
High-Throughput Molecular Dynamics-Based Alchemical Free Energy Calculations for Predicting the Binding Free Energy Change Associated with the Selected Omicron Mutations in the Spike Receptor-Binding Domain of SARS-CoV-2



Abstract
1. Introduction
2. Results and Discussion
2.1. Selection of Mutations for the Study
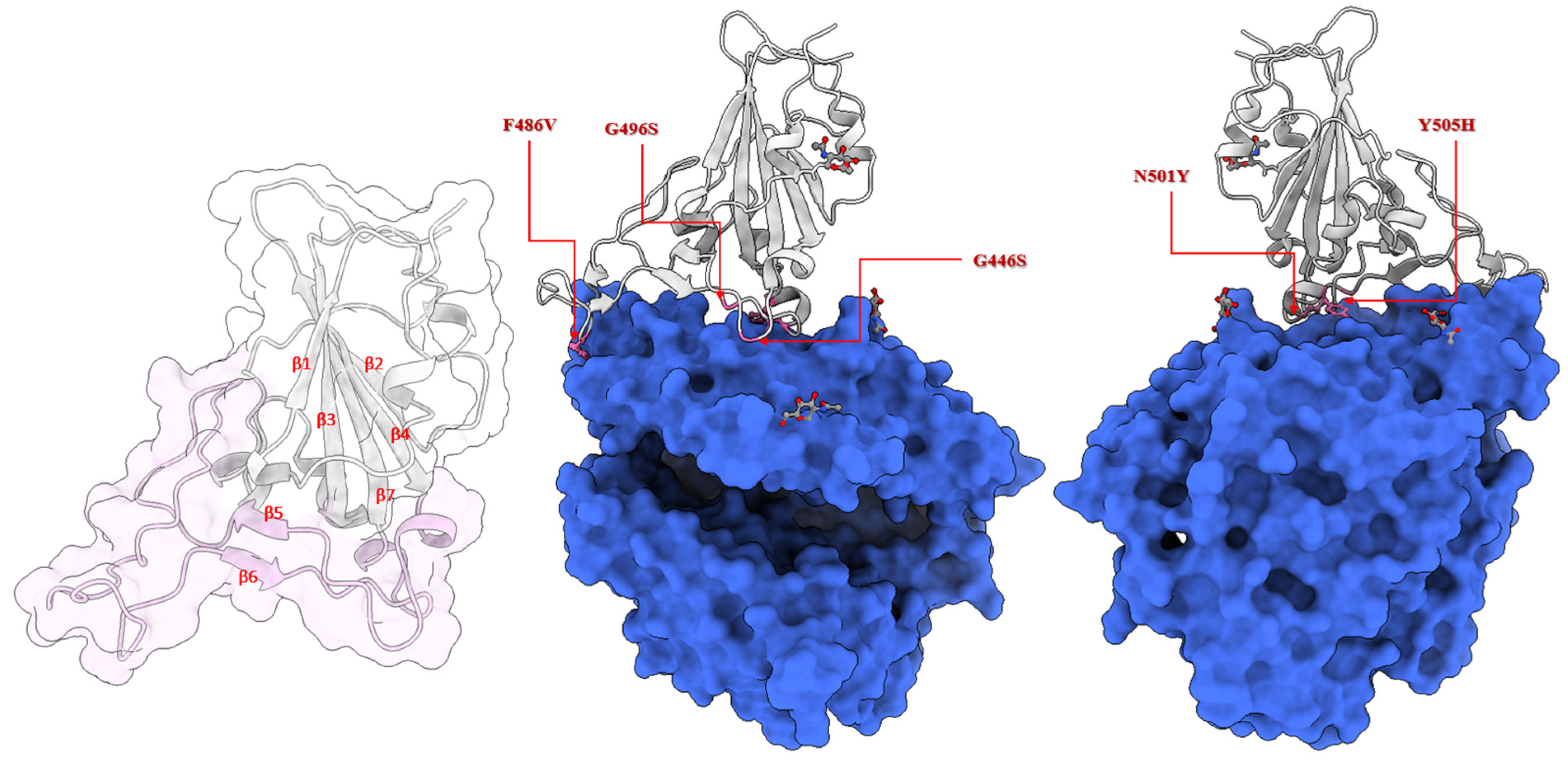
2.2. Location of the Selected Mutations at the Receptor-Binding Domain of the Spike Glycoprotein
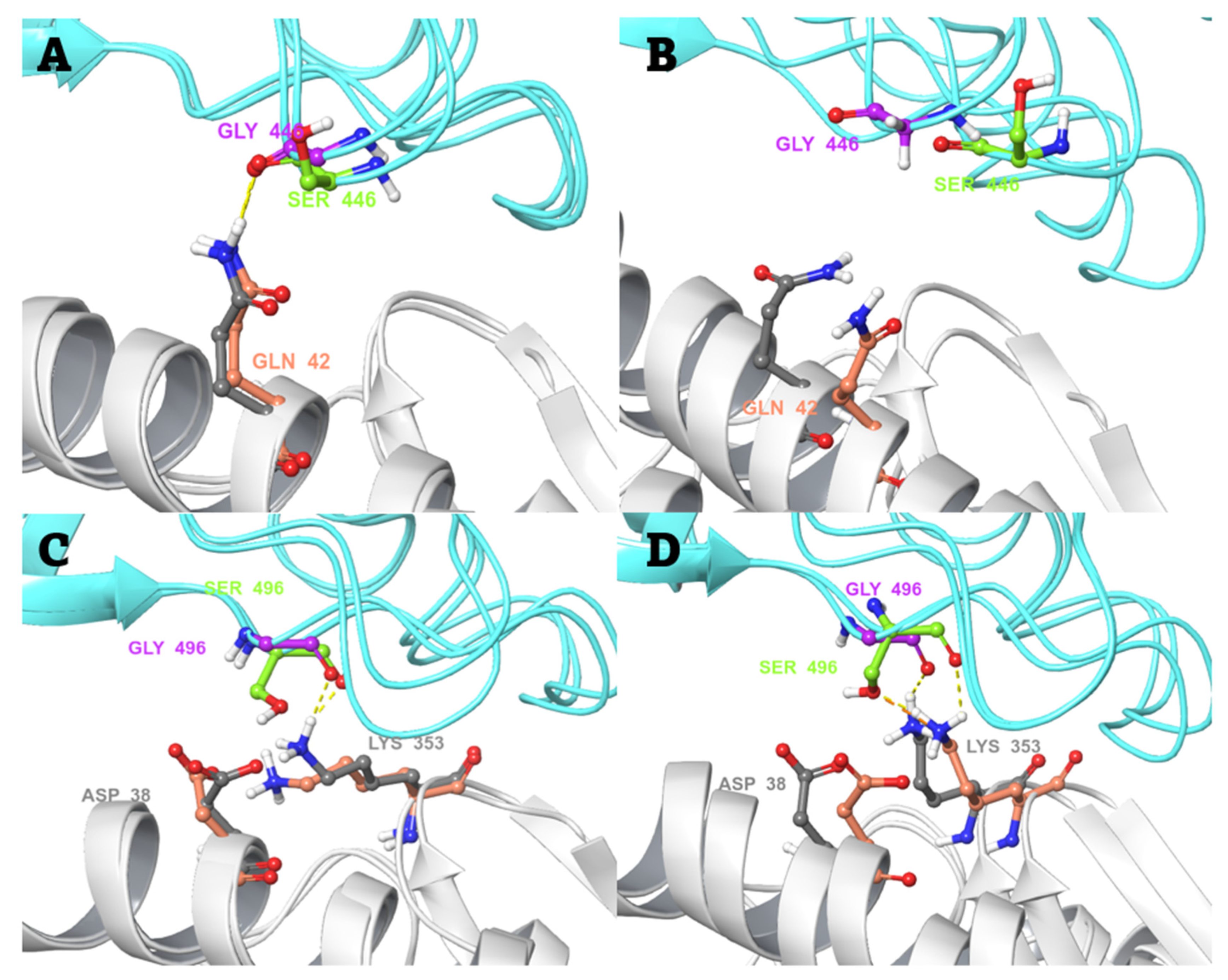
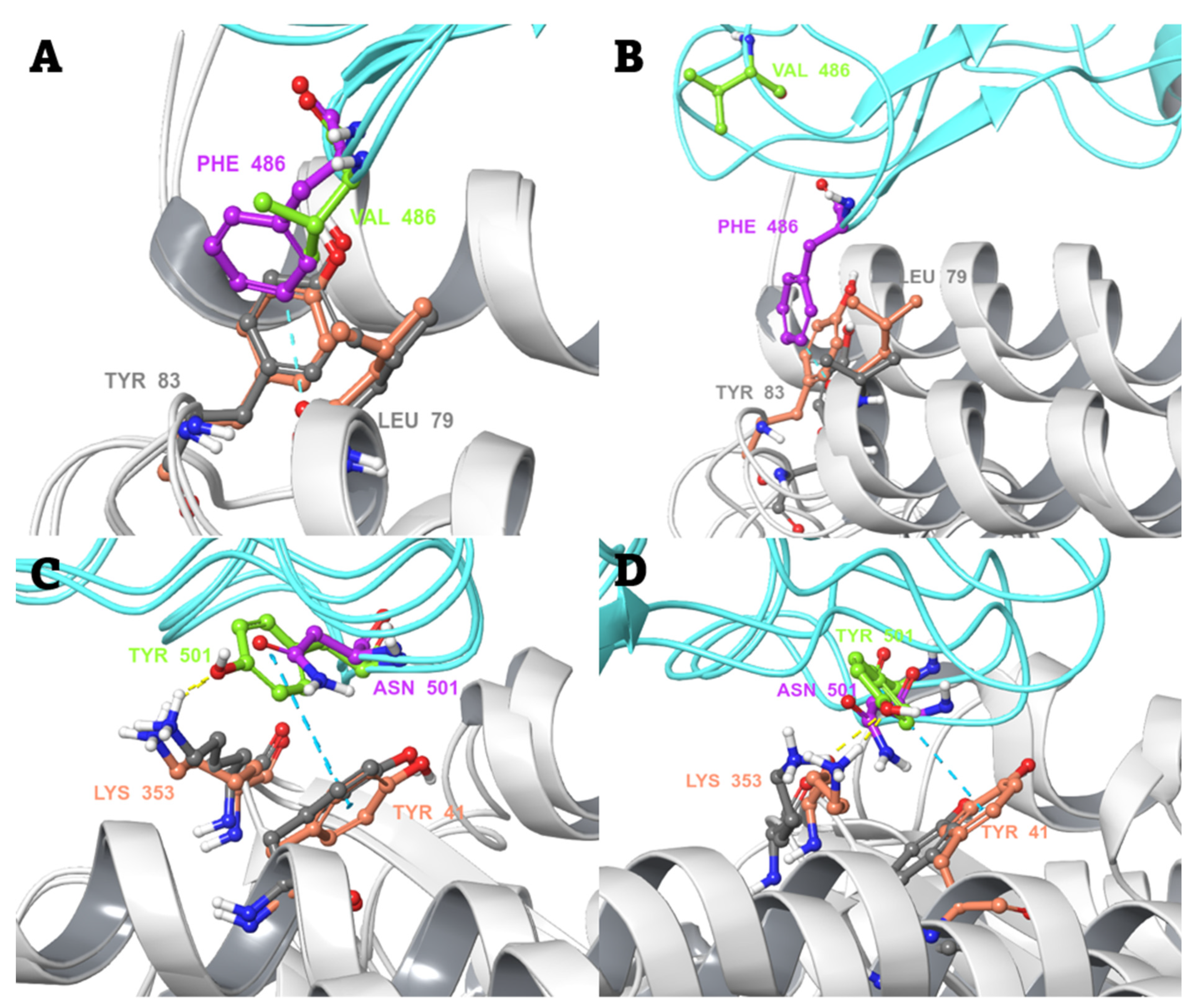
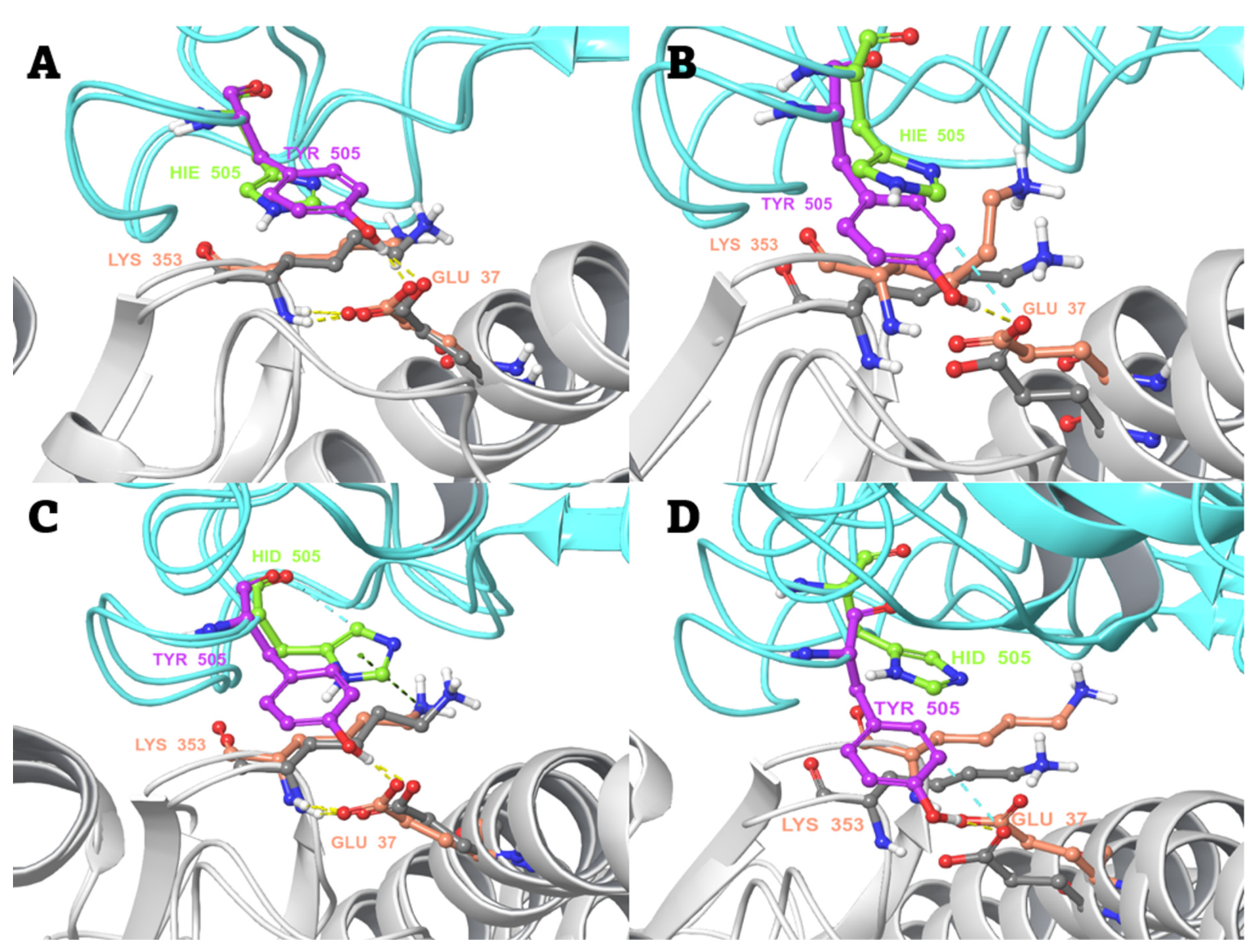
2.3. Conventional MD Simulation Studies
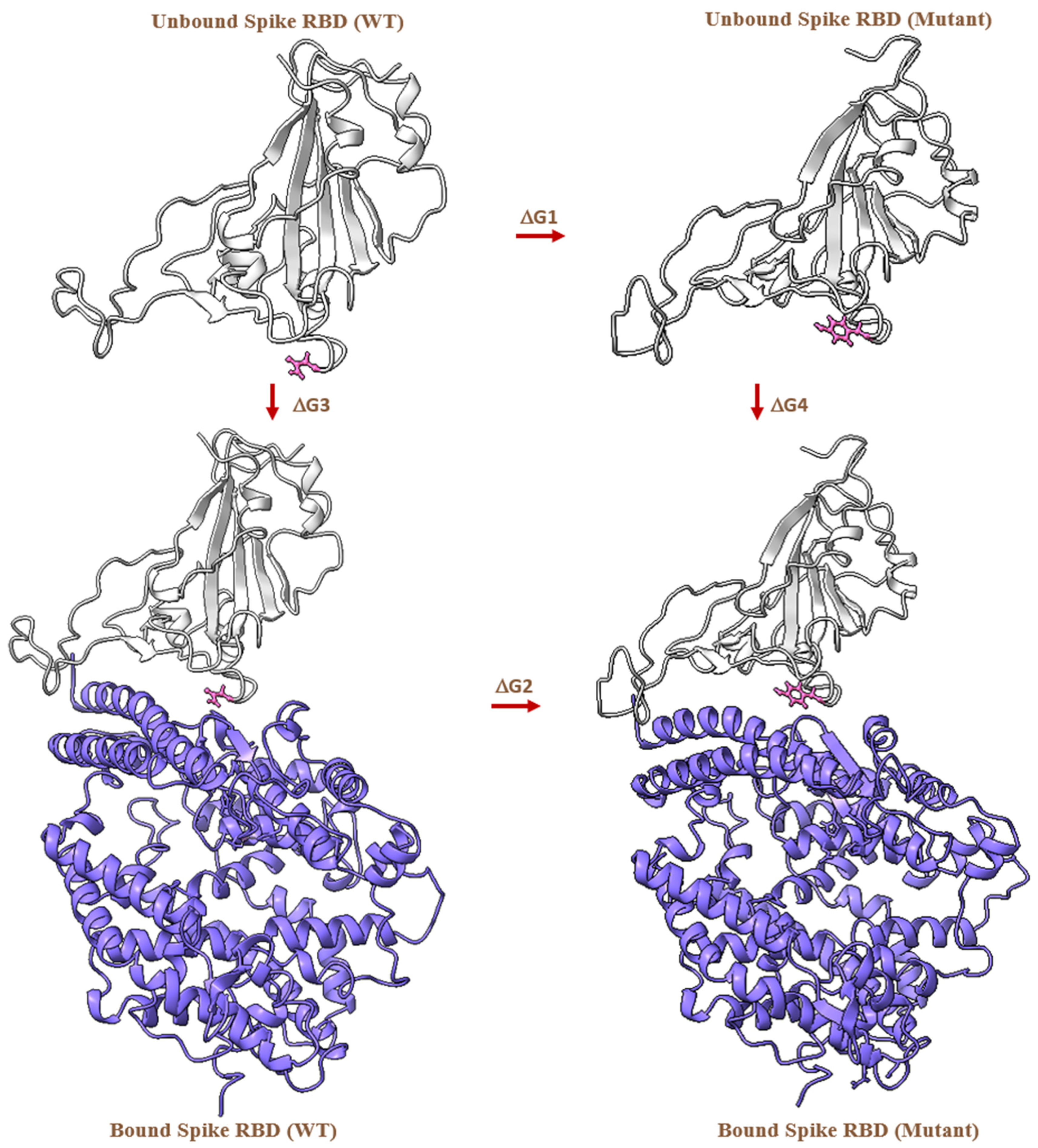
2.4. MD-Based Alchemical Free Energy Calculations
3. Conclusions
4. Materials and Methods
4.1. Protein Preparation
4.2. Conventional MD Simulations
4.3. Prime/MM-GBSA Binding Free Energy Analysis
4.4. MD-Based Alchemical Free Energy Calculations
4.5. Preparation of Hybrid Topology
4.6. Equilibrium and Non-Equilibrium MD Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Atzrodt, C.L.; Maknojia, I.; McCarthy, R.D.P.; Oldfield, T.M.; Po, J.; Ta, K.T.L.; Stepp, H.E.; Clements, T.P. A Guide to COVID-19: A Global Pandemic Caused by the Novel Coronavirus SARS-CoV-2. FEBS J. 2020, 287, 3633–3650. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of Viral Mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [PubMed]
