
Diversities in the Gut Microbial Patterns in Patients with Atherosclerotic Cardiovascular Diseases and Certain Heart Failure Phenotypes
 , , , , ,
, , , , ,  , , , , and add
Show full author list
, , , , and add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. Cohort Description
2.2. Blood and Stool Sample Collection and Preparation
2.3. Microbial Identification by MALDI-TOF-MS
2.4. DNA Extraction and Preparation of DNA Libraries and Sequencing
2.5. Metagenome Data Processing and Analysis
2.6. Real-Time qPCR Quantification of Bacterial DNA
2.6.1. Study Group
2.6.2. PCR Reaction
2.6.3. Bacterial DNA Quantification
2.7. Statistical Analysis
2.7.1. Software
2.7.2. Data Conversion
- (1)
- Raw clinical data contained sample ids, morphometric and demographic indicators, cardiovascular system state, and hematological and biochemical parameters.The table included binary and quantitative values which were transformed in accordance with the following formulas (Formulas (2)–(3)):where —initial value of the quantitative indicator presented at the i-th row, in the j-th column, —median value of j-th column, —interquartile range of the j-th column, —converted value.Taking into account the influence of outliers presented in the data often leads to the misleading interpretations and wrong conclusions. This explanation holds true for all further conversions during multivariate analysis.
- (2)
- MALDI-TOF mass spectra profiles of every sample were transformed in accordance with the following rule:
- -
- all zero elements were retained,
- -
- nonzero elements were converted by formula (Formula (4)):where —original non-zero value located in the i-th row, in the j-th column,—converted nonzero value.
- (3)
- PCR tables included sample ids columns and corresponding relative DNA-concentration values, which also were processed in accordance with the following rule (Formula (5)):where —the initial value of the quantitative indicator, located on the i-th line, in the j-th column, —median value of the j-th column, —interquartile range of the j-th column, tanh—hyperbolic tangent, —converted value.
- (4)
- We also provided custom normalization for 16S rRNA NGS data. During the bioinformatics read processing, we obtained a table which included sample ids and counts for every taxonomical string. Despite the validity of the taxonomical ratio provided by rarefaction curves, low abundant family groups (Brachyspiraceae, Burkholderiaceae, Dermatophilaceae, etc.) commonly reveal abundancy sampling biases. In order to minimize sampling effects, we ranged low abundant taxonomy groups in accordance with the following rule. All values of ASV-frequency table below 31 were changed to 0, and those from 31 to 99 were replaced by 75. Furthermore, values from 100 to 149 were changed to 125. Counts outside the specified ranges remained unchanged.
