
Adenovirus-Inspired Virus-like-Particles Displaying Melanoma Tumor Antigen Specifically Target Human DC Subsets and Trigger Antigen-Specific Immune Responses
 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Baculovirus Production
2.2. Protein Purification
2.2.1. ADDomers’ Purification
2.2.2. RBD-SC Purification
2.2.3. MelA-SC Purification
2.3. Complex Formation and Labelling
2.3.1. ADD-ST + MelA/RBD-SC (ADD-MelA, ADD KGE RBD and ADD RGD RBD) Complex Formation for Cross Priming and Cross Presentation Experiments
2.3.2. ADD-ST AF647 + MelA/RBD-SC (ADD-MelA AF647, ADD KGE/RGD +/− RBD AF647) Complex Formation for Internalization Experiments
2.4. Cell Preparation and Culture
2.4.1. Transformed Cell Lines
2.4.2. Peripheral Blood Mononuclear Cells (PBMCs) Isolation
2.5. Fixation, Internalization of ADDomer
2.5.1. Evaluation of the Fixation of ADDomers AF647 on DC Subsets by Flow Cytometry
2.5.2. Determination of the Internalization of ADDomers AF647 by DC Subsets by Confocal Microscopy
2.6. DC Subsets’ Activation and Cytokine Production upon Incubation with ADDomers
2.6.1. PBMCs Preparation/Cell Culture Conditions
2.6.2. Cell Viability and Costimulatory Molecules Expression Analysis by Flow Cytometry
2.6.3. Cytokine Secretion Analysis by Luminex
2.7. Analysis of DCs’ Ability to Perform the Cross-Presentation of ADDomers-Associated Epitopes/Antigens
2.7.1. PanDC Incubation with ADDomers
2.7.2. Conditions of Cross-Presentation
2.7.3. Measurement of Cytokines Production by CBA
2.8. Cross-Priming of Antigen (A2L) Specific CD8 T Cells Using ADDomers
2.8.1. Conditions of Cross-Priming
2.8.2. Determination of Anti-MelA26–35 CD8+ T Cells by Dextramer Staining
2.8.3. Amplification of Antigen Specific CD8+ T Cells
2.8.4. Determination of the Affinity of Antigen Specific CD8+ T Cells
2.9. Detargeting Retargeting Experiments
2.9.1. Evaluation of the Interactions between the ADDomers and CLR by Surface Plasmon Resonance
2.9.2. De-Targeting Evaluation Using ADD KGE versus ADD RGD on A549 Cell Line
2.9.3. ADD KGE and RGD Labeling with AF 488
2.9.4. ADD KGE and ADD RGD Internalization by A549 Evaluated by Flow Cytometry
2.9.5. ADD KGE and ADD RGD Internalization by A549 Evaluated by Time-Lapse Confocal Microscopy
3. Results
3.1. Human Melanoma Epitope A2L Can Be Displayed on the ADDomer Surface by Genetic Insertion
3.2. The Large Human Tumor Associated Antigen MelanA Can Be Displayed on the ADDomer’s Surface Using the SpyTag/SpyCatcher System
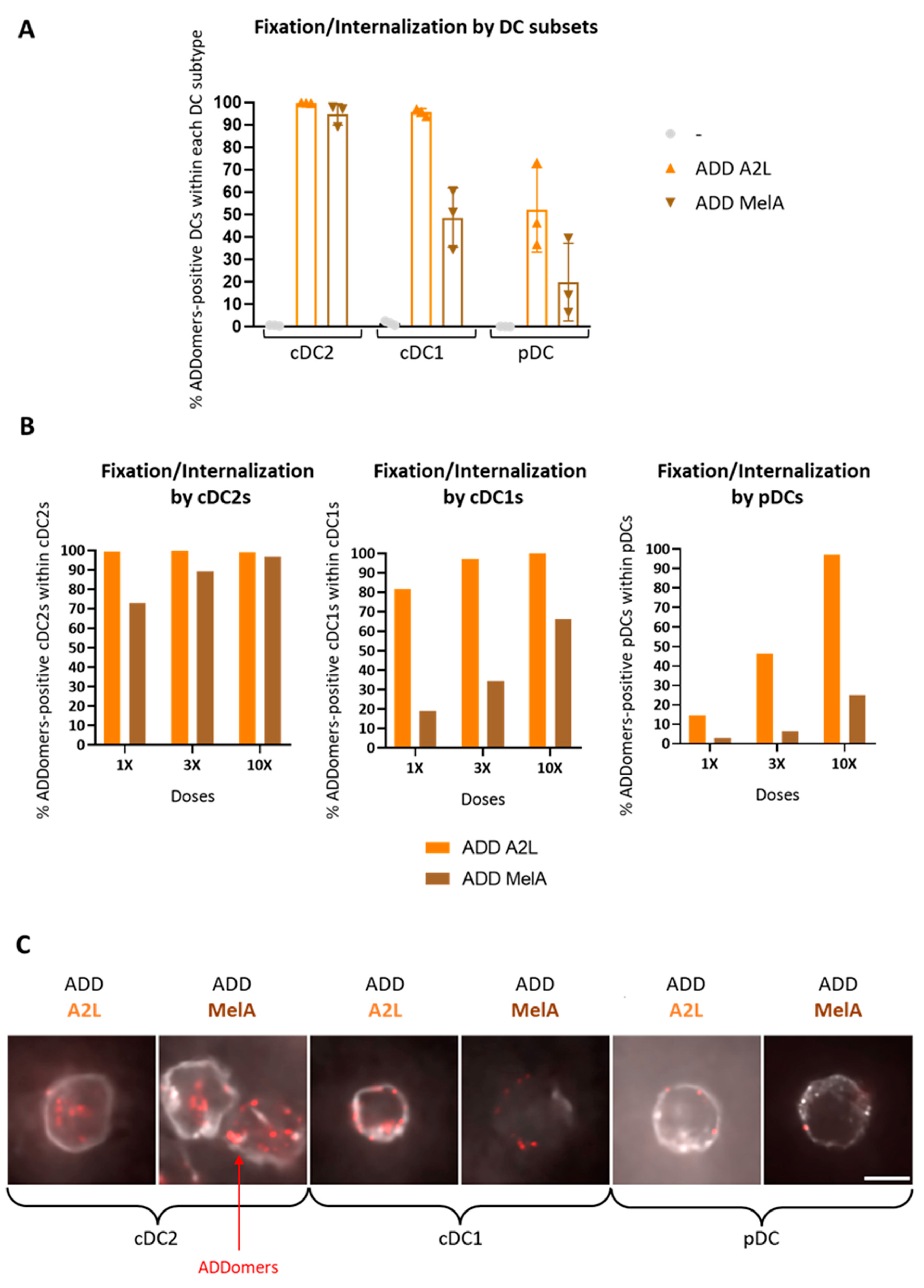
3.3. ADD A2L and ADD MelA Are Differently Internalized by DC Subsets
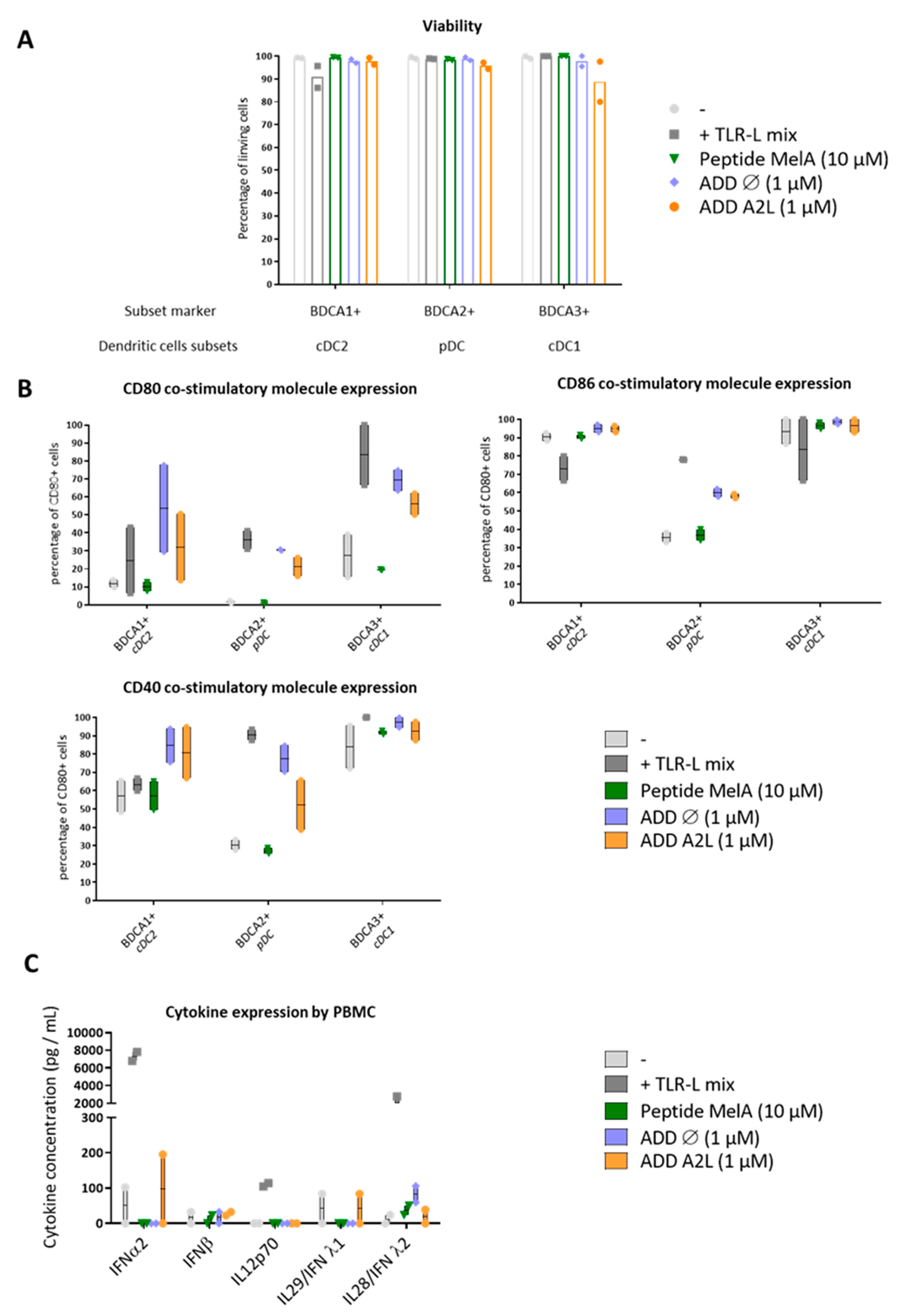
3.4. ADDomer Triggers a Strong Activation of All DC Subsets Together with Cytokine Secretion
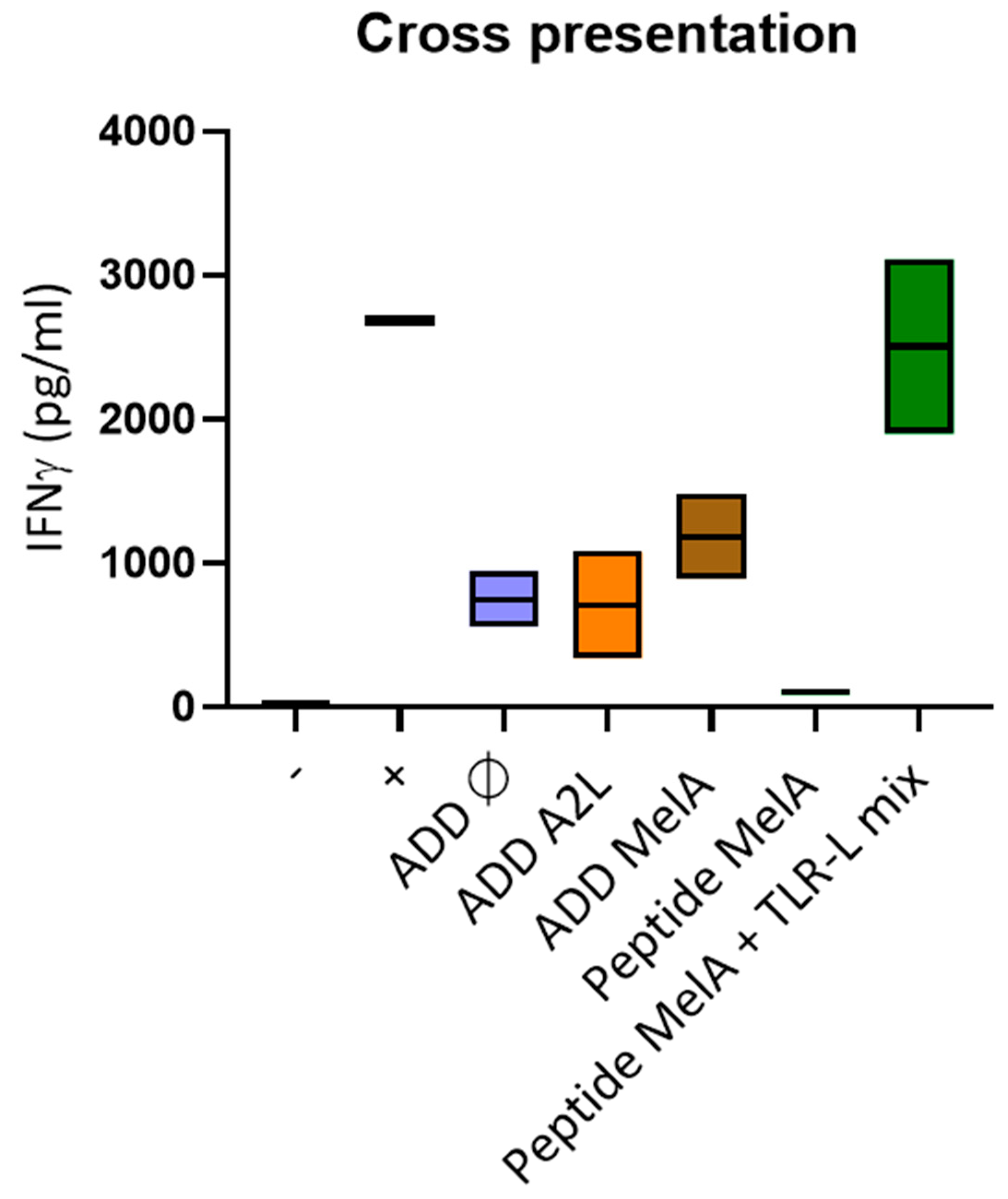
3.5. DCs Efficiently Cross-Present the A2L Epitope from ADDomer Displaying the Full MelanA Antigen
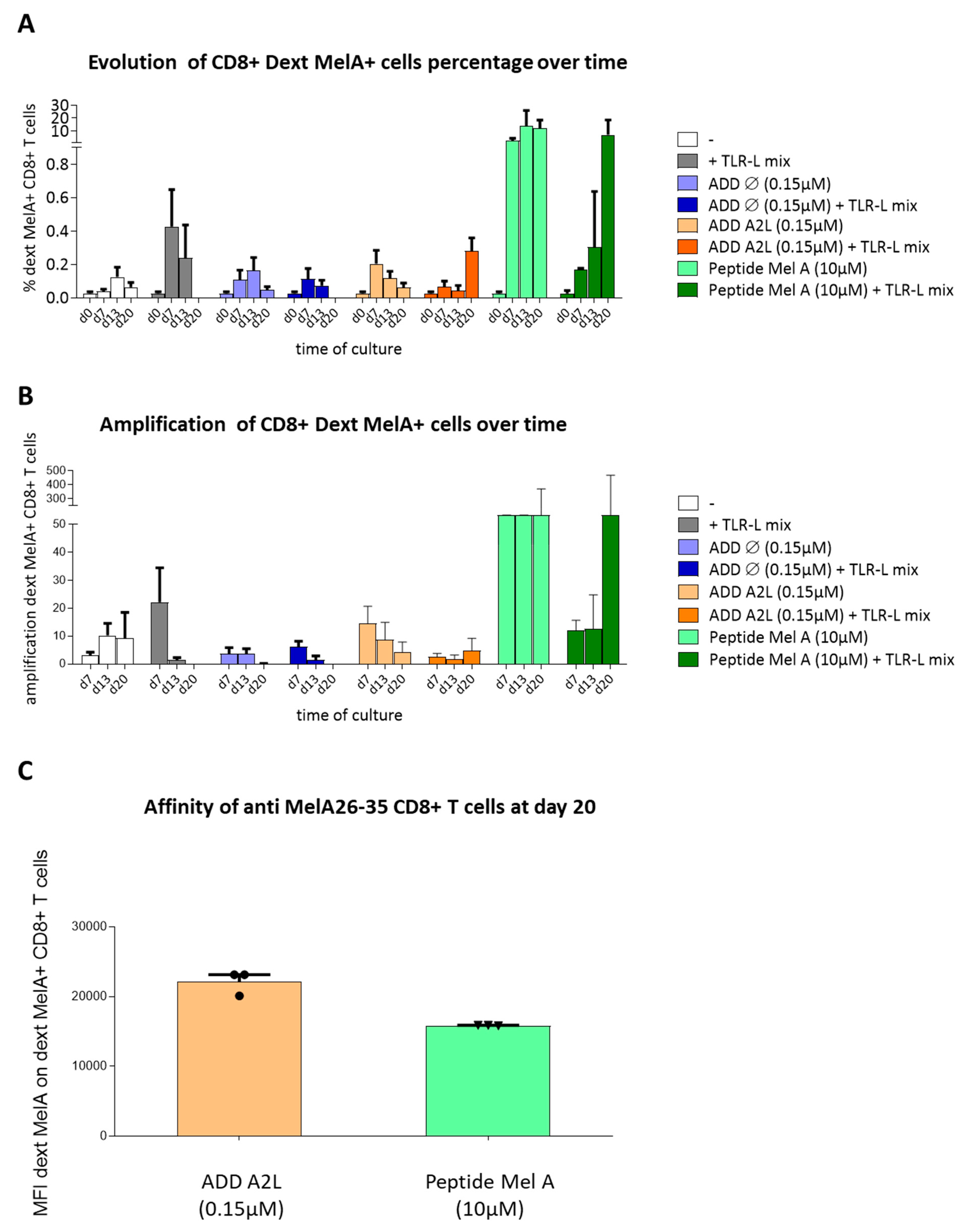
3.6. ADDomer Displaying A2L Epitope Cross-Prime High Affinity Peptide-Specific CD8+ T Cells
3.7. De-Targeting and Re-Targeting ADDomers Specifically to DC Subsets
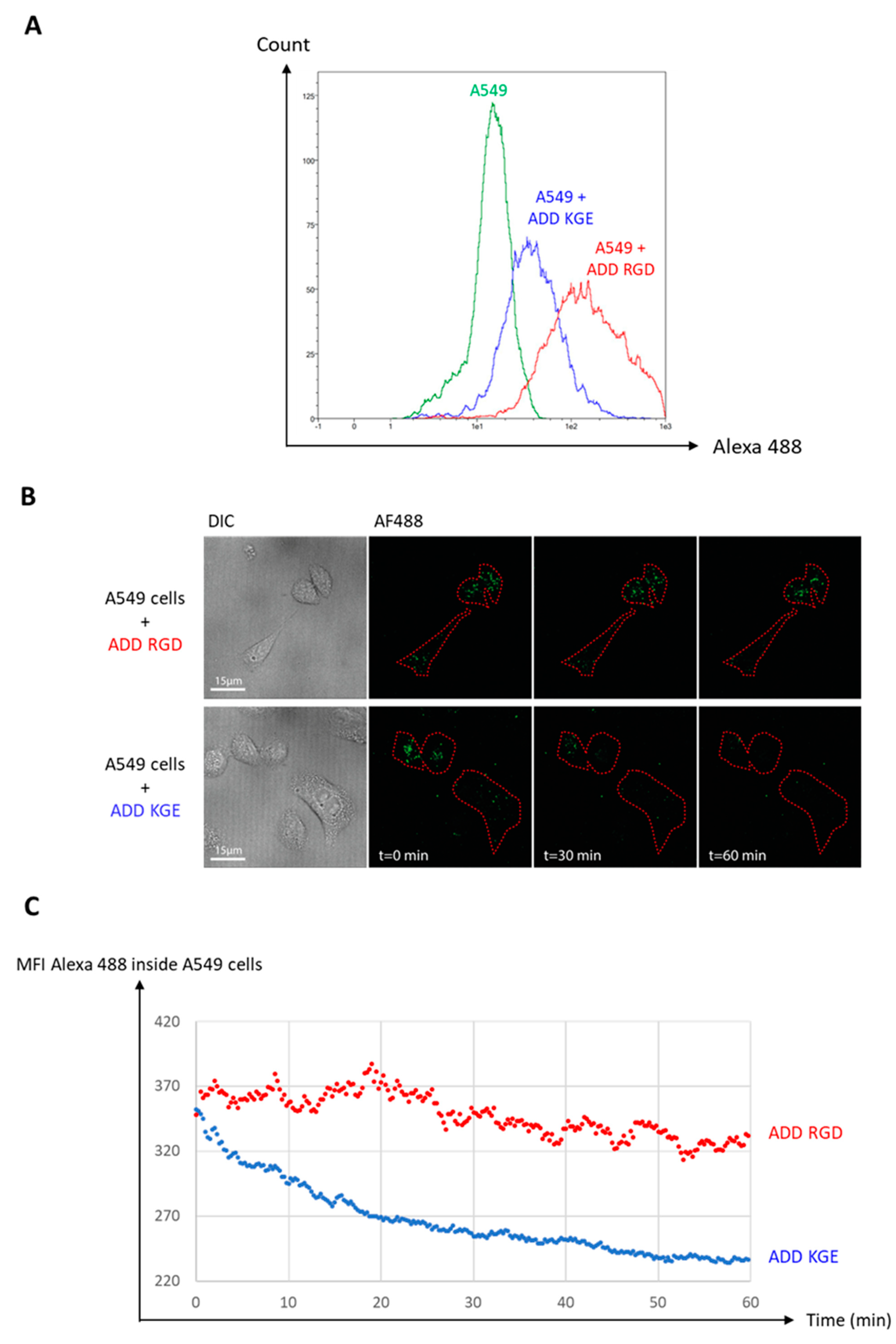
3.8. Functional De-Targeting of ADD KGE Compared to ADD RGD on A549 Cells
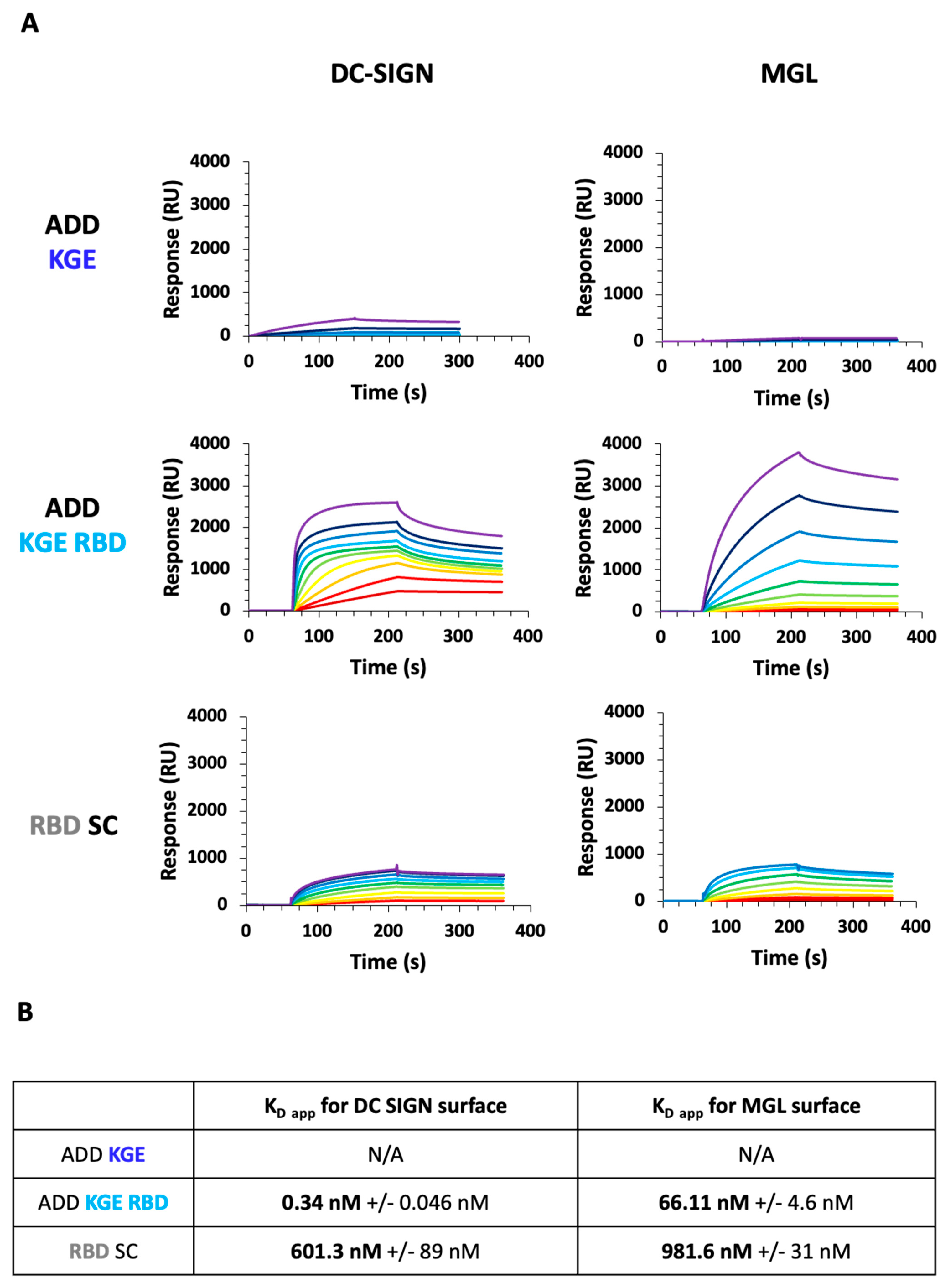
3.9. Assessment of ADDomer Targeting to C-Type Lectin Receptors (CLRs) by SPR
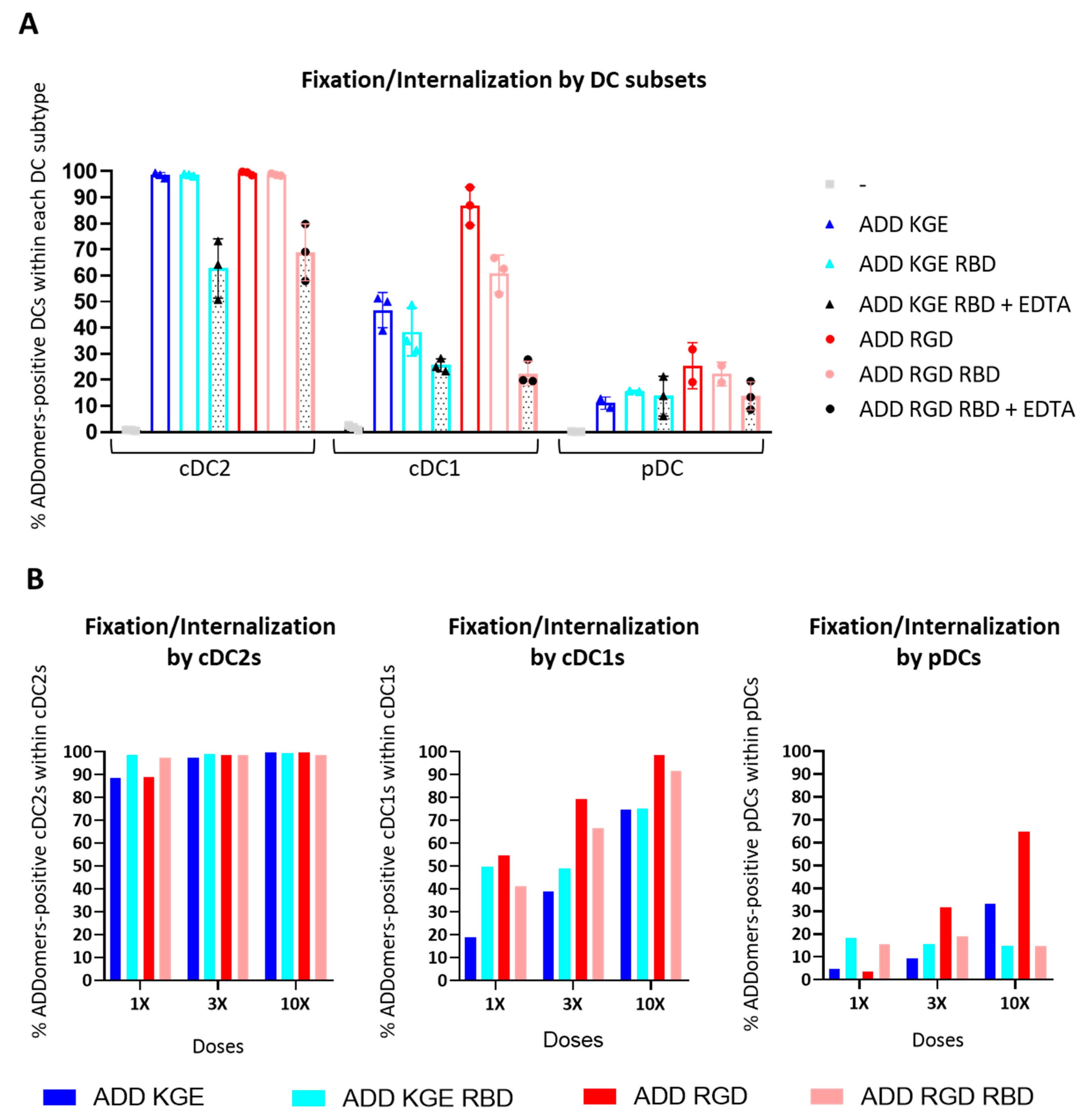
3.10. ADD KGE/RGD and ADD KGE/RGD RBD Are Successfully Fixed/Internalized by DC Subsets but Differentially Depending on the Subset
3.11. ADD KGE/RGD RBD Fixation/Internalization by Dendritic Cells Might Involve Binding to a CLR
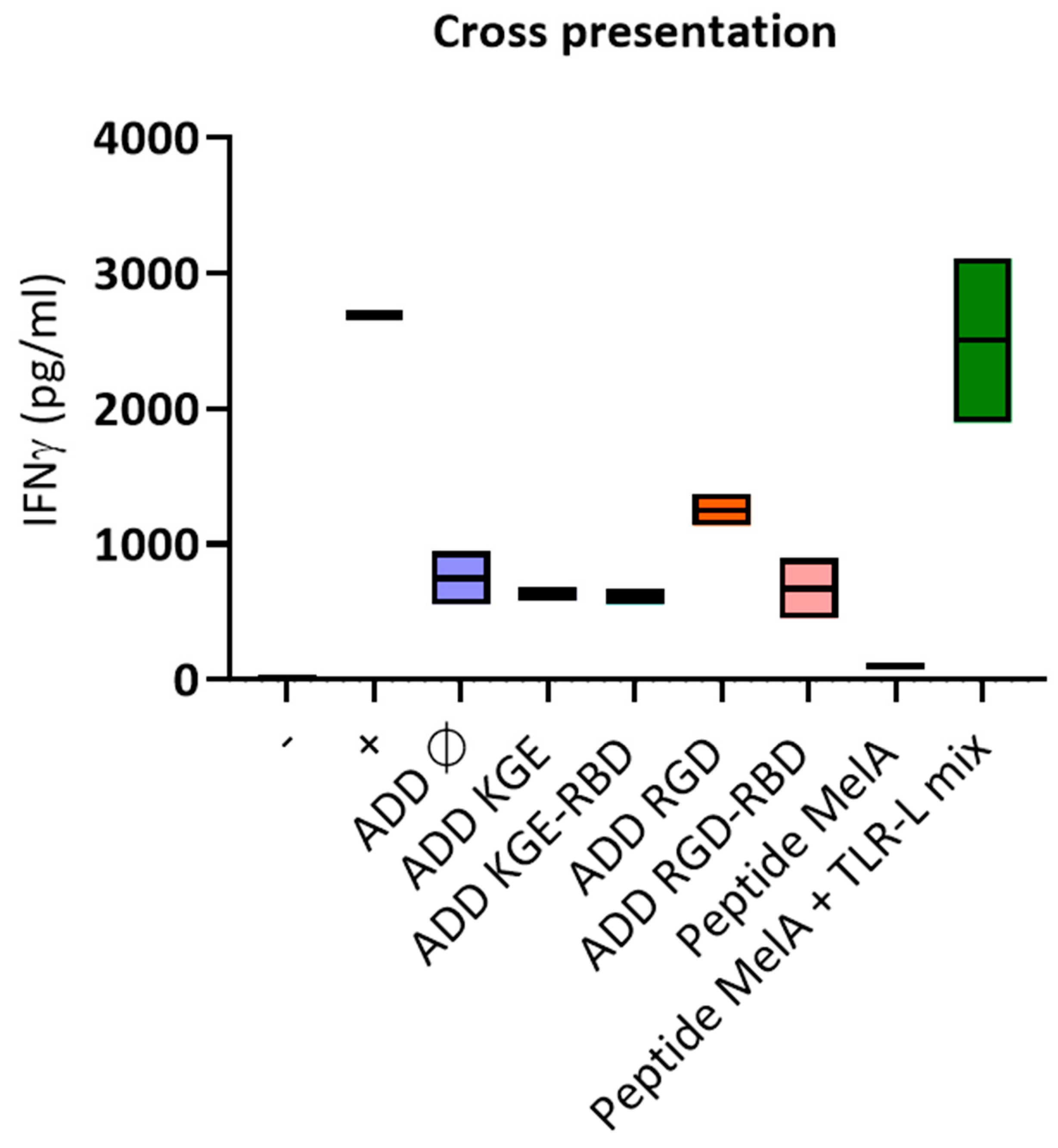
3.12. Only ADD RGD Is Cross-Presented by Dendritic Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vacchelli, E.; Bravo-San Pedro, J.M.; Buqué, A.; Senovilla, L.; Baracco, E.E.; Bloy, N.; Castoldi, F.; Abastado, J.-P.; Agostinis, P.; et al. Classification of current anticancer immunotherapies. Oncotarget 2014, 5, 12472–12508. [Google Scholar] [CrossRef]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, F.K.; Palucka, K. Understanding and activating immunity against human cancer. Curr. Opin. Immunol. 2010, 22, 212–214. [Google Scholar] [CrossRef]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar]
- Guo, Y.; Lei, K.; Tang, L. Neoantigen Vaccine Delivery for Personalized Anticancer Immunotherapy. Front. Immunol. 2018, 9, 1499. [Google Scholar] [CrossRef]
- Niemann, J.; Kühnel, F. Oncolytic viruses: Adenoviruses. Virus Genes 2017, 53, 700–706. [Google Scholar] [CrossRef]
- Hemminki, O.; Bauerschmitz, G.; Hemmi, S.; Lavilla-Alonso, S.; Diaconu, I.; Guse, K.; Koski, A.; Desmond, R.A.; Lappalainen, M.; Kanerva, A.; et al. Oncolytic adenovirus based on serotype 3. Cancer Gene Ther. 2011, 18, 288–296. [Google Scholar] [CrossRef]
- Hemminki, O.; dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef]
- Seymour, L.W.; Fisher, K.D. Oncolytic viruses: Finally delivering. Br. J. Cancer 2016, 114, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Vragniau, C.; Bufton, J.C.; Garzoni, F.; Stermann, E.; Rabi, F.; Terrat, C.; Guidetti, M.; Josserand, V.; Williams, M.; Woods, C.J.; et al. Synthetic self-assembling ADDomer platform for highly efficient vaccination by genetically encoded multiepitope display. Sci. Adv. 2019, 5, eaaw2853. [Google Scholar] [CrossRef] [PubMed]
- Fender, P.; Ruigrok, R.W.; Gout, E.; Buffet, S.; Chroboczek, J. Adenovirus dodecahedron, a new vector for human gene transfer. Nat. Biotechnol. 1997, 15, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Besson, S.; Vragniau, C.; Vassal-Stermann, E.; Dagher, M.C.; Fender, P. The Adenovirus Dodecahedron: Beyond the Platonic Story. Viruses 2020, 12, 718. [Google Scholar] [CrossRef]
- Chevillard, C.; Amen, A.; Besson, S.; Hannani, D.; Bally, I.; Dettling, V.; Gout, E.; Moreau, C.J.; Buisson, M.; Gallet, S.; et al. Elicitation of potent SARS-CoV-2 neutralizing antibody responses through immunization with a versatile adenovirus-inspired multimerization platform. Mol. Ther. 2022, 30, 1913–1925. [Google Scholar] [CrossRef]
- Villegas-Mendez, A.; Garin, M.I.; Pineda-Molina, E.; Veratti, E.; Bueren, J.A.; Fender, P.; Lenormand, J.-L. In vivo delivery of antigens by adenovirus dodecahedron induces cellular and humoral immune responses to elicit antitumor immunity. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1046–1053. [Google Scholar] [CrossRef]
- Reina, J.J.; Maldonado, O.S.; Tabarani, G.; Fieschi, F.; Rojo, J. Mannose Glycoconjugates Functionalized at Positions 1 and 6. Binding Analysis to DC-SIGN Using Biosensors. Bioconjug. Chem. 2007, 18, 963–969. [Google Scholar] [CrossRef]
- Bulteau, F.; Thépaut, M.; Henry, M.; Hurbin, A.; Vanwonterghem, L.; Vivès, C.; Le Roy, A.; Ebel, C.; Renaudet, O.; Fieschi, F.; et al. Targeting Tn-Antigen-Positive Human Tumors with a Recombinant Human Macrophage Galactose C-Type Lectin. Mol. Pharm. 2021, 19, 235–245. [Google Scholar] [CrossRef]
- Achilli, S.; Monteiro, J.T.; Serna, S.; Mayer-Lambertz, S.; Thépaut, M.; Le Roy, A.; Ebel, C.; Reichardt, N.-C.; Lepenies, B.; Fieschi, F.; et al. TETRALEC, Artificial Tetrameric Lectins: A Tool to Screen Ligand and Pathogen Interactions. Int. J. Mol. Sci. 2020, 21, 5290. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Gout, E.; Schoehn, G.; Fenel, D.; Lortat-Jacob, H.; Fender, P. The Adenovirus Type 3 Dodecahedron’s RGD Loop Comprisesan HSPG Binding Site That Influences Integrin Binding. J. Biomed. Biotechnol. 2010, 2010, e541939. [Google Scholar] [CrossRef] [PubMed]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins αvβ3 and αvβ5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Shi, X.; Jarvis, D.L. Protein N-Glycosylation in the Baculovirus-Insect Cell System. Curr. Drug Targets 2007, 8, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.L.; Jarvis, D.L. Protein N-glycosylation in the baculovirus-insect cell expression system and engineering of insect cells to produce ‘mammalianized’ recombinant glycoproteins. Adv. Virus Res. 2006, 68, 159–191. [Google Scholar]
- Gong, Y.; Qin, S.; Dai, L.; Tian, Z. The glycosylation in SARS-CoV-2 and its receptor ACE2. Signal Transduct. Target. Ther. 2021, 6, 396. [Google Scholar] [CrossRef]
- Thépaut, M.; Luczkowiak, J.; Vivès, C.; Labiod, N.; Bally, I.; Lasala, F.; Grimoire, Y.; Fenel, D.; Sattin, S.; Thielens, N.; et al. DC/L-SIGN recognition of spike glycoprotein promotes SARS-CoV-2 trans-infection and can be inhibited by a glycomimetic antagonist. PLOS Pathog. 2021, 17, e1009576. [Google Scholar] [CrossRef]
- Porkolab, V.; Pifferi, C.; Sutkeviciute, I.; Ordanini, S.; Taouai, M.; Thépaut, M.; Vivès, C.; Benazza, M.; Bernardi, A.; Renaudet, O.; et al. Development of C-type lectin-oriented surfaces for high avidity glycoconjugates: Towards mimicking multivalent interactions on the cell surface. Org. Biomol. Chem. 2020, 18, 4763–4772. [Google Scholar] [CrossRef]
- van Liempt, E.; Bank, C.M.; Mehta, P.; Garcı’a-Vallejoa, J.J.; Kawar, Z.S.; Geyer, R.; Alvarez, R.A.; Cummings, R.D.; van Kooyk, Y.; van Die, I. Specificity of DC-SIGN for mannose- and fucose-containing glycans. FEBS Lett. 2006, 580, 6123–6131. [Google Scholar] [CrossRef]
- Zizzari, I.G.; Napoletano, C.; Battisti, F.; Rahimi, H.; Caponnetto, S.; Pierelli, L.; Nuti, M.; Rughetti, A. MGL Receptor and Immunity: When the Ligand Can Make the Difference. J. Immunol. Res. 2015, 2015, 450695. [Google Scholar] [CrossRef]
- Boscheinen, J.B.; Thomann, S.; Knipe, D.M.; DeLuca, N.; Schuler-Thurner, B.; Gross, S.; Dörrie, J.; Schaft, N.; Bach, C.; Rohrhofer, A.; et al. Generation of an Oncolytic Herpes Simplex Virus 1 Expressing Human MelanA. Front. Immunol. 2019, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Tuettenberg, A.; Becker, C.; Huter, E.; Knop, J.; Enk, A.H.; Jonuleit, H. Induction of strong and persistent MelanA/MART-1-specific immune responses by adjuvant dendritic cell-based vaccination of stage II melanoma patients. Int. J. Cancer 2006, 118, 2617–2627. [Google Scholar] [CrossRef] [PubMed]
- Conniot, J.; Scomparin, A.; Peres, C.; Yeini, E.; Pozzi, S.; Matos, A.I.; Kleiner, R.; Moura, L.I.F.; Zupančič, E.; Viana, A.S.; et al. Immunization with mannosylated nanovaccines and inhibition of the immune-suppressing microenvironment sensitizes melanoma to immune checkpoint modulators. Nat. Nanotechnol. 2019, 14, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-M.; Larrieu, P.; Sarrabayrouse, G.; Prevost-Blondel, A.; Lengagne, R.; Desfrançois, J.; Labarrière, N.; Jotereau, F.; Shin, J.H.; Park, J.-Y.; et al. HLA anchor optimization of the melan-A-HLA-A2 epitope within a long peptide is required for efficient cross-priming of human tumor-reactive T cells. J. Immunol. Baltim. Md 1950 2012, 188, 2102–2110. [Google Scholar] [CrossRef]
- Del Val, M.; Schlicht, H.-J.; Ruppert, T.; Reddehase, M.J.; Koszinowski, U.H. Efficient processing of an antigenic sequence for presentation by MHC class I molecules depends on its neighboring residues in the protein. Cell 1991, 66, 1145–1153. [Google Scholar] [CrossRef]
- Kloetzel, P.-M. Antigen processing by the proteasome: Ubiquitin and proteasomes. Nat. Rev. Mol. Cell Biol. 2001, 2, 179–188. [Google Scholar] [CrossRef]
- Nussbaum, A.K.; Dick, T.P.; Keilholz, W.; Schirle, M.; Stevanović, S.; Dietz, K.; Heinemeyer, W.; Groll, M.; Wolf, D.H.; Huber, R.; et al. Cleavage motifs of the yeast 20S proteasome β subunits deduced from digests of enolase 1. Proc. Natl. Acad. Sci. USA 1998, 95, 12504–12509. [Google Scholar] [CrossRef]
- Zhao, W.; Wu, J.; Chen, S.; Zhou, Z. Shared neoantigens: Ideal targets for off-the-shelf cancer immunotherapy. Pharmacogenomics 2020, 21, 637–645. [Google Scholar] [CrossRef]
- Vragniau, C. Modification des Dodécaèdres Bases de L’adénovirus de Sérotype 3: Design et Caractérisation d’un Nouveau Vecteur Multi-épitopique Polyvalent. Ph.D. Thesis, Université Grenoble Alpes, Grenoble, France, 20 September 2018. Available online: https://hal.archives-ouvertes.fr/tel-02110396 (accessed on 20 September 2018).
- Villegas-Méndez, A.; Fender, P.; Garín, M.I.; Rothe, R.; Liguori, L.; Marques, B.; Lenormand, J.-L. Functional Characterisation of the WW Minimal Domain for Delivering Therapeutic Proteins by Adenovirus Dodecahedron. PLoS ONE 2012, 7, e45416. [Google Scholar] [CrossRef]
- Galinier, R.; Gout, E.; Lortat-Jacob, H.; Wood, J.; Chroboczek, J. Adenovirus Protein Involved in Virus Internalization Recruits Ubiquitin−Protein Ligases. Biochemistry 2002, 41, 14299–14305. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Besson, S.; Laurin, D.; Chauvière, C.; Thépaut, M.; Kleman, J.-P.; Pezet, M.; Manches, O.; Fieschi, F.; Aspord, C.; Fender, P. Adenovirus-Inspired Virus-like-Particles Displaying Melanoma Tumor Antigen Specifically Target Human DC Subsets and Trigger Antigen-Specific Immune Responses. Biomedicines 2022, 10, 2881. https://doi.org/10.3390/biomedicines10112881
Besson S, Laurin D, Chauvière C, Thépaut M, Kleman J-P, Pezet M, Manches O, Fieschi F, Aspord C, Fender P. Adenovirus-Inspired Virus-like-Particles Displaying Melanoma Tumor Antigen Specifically Target Human DC Subsets and Trigger Antigen-Specific Immune Responses. Biomedicines. 2022; 10(11):2881. https://doi.org/10.3390/biomedicines10112881
Chicago/Turabian StyleBesson, Solène, David Laurin, Cyrielle Chauvière, Michel Thépaut, Jean-Philippe Kleman, Mylène Pezet, Olivier Manches, Franck Fieschi, Caroline Aspord, and Pascal Fender. 2022. "Adenovirus-Inspired Virus-like-Particles Displaying Melanoma Tumor Antigen Specifically Target Human DC Subsets and Trigger Antigen-Specific Immune Responses" Biomedicines 10, no. 11: 2881. https://doi.org/10.3390/biomedicines10112881
APA StyleBesson, S., Laurin, D., Chauvière, C., Thépaut, M., Kleman, J.-P., Pezet, M., Manches, O., Fieschi, F., Aspord, C., & Fender, P. (2022). Adenovirus-Inspired Virus-like-Particles Displaying Melanoma Tumor Antigen Specifically Target Human DC Subsets and Trigger Antigen-Specific Immune Responses. Biomedicines, 10(11), 2881. https://doi.org/10.3390/biomedicines10112881