- Villa, T.G.; Abril, A.G.; Sánchez, S.; de Miguel, T.; Sánchez-Pérez, A. Animal and Human RNA Viruses: Genetic Variability and Ability to Overcome Vaccines. Arch. Microbiol. 2021, 203, 443–464. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, W.J.; Woods, R.J.; McCrone, J.T.; Woodman, A.; Arnold, J.J.; Yennawar, M.; Evans, R.; Cameron, C.E.; Lauring, A.S. A Speed–Fidelity Trade-off Determines the Mutation Rate and Virulence of an RNA Virus. PLoS Biol. 2018, 16, e2006459. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S. Why Are RNA Virus Mutation Rates so Damn High? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef]
- Burrell, C.J.; Howard, C.R.; Murphy, F.A. Pathogenesis of Virus Infections. In Fenner and White’s Medical Virology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 77–104. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell. 2020, 79, 710–727. [Google Scholar] [CrossRef]
- Frost, S.D.W.; Magalis, B.R.; Pond, S.L.K. Neutral Theory and Rapidly Evolving Viral Pathogens. Mol. Biol. Evol. 2018, 35, 1348–1354. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 21 June 2021).
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science 2021, 372, 3055. [Google Scholar] [CrossRef]
- Collier, D.A.; De Marco, A.; Ferreira, I.A.T.M.; Meng, B.; Datir, R.; Walls, A.C.; Kemp, S.S.A.; Bassi, J.; Pinto, D.; Fregni, C.S.; et al. SARS-CoV-2 B.1.1.7 Sensitivity to MRNA Vaccine-Elicited, Convalescent and Monoclonal Antibodies. medRxiv 2021. [Google Scholar] [CrossRef]
- Mallapaty, S. Where Did Omicron Come from? Three Key Theories. Nature 2022, 602, 26–28. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2022, 7, veab064. [Google Scholar] [CrossRef]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive Mapping of Mutations in the SARS-CoV-2 Receptor-Binding Domain That Affect Recognition by Polyclonal Human Plasma Antibodies. Cell Host Microbe 2021, 29, 463–476.e6. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain That Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef]
- Planas, D.; Bruel, T.; Grzelak, L.; Guivel-Benhassine, F.; Staropoli, I.; Porrot, F.; Planchais, C.; Buchrieser, J.; Rajah, M.M.; Bishop, E.; et al. Sensitivity of Infectious SARS-CoV-2 B.1.1.7 and B.1.351 Variants to Neutralizing Antibodies. Nat. Med. 2021, 27, 917–924. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef]
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Evidence of Escape of SARS-CoV-2 Variant B.1.351 from Natural and Vaccine-Induced Sera. Cell 2021, 184, 2348–2361.e6. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, Infectivity, and Neutralization of a Spike L452R SARS-CoV-2 Variant. Cell 2021, 184, 3426–3437.e8. [Google Scholar] [CrossRef]
- Federation Drug American (FDA). Fact Sheet for Health Care Providers Emergency Use Authorization of Bamlanivimab and Etesevimab; FDA: Silver Spring, MD, USA, 2020. [Google Scholar]
- SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html#Interest (accessed on 26 June 2021).
- Mohammad, T.; Choudhury, A.; Habib, I.; Asrani, P.; Mathur, Y.; Umair, M.; Anjum, F.; Shafie, A.; Yadav, D.K.; Hassan, M.I. Genomic Variations in the Structural Proteins of SARS-CoV-2 and Their Deleterious Impact on Pathogenesis: A Comparative Genomics Approach. Front. Cell. Infect. Microbiol. 2021, 11, 951. [Google Scholar] [CrossRef]
- Jangra, S.; Ye, C.; Rathnasinghe, R.; Stadlbauer, D.; Alshammary, H.; Amoako, A.A.; Awawda, M.H.; Beach, K.F.; Bermúdez-González, M.C.; Chernet, R.L.; et al. SARS-CoV-2 Spike E484K Mutation Reduces Antibody Neutralisation. Lancet Microbe 2021, 2, e283–e284. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Lam, E.C.; St. Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 Variants Escape Neutralization by Vaccine-Induced Humoral Immunity. Cell 2021, 184, 2372–2383.e9. [Google Scholar] [CrossRef] [PubMed]
- Estimates of Severity and Transmissibility of Novel SARS-CoV-2 Variant 501Y.V2 in South Africa. CMMID Repository. Available online: https://cmmid.github.io/topics/covid19/sa-novel-variant.html (accessed on 21 June 2021).
- How Dangerous Is the Delta Variant (B.1.617.2)? Available online: https://asm.org/Articles/2021/July/How-Dangerous-is-the-Delta-Variant-B-1-617-2 (accessed on 12 August 2021).
- Issa, E.; Merhi, G.; Panossian, B.; Salloum, T.; Tokajian, S. SARS-CoV-2 and ORF3a: Nonsynonymous Mutations, Functional Domains, and Viral Pathogenesis. mSystems 2020, 5, e00266-20. [Google Scholar] [CrossRef] [PubMed]
- Moghaddar, M.; Radman, R.; Macreadie, I. Severity, Pathogenicity and Transmissibility of Delta and Lambda Variants of SARS-CoV-2, Toxicity of Spike Protein and Possibilities for Future Prevention of COVID-19. Microorganisms 2021, 9, 2167. [Google Scholar] [CrossRef] [PubMed]
- Saville, J.W.; Mannar, D.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Demers, J.-P.; Zhou, S.; Tuttle, K.S.; Sekirov, I.; Kim, A.; et al. Structural and Biochemical Rationale for Enhanced Spike Protein Fitness in Delta and Kappa SARS-CoV-2 Variants. Nat. Commun. 2022, 13, 1–10. [Google Scholar] [CrossRef]
- Uriu, K.; Kimura, I.; Shirakawa, K.; Takaori-Kondo, A.; Nakada, T.; Kaneda, A.; Nakagawa, S.; Sato, K. Neutralization of the SARS-CoV-2 Mu Variant by Convalescent and Vaccine Serum. N. Engl. J. Med. 2021, 385, 2397–2399. [Google Scholar] [CrossRef]
- Kwon, D. Omicron’s Molecular Structure Could Help Explain Its Global Takeover. Nature 2022, 602, 373–374. [Google Scholar] [CrossRef]
- Lu, L.; Chu, A.W.H.; Zhang, R.R.; Chan, W.M.; Ip, J.D.; Tsoi, H.W.; Chen, L.-l.; Cai, J.P.; Lung, D.C.; Tam, A.R.; et al. The Impact of Spike N501Y Mutation on Neutralizing Activity and RBD Binding of SARS-CoV-2 Convalescent Serum. EBioMedicine 2021, 71, 103544. [Google Scholar] [CrossRef]
- Bate, N.; Savva, C.G.; Moody, P.C.E.; Brown, E.A.; Evans, S.E.; Ball, J.K.; Schwabe, J.W.R.; Sale, J.E.; Brindle, N.P.J. In Vitro Evolution Predicts Emerging SARS-CoV-2 Mutations with High Affinity for ACE2 and Cross-Species Binding. PLoS Pathog. 2022, 18, e1010733. [Google Scholar] [CrossRef]
- Rodriguez-Rivas, J.; Croce, G.; Muscat, M.; Weigt, M. Epistatic Models Predict Mutable Sites in SARS-CoV-2 Proteins and Epitopes. Proc. Natl. Acad. Sci. USA 2022, 119, e2113118119. [Google Scholar] [CrossRef]
- Hossen, M.L.; Baral, P.; Sharma, T.; Gerstman, B.; Chapagain, P. Significance of the RBD Mutations in the SARS-CoV-2 Omicron: From Spike Opening to Antibody Escape and Cell Attachment. Phys. Chem. Chem. Phys. 2022, 24, 9123–9129. [Google Scholar] [CrossRef]
- Ou, J.; Zhou, Z.; Dai, R.; Zhang, J.; Zhao, S.; Wu, X.; Lan, W.; Ren, Y.; Cui, L.; Lan, Q.; et al. V367F Mutation in SARS-CoV-2 Spike RBD Emerging during the Early Transmission Phase Enhances Viral Infectivity through Increased Human ACE2 Receptor Binding Affinity. J. Virol. 2021, 95, 617–638. [Google Scholar] [CrossRef]
- Li, T.; Cui, Z.; Jia, Y.; Liang, Z.; Nie, J.; Zhang, L.; Wang, M.; Li, Q.; Wu, J.; Xu, N.; et al. Aggregation of High-Frequency RBD Mutations of SARS-CoV-2 with Three VOCs Did Not Cause Significant Antigenic Drift. J. Med. Virol. 2022, 94, 2108–2125. [Google Scholar] [CrossRef]
- Barton, M.I.; Macgowan, S.; Kutuzov, M.; Dushek, O.; Barton, G.J.; Anton Van Der Merwe, P. Effects of Common Mutations in the Sars-Cov-2 Spike Rbd and Its Ligand the Human Ace2 Receptor on Binding Affinity and Kinetics. eLife 2021, 10, e70658. [Google Scholar] [CrossRef]
- Lupala, C.S.; Ye, Y.; Chen, H.; Su, X.D.; Liu, H. Mutations on RBD of SARS-CoV-2 Omicron Variant Result in Stronger Binding to Human ACE2 Receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34–41. [Google Scholar] [CrossRef]
- da Costa, C.H.S.; de Freitas, C.A.B.; Alves, C.N.; Lameira, J. Assessment of Mutations on RBD in the Spike Protein of SARS-CoV-2 Alpha, Delta and Omicron Variants. Sci. Rep. 2022, 12, 8540. [Google Scholar] [CrossRef]
- Othman, H.; Bouslama, Z.; Brandenburg, J.T.; da Rocha, J.; Hamdi, Y.; Ghedira, K.; Srairi-Abid, N.; Hazelhurst, S. Interaction of the Spike Protein RBD from SARS-CoV-2 with ACE2: Similarity with SARS-CoV, Hot-Spot Analysis and Effect of the Receptor Polymorphism. Biochem. Biophys. Res. Commun. 2020, 527, 702–708. [Google Scholar] [CrossRef]
- Blanco, J.D.; Hernandez-Alias, X.; Cianferoni, D.; Serrano, L. In Silico Mutagenesis of Human ACE2 with S Protein and Translational Efficiency Explain SARS-CoV-2 Infectivity in Different Species. PLoS Comput. Biol. 2020, 16, e1008450. [Google Scholar] [CrossRef]
- Verma, S.; Patil, V.M.; Gupta, M.K. Mutation Informatics: SARS-CoV-2 Receptor-Binding Domain of the Spike Protein. Drug Discov. Today 2022, 27, 103312. [Google Scholar] [CrossRef]
- Mehra, R.; Kepp, K.P. Structural Heterogeneity and Precision of Implications Drawn from Cryo-Electron Microscopy Structures: SARS-CoV-2 Spike-Protein Mutations as a Test Case. Eur. Biophys. J. 2022, 1, 1–14. [Google Scholar] [CrossRef]
- Ray, D.; Le, L.; Andricioaei, I. Distant Residues Modulate Conformational Opening in SARS-CoV-2 Spike Protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2100943118. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.; Sobitan, A.; Rhoades, R.; Liu, D.; Tang, Q. Systemic Effects of Missense Mutations on SARS-CoV-2 Spike Glycoprotein Stability and Receptor-Binding Affinity. Brief Bioinform. 2021, 22, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- Sobitan, A.; Mahase, V.; Rhoades, R.; Williams, D.; Liu, D.; Xie, Y.; Li, L.; Tang, Q.; Teng, S. Computational Saturation Mutagenesis of SARS-CoV-1 Spike Glycoprotein: Stability, Binding Affinity, and Comparison With SARS-CoV-2. Front. Mol. Biosci. 2021, 8, 1212. [Google Scholar] [CrossRef] [PubMed]
- Celik, I.; Khan, A.; Dwivany, F.M.; Fatimawali; Wei, D.Q.; Tallei, T.E. Computational Prediction of the Effect of Mutations in the Receptor-Binding Domain on the Interaction between SARS-CoV-2 and Human ACE2. Mol. Divers. 2022, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.A.; Jha, R.K. Identifying Key Determinants and Dynamics of SARS-CoV-2/ACE2 Tight Interaction. PLoS ONE 2021, 16, e0257905. [Google Scholar] [CrossRef]
- Siebenmorgen, T.; Zacharias, M. Computational Prediction of Protein–Protein Binding Affinities. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1448. [Google Scholar] [CrossRef]
- Fratev, F. R346K Mutation in the Mu Variant of SARS-CoV-2 Alters the Interactions with Monoclonal Antibodies from Class 2: A Free Energy Perturbation Study. J. Chem. Inf. Model. 2022, 62, 627–631. [Google Scholar] [CrossRef]
- Zou, J.; Yin, J.; Fang, L.; Yang, M.; Wang, T.; Wu, W.; Bellucci, M.A.; Zhang, P. Computational Prediction of Mutational Effects on SARS-CoV-2 Binding by Relative Free Energy Calculations. J. Chem. Inf. Model. 2020, 60, 5794–5802. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Gao, J. Enhanced Receptor Binding of SARS-CoV-2 through Networks of Hydrogen-Bonding and Hydrophobic Interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef]
- Zhu, F.; Bourguet, F.A.; Bennett, W.F.D.; Lau, E.Y.; Arrildt, K.T.; Segelke, B.W.; Zemla, A.T.; Desautels, T.A.; Faissol, D.M. Large-Scale Application of Free Energy Perturbation Calculations for Antibody Design. Sci. Rep. 2022, 12, 12489. [Google Scholar] [CrossRef]
- Hou, X.; Zhang, Z.; Gao, J.; Wang, Y. SARS-CoV-2 Spike Protein N501Y Mutation Causes Differential Species Transmissibility and Antibody Sensitivity: A Molecular Dynamics and Alchemical Free Energy Study. Mol. Syst. Des. Eng. 2021, 6, 964–974. [Google Scholar] [CrossRef]
- Simões, I.C.M.; Costa, I.P.D.; Coimbra, J.T.S.; Ramos, M.J.; Fernandes, P.A. New Parameters for Higher Accuracy in the Computation of Binding Free Energy Differences upon Alanine Scanning Mutagenesis on Protein-Protein Interfaces. J. Chem. Inf. Model. 2017, 57, 60–72. [Google Scholar] [CrossRef]
- Zwanzig, R.W. High-Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases. J. Chem. Phys. 2004, 22, 1420. [Google Scholar] [CrossRef]
- Straatsma, T.P.; Berendsen, H.J.C. Free Energy of Ionic Hydration: Analysis of a Thermodynamic Integration Technique to Evaluate Free Energy Differences by Molecular Dynamics Simulations. J. Chem. Phys. 1998, 89, 5876. [Google Scholar] [CrossRef]
- Bennett, C.H. Efficient Estimation of Free Energy Differences from Monte Carlo Data. J. Comput. Phys. 1976, 22, 245–268. [Google Scholar] [CrossRef]
- Goette, M.; Grubmüller, H. Accuracy and Convergence of Free Energy Differences Calculated from Nonequilibrium Switching Processes. J. Comput. Chem. 2009, 30, 447–456. [Google Scholar] [CrossRef]
- Lee, T.S.; Allen, B.K.; Giese, T.J.; Guo, Z.; Li, P.; Lin, C.; Dwight McGee, T.; Pearlman, D.A.; Radak, B.K.; Tao, Y.; et al. Alchemical Binding Free Energy Calculations in AMBER20: Advances and Best Practices for Drug Discovery. J. Chem. Inf. Model. 2020, 60, 5595–5623. [Google Scholar] [CrossRef]
- Gapsys, V.; Michielssens, S.; Seeliger, D.; de Groot, B.L. Accurate and Rigorous Prediction of the Changes in Protein Free Energies in a Large-Scale Mutation Scan. Angew. Chem. Int. Ed. 2016, 55, 7364–7368. [Google Scholar] [CrossRef]
- Clark, A.J.; Negron, C.; Hauser, K.; Sun, M.; Wang, L.; Abel, R.; Friesner, R.A. Relative Binding Affinity Prediction of Charge-Changing Sequence Mutations with FEP in Protein–Protein Interfaces. J. Mol. Biol. 2019, 431, 1481. [Google Scholar] [CrossRef]
- Haycroft, E.R.; Davis, S.K.; Ramanathan, P.; Lopez, E.; Purcell, R.A.; Lynn Tan, L.; Pymm, P.; Wines, B.D.; Mark Hogarth, P.; Wheatley, K.; et al. Altered Affinity to ACE2 and Reduced Fc Functional Antibodies to SARS-CoV-2 RBD Variants. medRxiv 2022, 7364. [Google Scholar] [CrossRef]
- Nutalai, R.; Zhou, D.; Tuekprakhon, A.; Ginn, H.M.; Supasa, P.; Liu, C.; Huo, J.; Mentzer, A.J.; Duyvesteyn, H.M.E.; Dijokaite-Guraliuc, A.; et al. Potent Cross-Reactive Antibodies Following Omicron Breakthrough in Vaccinees. Cell 2022, 185, 2116–2131.e18. [Google Scholar] [CrossRef] [PubMed]
- Rössler, A.; Knabl, L.; von Laer, D.; Kimpel, J. Neutralization Profile after Recovery from SARS-CoV-2 Omicron Infection. N. Engl. J. Med. 2022, 386, 1764–1766. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Huo, J.; Zhou, D.; Zahradník, J.; Supasa, P.; Liu, C.; Duyvesteyn, H.M.E.; Ginn, H.M.; Mentzer, A.J.; Tuekprakhon, A.; et al. SARS-CoV-2 Omicron-B.1.1.529 Leads to Widespread Escape from Neutralizing Antibody Responses. Cell 2022, 185, 467. [Google Scholar] [CrossRef] [PubMed]
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliuc, A.; Ren, J.; Stuart, D.I.; Screaton Correspondence, G.R. Antibody Escape of SARS-CoV-2 Omicron BA.4 and BA.5 from Vaccine and BA.1 Serum. Cell 2022, 185, 2422–2433.e13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody Evasion by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4 and BA.5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef]
- Lyngse, F.P.; Kirkeby, C.T.; Denwood, M.; Christiansen, L.E.; Mølbak, K.; Møller, C.H.; Skov, R.L.; Krause, T.G.; Rasmussen, M.; Sieber, R.N.; et al. Household transmission of SARS-CoV-2 Omicron variant of concern subvariants BA.1 and BA.2 in Denmark. Nat. Commun. 2022, 13, 5760. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 Spike L452R Variant Evades Cellular Immunity and Increases Infectivity. Cell Host Microbe 2021, 29, 1124–1136.e11. [Google Scholar] [CrossRef]
- Laffeber, C.; de Koning, K.; Kanaar, R.; Lebbink, J.H.G. Experimental Evidence for Enhanced Receptor Binding by Rapidly Spreading SARS-CoV-2 Variants. J. Mol. Biol. 2021, 433, 167058. [Google Scholar] [CrossRef]
- Bai, C.; Wang, J.; Chen, G.; Zhang, H.; An, K.; Xu, P.; Du, Y.; Ye, R.D.; Saha, A.; Zhang, A.; et al. Predicting Mutational Effects on Receptor Binding of the Spike Protein of SARS-CoV-2 Variants. J. Am. Chem. Soc. 2021, 143, 17646–17654. [Google Scholar] [CrossRef]
- Upadhyay, V.; Lucas, A.; Panja, S.; Miyauchi, R.; Mallela, K.M.G. Receptor Binding, Immune Escape, and Protein Stability Direct the Natural Selection of SARS-CoV-2 Variants. J. Biol. Chem. 2021, 297, 101208. [Google Scholar] [CrossRef]
- Supasa, P.; Zhou, D.; Dejnirattisai, W.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Nutalai, R.; Tuekprakhon, A.; et al. Reduced Neutralization of SARS-CoV-2 B.1.1.7 Variant by Convalescent and Vaccine Sera. Cell 2021, 184, 2201–2211.e7. [Google Scholar] [CrossRef]
- Williams, A.H.; Zhan, C.G. Fast Prediction of Binding Affinities of the Sars-Cov-2 Spike Protein Mutant N501y (UK Variant) with Ace2 and Miniprotein Drug Candidates. J. Phys. Chem. B 2021, 125, 4330–4336. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Beard, H.; Cholleti, A.; Pearlman, D.; Sherman, W.; Loving, K.A. Applying Physics-Based Scoring to Calculate Free Energies of Binding for Single Amino Acid Mutations in Protein-Protein Complexes. PLoS ONE 2013, 8, e82849. [Google Scholar] [CrossRef]
- Chen, F.; Liu, H.; Sun, H.; Pan, P.; Li, Y.; Li, D.; Hou, T. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 6. Capability to Predict Protein–Protein Binding Free Energies and Re-Rank Binding Poses Generated by Protein–Protein Docking. Phys. Chem. Chem. Phys. 2016, 18, 22129–22139. [Google Scholar] [CrossRef]
- Loo, J.S.E.; Yong, A.Y.Y.; Yong, Y.N. The Effect of Multiple Simulation Parameters on MM/PBSA Performance for Binding Affinity Prediction of CB1 Cannabinoid Receptor Agonists and Antagonists. Chem. Biol. Drug Des. 2020, 96, 1244–1254. [Google Scholar] [CrossRef]
- Bhati, A.P.; Wan, S.; Hu, Y.; Sherborne, B.; Coveney, P.V. Uncertainty Quantification in Alchemical Free Energy Methods. J. Chem. Theory Comput. 2018, 14, 2867–2880. [Google Scholar] [CrossRef]
- Kuhn, B.; Gerber, P.; Schulz-Gasch, T.; Stahl, M. Validation and Use of the MM-PBSA Approach for Drug Discovery. J. Med. Chem. 2005, 48, 4040–4048. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Aldeghi, M.; de Groot, B.L.; Gapsys, V. Accurate Calculation of Free Energy Changes upon Amino Acid Mutation. In Methods in Molecular Biology; Sikosek, T., Ed.; Humana Press: New York, NY, USA, 2019; Volume 1851, pp. 19–47. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Bnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, A.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef] [PubMed]
- Gundelach, L.; Fox, T.; Tautermann, C.S.; Skylaris, C.K. Protein–Ligand Free Energies of Binding from Full-Protein DFT Calculations: Convergence and Choice of Exchange–Correlation Functional. Phys. Chem. Chem. Phys. 2021, 23, 9381–9393. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, SC’06, Tampa, FL, USA, 11 November 2006; ACM Press: New York, NY, USA, 2006; p. 84. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces; Pullman, B., Ed.; Springer: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar] [CrossRef]
- Nosé, S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Nosé, S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant Pressure Molecular Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Predescu, C.; Lerer, A.K.; Lippert, R.A.; Towles, B.; Grossman, J.P.; Dirks, R.M.; Shaw, D.E. The u -Series: A Separable Decomposition for Electrostatics Computation with Improved Accuracy. J. Chem. Phys. 2020, 152, 084113. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Genet. 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-Chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 Model: A next Generation Energy Model for High Resolution Protein Structure Modeling. Proteins Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Mey, A.S.J.S.; Allen, B.K.; Bruce Macdonald, H.E.; Chodera, J.D.; Hahn, D.F.; Kuhn, M.; Michel, J.; Mobley, D.L.; Naden, L.N.; Prasad, S.; et al. Best Practices for Alchemical Free Energy Calculations [Article v1.0]. Living J. Comput. Mol. Sci. 2020, 2, 18378. [Google Scholar] [CrossRef]
- Gapsys, V.; Michielssens, S.; Seeliger, D.; de Groot, B.L. Insights from the First Principles Based Large Scale Protein Thermostability Calculations. Biophys. J. 2016, 110, 368a. [Google Scholar] [CrossRef]
- Seeliger, D.; de Groot, B.L. Protein Thermostability Calculations Using Alchemical Free Energy Simulations. Biophys. J. 2010, 98, 2309–2316. [Google Scholar] [CrossRef]
- Gapsys, V.; De Groot, B.L. Pmx Webserver: A User Friendly Interface for Alchemistry. J. Chem. Inf. Model. 2017, 57, 109–114. [Google Scholar] [CrossRef]
- Gapsys, V.; Michielssens, S.; Seeliger, D.; De Groot, B.L. Pmx: Automated Protein Structure and Topology Generation for Alchemical Perturbations. J. Comput. Chem. 2015, 36, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins Struct. Funct. Genet. 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Puhti—Puhti—Services for Research—CSC Company Site. Available online: https://research.csc.fi/-/puhti (accessed on 26 June 2021).
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A Package for Molecular Simulation and Trajectory Analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GRGMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Solving Software Challenges for Exascale: Proceedings of the International Conference on Exascale Applications and Software, EASC 2014, Stockholm, Sweden, 2–3 April 2014; Springer: Cham, Switzerland, 2015; Volume 8759, pp. 3–27. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef]
- Petrova, S.S.; Solov’Ev, A.D. The Origin of the Method of Steepest Descent. Hist. Math. 1997, 24, 361–375. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1998, 81, 3684. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Berendsen, H.J.C. A Leap-Frog Algorithm for Stochastic Dynamics. Mol. Simul. 1988, 1, 173–185. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations; John Wiley & Sons: Hoboken, NJ, USA, 1997; Volume 18. [Google Scholar]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Identified in Variants | Increases hACE2 Binding | Evading Antibodies | Experimental Binding Affinity of Spike Glycoprotein to hACE2 (KD in nM) |
|---|---|---|---|---|
| G446S | O (BA.1) | No [66,67] | Yes [68] | 46.9 [66] |
| F486V | O (BA.4, BA.5) | No [69,70,71] | Yes [72] | - |
| G496S | O (BA.1) | No [69] | Yes [68] | - |
| N501Y | β, α, γ, µ, θ, O | Yes [73,74] | Yes (slightly) [75] | 2.4 ± 0.4 [74] 3.0 ± 2.1 [76] 5.5 ± 2.4 [40] 10.7 [77] 0.4 [78] |
| Y505H | O | No [69,70,71] | Yes [68] | - |
| Spike RBD Mutant | Bound ΔG (kJ/mol) | Unbound ΔG (kJ/mol) | ΔΔG (kJ/mol) | ΔΔG (kcal/mol) |
|---|---|---|---|---|
| G446S | 1.80± 0.28 | 2.97 ± 0.09 | −1.17 ± 0.29 | −0.28 ± 0.07 |
| F486V | −95.80 ± 0.27 | −99.99 ± 0.18 | 4.19 ± 0.32 | 1.00 ± 0.08 |
| G496S | 24.99 ± 1.65 | 12.57 ± 0.70 | 12.42 ± 1.79 | 2.97 ± 0.43 |
| N501Y | 290.22 ± 0.84 | 304.24 ± 0.46 | −14.02 ± 0.96 | −3.35 ± 0.23 |
| Y505Hɛ | −2.57 ± 0.44 | −6.05 ± 0.20 | 3.48 ± 0.48 | 0.83 ± 0.12 |
| Y505Hδ | 2.87 ± 0.41 | 0.62 ± 0.14 | 2.25 ± 0.43 | 0.54 ± 0.10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhadane, R.; Salo-Ahen, O.M.H. High-Throughput Molecular Dynamics-Based Alchemical Free Energy Calculations for Predicting the Binding Free Energy Change Associated with the Selected Omicron Mutations in the Spike Receptor-Binding Domain of SARS-CoV-2. Biomedicines 2022, 10, 2779. https://doi.org/10.3390/biomedicines10112779
Bhadane R, Salo-Ahen OMH. High-Throughput Molecular Dynamics-Based Alchemical Free Energy Calculations for Predicting the Binding Free Energy Change Associated with the Selected Omicron Mutations in the Spike Receptor-Binding Domain of SARS-CoV-2. Biomedicines. 2022; 10(11):2779. https://doi.org/10.3390/biomedicines10112779
Chicago/Turabian StyleBhadane, Rajendra, and Outi M. H. Salo-Ahen. 2022. "High-Throughput Molecular Dynamics-Based Alchemical Free Energy Calculations for Predicting the Binding Free Energy Change Associated with the Selected Omicron Mutations in the Spike Receptor-Binding Domain of SARS-CoV-2" Biomedicines 10, no. 11: 2779. https://doi.org/10.3390/biomedicines10112779
APA StyleBhadane, R., & Salo-Ahen, O. M. H. (2022). High-Throughput Molecular Dynamics-Based Alchemical Free Energy Calculations for Predicting the Binding Free Energy Change Associated with the Selected Omicron Mutations in the Spike Receptor-Binding Domain of SARS-CoV-2. Biomedicines, 10(11), 2779. https://doi.org/10.3390/biomedicines10112779