2.7.3. Data Filtering and Exploratory Analysis
- Control group and ASCVD
- Control group and HFpEF
- Control group and HFrEF
2.7.4. Statistical Analysis
2.7.5. Statistical Analysis of Clinical Diagnostic Data
3. Results
3.1. Bacterial Culturation Study
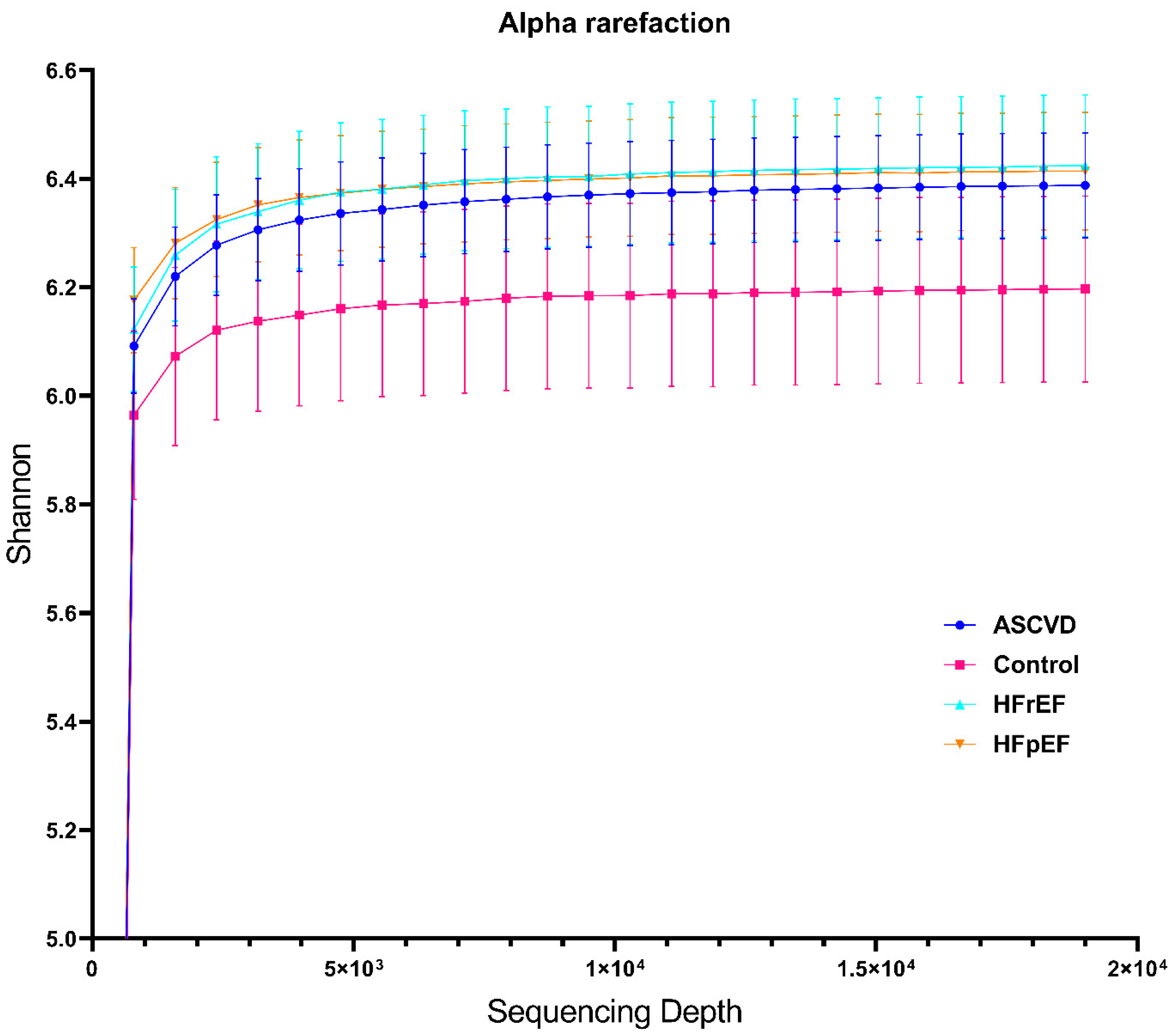
3.2. Metagenome Data
3.3. Real-Time qPCR Analysis
4. Discussion
4.1. Comparison of qPCR Data between Extraction Methods and with the NGS Data
4.2. Analysis of NGS Data and Trends in Prior International Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kassebaum, N.J.; Arora, M.; Barber, R.M.; Bhutta, Z.A.; Brown, J.; Carter, A.; Casey, D.C.; Charlson, F.J.; Coates, M.M.; Coggeshall, M.; et al. Global, regional, and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1603–1658. [Google Scholar] [CrossRef]
- Wang, H.; Naghavi, M.; Allen, C.; Barber, R.M.; Bhutta, Z.A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; Coates, M.M.; et al. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef]
- Vos, T.; Lim, S.; Abbafati, C.; Abbas, K.; Abbasi, M.; Abbasifard, M. GBD 2019 Diseases and Injuries Collaborators.Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Khatana, C.; Saini, N.K.; Chakrabarti, S.; Saini, V.; Sharma, A.; Saini, R.V.; Saini, A.K. Mechanistic Insights into the Oxidized Low-Density Lipoprotein-Induced Atherosclerosis. Oxid. Med. Cell. Longev. 2020, 2020, 5245308. [Google Scholar] [CrossRef]
- Mathers, C.D.; Loncar, D. Projections of Global Mortality and Burden of Disease from 2002 to 2030. PLOS Med. 2006, 3, e442. [Google Scholar] [CrossRef]
- Cowie, M.; Wood, D.; Coats, A.S.; Thompson, S.; Poole-Wilson, P.; Suresh, V.; Sutton, G. Incidence and aetiology of heart failure; a population-based study. Eur. Heart J. 1999, 20, 421–428. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- He, J.; Ogden, L.G.; Bazzano, L.A.; Vupputuri, S.; Loria, C.; Whelton, P.K. Risk factors for congestive heart failure in US men and women: NHANES I epidemiologic follow-up study. Arch. Intern. Med. 2001, 161, 996–1002. [Google Scholar] [CrossRef]
- Borlaug, B.A.; Melenovsky, V.; Russell, S.D.; Kessler, K.; Pacak, K.; Becker, L.C.; Kass, D.A. Impaired Chronotropic and Vasodilator Reserves Limit Exercise Capacity in Patients with Heart Failure and a Preserved Ejection Fraction. Circulation 2006, 114, 2138–2147. [Google Scholar] [CrossRef]
- Jiang, S.; Shui, Y.; Cui, Y.; Tang, C.; Wang, X.; Qiu, X.; Hu, W.; Fei, L.; Li, Y.; Zhang, S.; et al. Gut microbiota dependent trimethylamine N-oxide aggravates angiotensin II–induced hypertension. Redox Biol. 2021, 46, 102115. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, G.; Jin, X.; Fan, T.; Chen, Z.; Sheng, X. Influence of Gut Microbiota and Trimethylamine N-Oxide in Patients with Coronary Heart Disease. Int. Heart J. 2022, 63, 683–691. [Google Scholar] [CrossRef]
- Madan, S.; Mehra, M.R. Gut dysbiosis and heart failure: Navigating the universe within. Eur. J. Heart Fail. 2020, 22, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Deluca, J.; Allred, K.F.; Menon, R.; Riordan, R.; Weeks, B.; Jayaraman, A.; Allred, C.D. Bisphenol-A alters microbiota metabolites derived from aromatic amino acids and worsens disease activity during colitis. Exp. Biol. Med. 2018, 243, 864–875. [Google Scholar] [CrossRef]
- Quagliariello, V.; Coppola, C.; Mita, D.; Piscopo, G.; Iaffaioli, R.; Botti, G.; Maurea, N. Low doses of Bisphenol A have pro-inflammatory and pro-oxidant effects, stimulate lipid peroxidation and increase the cardiotoxicity of Doxorubicin in cardiomyoblasts. Environ. Toxicol. Pharmacol. 2019, 69, 1–8. [Google Scholar] [CrossRef]
- Spehlmann, M.E.; Rangrez, A.Y.; Dhotre, D.P.; Schmiedel, N.; Chavan, N.; Bang, C.; Müller, O.J.; Shouche, Y.S.; Franke, A.; Frank, D.; et al. Heart Failure Severity Closely Correlates with Intestinal Dysbiosis and Subsequent Metabolomic Alterations. Biomedicines 2022, 10, 809. [Google Scholar] [CrossRef]
- Huang, Z.; Mei, X.; Jiang, Y.; Chen, T.; Zhou, Y. Gut Microbiota in Heart Failure Patients with Preserved Ejection Fraction (GUMPTION Study). Front. Cardiovasc. Med. 2022, 8, 803744. [Google Scholar] [CrossRef] [PubMed]
- Beale, A.L.; O’Donnell, J.A.; Nakai, M.E.; Nanayakkara, S.; Vizi, D.; Carter, K.; Dean, E.; Ribeiro, R.V.; Yiallourou, S.; Carrington, M.J.; et al. The Gut Microbiome of Heart Failure with Preserved Ejection Fraction. J. Am. Heart. Assoc. 2021, 10, e020654. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.S.; Jordan, C.O.; Lloyd-Jones, D.; Blumenthal, R.S. Screening for Cardiovascular Risk in Asymptomatic Patients. J. Am. Coll. Cardiol. 2010, 55, 1169–1177. [Google Scholar] [CrossRef]
- Cooney, M.T.; Dudina, A.L.; Graham, I.M. Value and Limitations of Existing Scores for the Assessment of Cardiovascular Risk: A Review for Clinicians. J. Am. Coll. Cardiol. 2009, 54, 1209–1227. [Google Scholar] [CrossRef]
- Tunstall-Pedoe, H. Cardiovascular Risk and Risk Scores: ASSIGN, