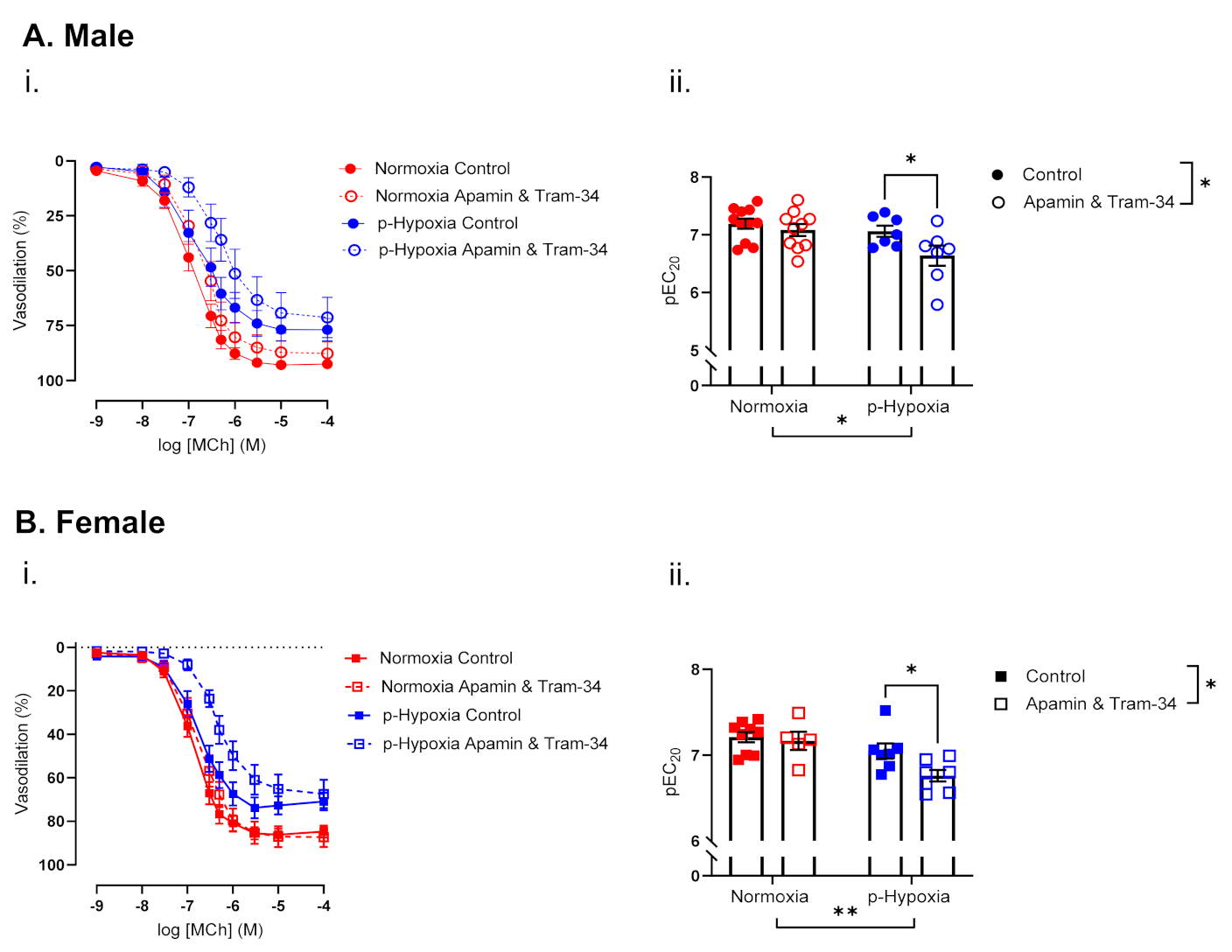
3.2.3. The Contribution of Endothelium-Derived Hyperpolarization (EDH) to Endothelium-Dependent Vasodilation Is Enhanced in Coronary Arteries of Prenatally Hypoxia Male and Female Offspring
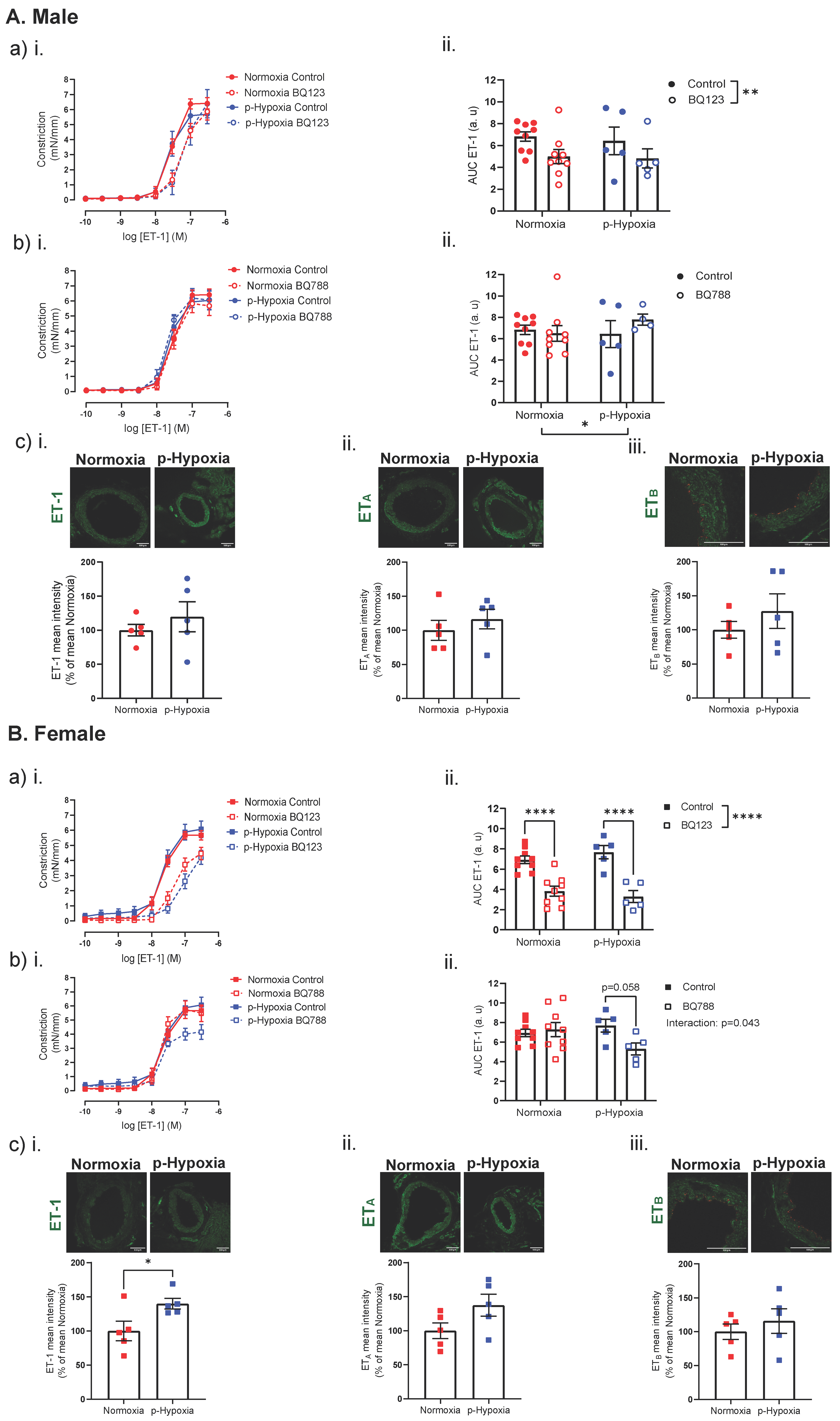
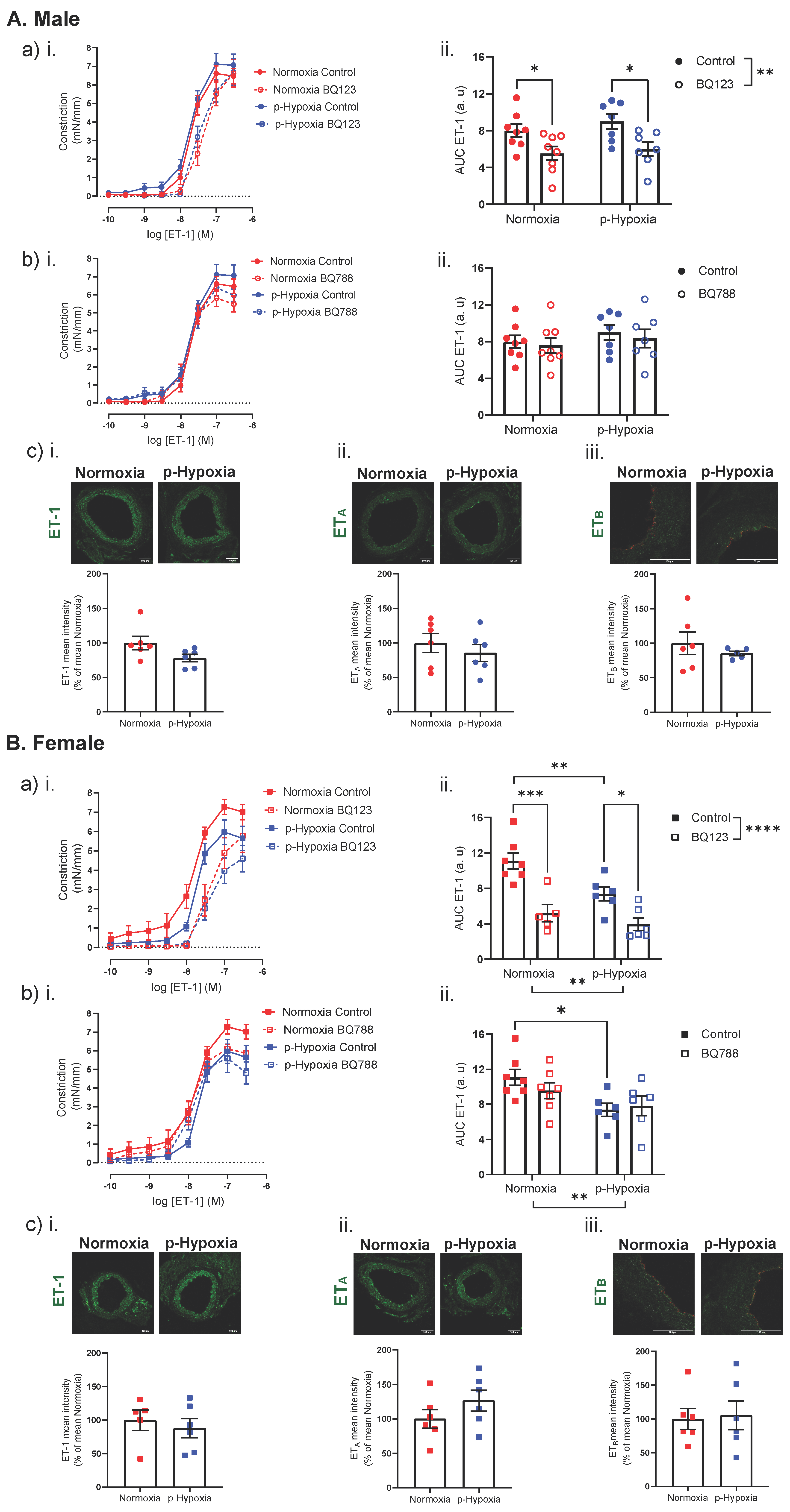
3.3. Coronary Artery Responses to ET-1 and the Contribution of ETA and ETB
3.3.1. Increased Contribution of ETB Receptors to ET-1 Mediated Vasoconstriction in 4-Month-Old Female Offspring
3.3.2. An Impaired ET-1 Mediated Vasoconstriction in 9.5-Month-Old Female Offspring
4. Discussion
4.1. The Effect of Prenatal Hypoxia on Coronary Artery Endothelium-Dependent and Endothelium-Independent Vasodilation in Adult Male and Female Offspring
4.2. The Effect of Prenatal Hypoxia on Coronary Artery ET-1 System in Male and Female Offspring
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheong, J.N.; Wlodek, M.E.; Moritz, K.M.; Cuffe, J.S. Programming of maternal and offspring disease: Impact of growth restriction, fetal sex and transmission across generations. J. Physiol. 2016, 594, 4727–4740. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.P.; Crimmins, S.; Telugu, B.; Turan, S. Intrauterine hypoxia: Clinical consequences and therapeutic perspectives. Res. Rep. Neonatol. 2015, 5, 79–89. [Google Scholar] [CrossRef]
- Giussani, D.A.; Davidge, S.T. Developmental programming of cardiovascular disease by prenatal hypoxia. J. Dev. Orig. Health Dis. 2013, 4, 328–337. [Google Scholar] [CrossRef]
- Bourque, S.L.; Gragasin, F.S.; Quon, A.L.; Mansour, Y.; Morton, J.S.; Davidge, S.T. Prenatal hypoxia causes long-term alterations in vascular endothelin-1 function in aged male, but not female, offspring. Hypertension 2013, 62, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Ream, M.; Ray, A.M.; Chandra, R.; Chikaraishi, D.M. Early fetal hypoxia leads to growth restriction and myocardial thinning. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R583–R595. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, L.; Wang, C. Prenatal hypoxia-induced epigenomic and transcriptomic reprogramming in rat fetal and adult offspring hearts. Sci. Data 2019, 6, 238. [Google Scholar] [CrossRef] [PubMed]
- Crispi, F.; Miranda, J.; Gratacos, E. Long-term cardiovascular consequences of fetal growth restriction: Biology, clinical implications, and opportunities for prevention of adult disease. Am. J. Obstet Gynecol. 2018, 218, S869–S879. [Google Scholar] [CrossRef] [PubMed]
- Giussani, D.A.; Camm, E.J.; Niu, Y.; Richter, H.G.; Blanco, C.E.; Gottschalk, R.; Blake, E.Z.; Horder, K.A.; Thakor, A.S.; Hansell, J.A.; et al. Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS ONE 2012, 7, e31017. [Google Scholar] [CrossRef]
- Zhang, L. Prenatal hypoxia and cardiac programming. J. Soc. Gynecol. Investig. 2005, 12, 2–13. [Google Scholar] [CrossRef]
- Barker, D.J.; Winter, P.D.; Osmond, C.; Margetts, B.; Simmonds, S.J. Weight in infancy and death from ischaemic heart disease. Lancet 1989, 2, 577–580. [Google Scholar] [CrossRef]
- Stein, C.E.; Fall, C.H.; Kumaran, K.; Osmond, C.; Cox, V.; Barker, D.J. Fetal growth and coronary heart disease in south India. Lancet 1996, 348, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Forsen, T.; Eriksson, J.G.; Tuomilehto, J.; Osmond, C.; Barker, D.J. Growth in utero and during childhood among women who develop coronary heart disease: Longitudinal study. BMJ 1999, 319, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Leon, D.A.; Lithell, H.O.; Vagero, D.; Koupilova, I.; Mohsen, R.; Berglund, L.; Lithell, U.B.; McKeigue, P.M. Reduced fetal growth rate and increased risk of death from ischaemic heart disease: Cohort study of 15 000 Swedish men and women born 1915-29. BMJ 1998, 317, 241–245. [Google Scholar] [CrossRef]
- Aburawi, E.H.; Malcus, P.; Thuring, A.; Fellman, V.; Pesonen, E. Coronary flow in neonates with impaired intrauterine growth. J. Am. Soc. Echocardiogr. 2012, 25, 313–318. [Google Scholar] [CrossRef]
- Baschat, A.A.; Gembruch, U.; Harman, C.R. Coronary blood flow in fetuses with intrauterine growth restriction. J. Perinat Med. 1998, 26, 143–156. [Google Scholar]
- Baschat, A.A.; Gembruch, U.; Reiss, I.; Gortner, L.; Diedrich, K. Demonstration of fetal coronary blood flow by Doppler ultrasound in relation to arterial and venous flow velocity waveforms and perinatal outcome--the ‘heart-sparing effect’. Ultrasound Obstet. Gynecol. 1997, 9, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Cleal, J.K.; Poore, K.R.; Boullin, J.P.; Khan, O.; Chau, R.; Hambidge, O.; Torrens, C.; Newman, J.P.; Poston, L.; Noakes, D.E.; et al. Mismatched pre- and postnatal nutrition leads to cardiovascular dysfunction and altered renal function in adulthood. Proc. Natl. Acad. Sci. USA 2007, 104, 9529–9533. [Google Scholar] [CrossRef]
- Bubb, K.J.; Cock, M.L.; Black, M.J.; Dodic, M.; Boon, W.M.; Parkington, H.C.; Harding, R.; Tare, M. Intrauterine growth restriction delays cardiomyocyte maturation and alters coronary artery function in the fetal sheep. J. Physiol. 2007, 578, 871–881. [Google Scholar] [CrossRef]
- Schipke, J.; Gonzalez-Tendero, A.; Cornejo, L.; Willfuhr, A.; Bijnens, B.; Crispi, F.; Muhlfeld, C.; Gratacos, E. Experimentally induced intrauterine growth restriction in rabbits leads to differential remodelling of left versus right ventricular myocardial microstructure. Histochem. Cell Biol. 2017, 148, 557–567. [Google Scholar] [CrossRef]
- Sutherland, M.R.; Ng, K.W.; Drenckhahn, J.D.; Wlodek, M.E.; Black, M.J. Impact of Intrauterine Growth Restriction on the Capillarization of the Early Postnatal Rat Heart. Anat. Rec. (Hoboken) 2019, 302, 1580–1586. [Google Scholar] [CrossRef]
- Tare, M.; Parkington, H.C.; Wallace, E.M.; Sutherland, A.E.; Lim, R.; Yawno, T.; Coleman, H.A.; Jenkin, G.; Miller, S.L. Maternal melatonin administration mitigates coronary stiffness and endothelial dysfunction, and improves heart resilience to insult in growth restricted lambs. J. Physiol. 2014, 592, 2695–2709. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Canadilla, P.; de Vries, T.; Gonzalez-Tendero, A.; Bonnin, A.; Gratacos, E.; Crispi, F.; Bijnens, B.; Zhang, C. Structural coronary artery remodelling in the rabbit fetus as a result of intrauterine growth restriction. PLoS ONE 2019, 14, e0218192. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Yang, S.; Zhang, L. Prenatal cocaine exposure causes sex-dependent impairment in the myogenic reactivity of coronary arteries in adult offspring. Hypertension 2009, 54, 1123–1128. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, X.; Qi, L.; Su, H.; He, Y.; Li, N.; Gao, Q.; Li, H.; Xu, T.; Lu, L.; Xu, Z.; et al. Prenatal hypoxia attenuated contraction of offspring coronary artery associated with decreased PKCbeta Ser(660) phosphorylation and intracellular calcium. Life Sci. 2020, 261, 118364. [Google Scholar] [CrossRef]
- Deussen, A.; Ohanyan, V.; Jannasch, A.; Yin, L.; Chilian, W. Mechanisms of metabolic coronary flow regulation. J. Mol. Cell Cardiol 2012, 52, 794–801. [Google Scholar] [CrossRef]
- Radico, F.; Zimarino, M.; Fulgenzi, F.; Ricci, F.; Di Nicola, M.; Jespersen, L.; Chang, S.M.; Humphries, K.H.; Marzilli, M.; De Caterina, R. Determinants of long-term clinical outcomes in patients with angina but without obstructive coronary artery disease: A systematic review and meta-analysis. Eur. Heart J. 2018, 39, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Sandoo, A.; van Zanten, J.J.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef]
- Mensah, G.A. Healthy endothelium: The scientific basis for cardiovascular health promotion and chronic disease prevention. Vascul. Pharmacol. 2007, 46, 310–314. [Google Scholar] [CrossRef]
- Grover-Paez, F.; Zavalza-Gomez, A.B. Endothelial dysfunction and cardiovascular risk factors. Diabetes Res. Clin. Pract. 2009, 84, 1–10. [Google Scholar] [CrossRef]
- Brain, K.L.; Allison, B.J.; Niu, Y.; Cross, C.M.; Itani, N.; Kane, A.D.; Herrera, E.A.; Skeffington, K.L.; Botting, K.J.; Giussani, D.A. Intervention against hypertension in the next generation programmed by developmental hypoxia. PLoS Biol. 2019, 17, e2006552. [Google Scholar] [CrossRef]
- Garcia-Villalon, A.L.; Fernandez, N.; Monge, L.; Salcedo, A.; Dieguez, G. Effects of endothelin-1 on the relaxation of rat coronary arteries. J. Cardiovasc. Pharmacol. 2009, 54, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.E.; Greenwood, I.A.; Large, W.A. Tonic regulation of vascular tone by nitric oxide and chloride ions in rat isolated small coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2604–H2611. [Google Scholar] [CrossRef] [PubMed]
- Climent, B.; Moreno, L.; Martinez, P.; Contreras, C.; Sanchez, A.; Perez-Vizcaino, F.; Garcia-Sacristan, A.; Rivera, L.; Prieto, D. Upregulation of SK3 and IK1 channels contributes to the enhanced endothelial calcium signaling and the preserved coronary relaxation in obese Zucker rats. PLoS ONE 2014, 9, e109432. [Google Scholar] [CrossRef] [PubMed]
- Mazzuca, M.Q.; Khalil, R.A. Vascular endothelin receptor type B: Structure, function and dysregulation in vascular disease. Biochem. Pharmacol. 2012, 84, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Lerman, A.; Burnett, J.C., Jr. Intact and altered endothelium in regulation of vasomotion. Circulation 1992, 86, III12–III19. [Google Scholar]
- Panza, J.A.; Quyyumi, A.A.; Brush, J.E., Jr.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990, 323, 22–27. [Google Scholar] [CrossRef]
- Vita, J.A.; Treasure, C.B.; Nabel, E.G.; McLenachan, J.M.; Fish, R.D.; Yeung, A.C.; Vekshtein, V.I.; Selwyn, A.P.; Ganz, P. Coronary vasomotor response to acetylcholine relates to risk factors for coronary artery disease. Circulation 1990, 81, 491–497. [Google Scholar] [CrossRef]
- Chong, A.Y.; Freestone, B.; Patel, J.; Lim, H.S.; Hughes, E.; Blann, A.D.; Lip, G.Y. Endothelial activation, dysfunction, and damage in congestive heart failure and the relation to brain natriuretic peptide and outcomes. Am. J. Cardiol. 2006, 97, 671–675. [Google Scholar] [CrossRef]
- Niu, Y.; Kane, A.D.; Lusby, C.M.; Allison, B.J.; Chua, Y.Y.; Kaandorp, J.J.; Nevin-Dolan, R.; Ashmore, T.J.; Blackmore, H.L.; Derks, J.B.; et al. Maternal Allopurinol Prevents Cardiac Dysfunction in Adult Male Offspring Programmed by Chronic Hypoxia During Pregnancy. Hypertension 2018, 72, 971–978. [Google Scholar] [CrossRef]
- Rueda-Clausen, C.F.; Morton, J.S.; Davidge, S.T. Effects of hypoxia-induced intrauterine growth restriction on cardiopulmonary structure and function during adulthood. Cardiovasc. Res. 2009, 81, 713–722. [Google Scholar] [CrossRef]
- Xu, Y.; Williams, S.J.; O’Brien, D.; Davidge, S.T. Hypoxia or nutrient restriction during pregnancy in rats leads to progressive cardiac remodeling and impairs postischemic recovery in adult male offspring. FASEB J. 2006, 20, 1251–1253. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Zhang, L. Prenatal hypoxia causes a sex-dependent increase in heart susceptibility to ischemia and reperfusion injury in adult male offspring: Role of protein kinase C epsilon. J. Pharmacol. Exp. Ther. 2009, 330, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Hasdai, D.; Gibbons, R.J.; Holmes, D.R., Jr.; Higano, S.T.; Lerman, A. Coronary endothelial dysfunction in humans is associated with myocardial perfusion defects. Circulation 1997, 96, 3390–3395. [Google Scholar] [CrossRef] [PubMed]
- Quyyumi, A.A.; Dakak, N.; Andrews, N.P.; Husain, S.; Arora, S.; Gilligan, D.M.; Panza, J.A.; Cannon, R.O., 3rd. Nitric oxide activity in the human coronary circulation. Impact of risk factors for coronary atherosclerosis. J. Clin. Invest. 1995, 95, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Kelm, M.; Schrader, J. Control of coronary vascular tone by nitric oxide. Circ. Res. 1990, 66, 1561–1575. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.B.; Punihaole, D.; Levine, T.B. Characterization of the role of nitric oxide and its clinical applications. Cardiology 2012, 122, 55–68. [Google Scholar] [CrossRef]
- Garcia, V.; Sessa, W.C. Endothelial NOS: Perspective and recent developments. Br. J. Pharmacol 2019, 176, 189–196. [Google Scholar] [CrossRef]
- Berges, A.; Van Nassauw, L.; Timmermans, J.P.; Vrints, C. Role of nitric oxide during coronary endothelial dysfunction after myocardial infarction. Eur. J. Pharmacol. 2005, 516, 60–70. [Google Scholar] [CrossRef]
- Chen, C.; Ochoa, L.N.; Kagan, A.; Chai, H.; Liang, Z.; Lin, P.H.; Yao, Q. Lysophosphatidic acid causes endothelial dysfunction in porcine coronary arteries and human coronary artery endothelial cells. Atherosclerosis 2012, 222, 74–83. [Google Scholar] [CrossRef]
- Ramaswami, G.; Chai, H.; Yao, Q.; Lin, P.H.; Lumsden, A.B.; Chen, C. Curcumin blocks homocysteine-induced endothelial dysfunction in porcine coronary arteries. J. Vasc Surg 2004, 40, 1216–1222. [Google Scholar] [CrossRef]
- Davidge, S.T. Prostaglandin H synthase and vascular function. Circ. Res. 2001, 89, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Goodwill, A.G.; Dick, G.M.; Kiel, A.M.; Tune, J.D. Regulation of Coronary Blood Flow. Compr. Physiol. 2017, 7, 321–382. [Google Scholar] [CrossRef] [PubMed]
- Loftin, C.D.; Trivedi, D.B.; Tiano, H.F.; Clark, J.A.; Lee, C.A.; Epstein, J.A.; Morham, S.G.; Breyer, M.D.; Nguyen, M.; Hawkins, B.M.; et al. Failure of ductus arteriosus closure and remodeling in neonatal mice deficient in cyclooxygenase-1 and cyclooxygenase-2. Proc. Natl. Acad. Sci. USA 2001, 98, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Norwood, V.F.; Morham, S.G.; Smithies, O. Postnatal development and progression of renal dysplasia in cyclooxygenase-2 null mice. Kidney Int. 2000, 58, 2291–2300. [Google Scholar] [CrossRef] [PubMed]
- Baserga, M.; Hale, M.A.; Wang, Z.M.; Yu, X.; Callaway, C.W.; McKnight, R.A.; Lane, R.H. Uteroplacental insufficiency alters nephrogenesis and downregulates cyclooxygenase-2 expression in a model of IUGR with adult-onset hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1943–R1955. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ellinsworth, D.C.; Sandow, S.L.; Shukla, N.; Liu, Y.; Jeremy, J.Y.; Gutterman, D.D. Endothelium-Derived Hyperpolarization and Coronary Vasodilation: Diverse and Integrated Roles of Epoxyeicosatrienoic Acids, Hydrogen Peroxide, and Gap Junctions. Microcirculation 2016, 23, 15–32. [Google Scholar] [CrossRef]
- Edwards, G.; Feletou, M.; Weston, A.H. Endothelium-derived hyperpolarising factors and associated pathways: A synopsis. Pflugers Arch. 2010, 459, 863–879. [Google Scholar] [CrossRef]
- Sandow, S.L.; Neylon, C.B.; Chen, M.X.; Garland, C.J. Spatial separation of endothelial small- and intermediate-conductance calcium-activated potassium channels (K(Ca)) and connexins: Possible relationship to vasodilator function? J. Anat. 2006, 209, 689–698. [Google Scholar] [CrossRef]
- Socha, M.J.; Behringer, E.J.; Segal, S.S. Calcium and electrical signalling along endothelium of the resistance vasculature. Basic Clin. Pharmacol. Toxicol. 2012, 110, 80–86. [Google Scholar] [CrossRef]
- Grgic, I.; Kaistha, B.P.; Hoyer, J.; Kohler, R. Endothelial Ca+-activated K+ channels in normal and impaired EDHF-dilator responses--relevance to cardiovascular pathologies and drug discovery. Br. J. Pharmacol. 2009, 157, 509–526. [Google Scholar] [CrossRef]
- Feng, J.; Liu, Y.; Clements, R.T.; Sodha, N.R.; Khabbaz, K.R.; Senthilnathan, V.; Nishimura, K.K.; Alper, S.L.; Sellke, F.W. Calcium-activated potassium channels contribute to human coronary microvascular dysfunction after cardioplegic arrest. Circulation 2008, 118, S46–S51. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Huang, J.H.; Man, Y.B.; Yao, X.Q.; He, G.W. Use of intermediate/small conductance calcium-activated potassium-channel activator for endothelial protection. J. Thorac. Cardiovasc. Surg. 2011, 141, 501–510.e1. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Li, A.Y.; Guo, Q.H.; Guo, Y.J.; Weiss, J.W.; Ji, E.S. Endothelin-1 and ET receptors impair left ventricular function by mediated coronary arteries dysfunction in chronic intermittent hypoxia rats. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Dagassan, P.H.; Breu, V.; Clozel, M.; Kunzli, A.; Vogt, P.; Turina, M.; Kiowski, W.; Clozel, J.P. Up-regulation of endothelin-B receptors in atherosclerotic human coronary arteries. J. Cardiovasc. Pharmacol. 1996, 27, 147–153. [Google Scholar] [CrossRef]
- Wackenfors, A.; Emilson, M.; Ingemansson, R.; Hortobagyi, T.; Szok, D.; Tajti, J.; Vecsei, L.; Edvinsson, L.; Malmsjo, M. Ischemic heart disease induces upregulation of endothelin receptor mRNA in human coronary arteries. Eur. J. Pharmacol. 2004, 484, 103–109. [Google Scholar] [CrossRef]
- Kellogg, D.L., Jr.; Liu, Y.; Pergola, P.E. Selected contribution: Gender differences in the endothelin-B receptor contribution to basal cutaneous vascular tone in humans. J. Appl Physiol. (1985) 2001, 91, 2407–2411; discussion 2389–2490. [Google Scholar] [CrossRef]
- Katakam, P.V.; Snipes, J.A.; Tulbert, C.D.; Mayanagi, K.; Miller, A.W.; Busija, D.W. Impaired endothelin-induced vasoconstriction in coronary arteries of Zucker obese rats is associated with uncoupling of [Ca2+]i signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol 2006, 290, R145–R153. [Google Scholar] [CrossRef]
- Giulumian, A.D.; Molero, M.M.; Reddy, V.B.; Pollock, J.S.; Pollock, D.M.; Fuchs, L.C. Role of ET-1 receptor binding and [Ca(2+)](i) in contraction of coronary arteries from DOCA-salt hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1944–H1949. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hula, N.; Liu, R.; Spaans, F.; Pasha, M.; Quon, A.; Kirschenman, R.; Cooke, C.-L.M.; Davidge, S.T. The Long-Term Effects of Prenatal Hypoxia on Coronary Artery Function of the Male and Female Offspring. Biomedicines 2022, 10, 3019. https://doi.org/10.3390/biomedicines10123019
Hula N, Liu R, Spaans F, Pasha M, Quon A, Kirschenman R, Cooke C-LM, Davidge ST. The Long-Term Effects of Prenatal Hypoxia on Coronary Artery Function of the Male and Female Offspring. Biomedicines. 2022; 10(12):3019. https://doi.org/10.3390/biomedicines10123019
Chicago/Turabian StyleHula, Nataliia, Ricky Liu, Floor Spaans, Mazhar Pasha, Anita Quon, Raven Kirschenman, Christy-Lynn M. Cooke, and Sandra T. Davidge. 2022. "The Long-Term Effects of Prenatal Hypoxia on Coronary Artery Function of the Male and Female Offspring" Biomedicines 10, no. 12: 3019. https://doi.org/10.3390/biomedicines10123019
APA StyleHula, N., Liu, R., Spaans, F., Pasha, M., Quon, A., Kirschenman, R., Cooke, C.-L. M., & Davidge, S. T. (2022). The Long-Term Effects of Prenatal Hypoxia on Coronary Artery Function of the Male and Female Offspring. Biomedicines, 10(12), 3019. https://doi.org/10.3390/biomedicines10123019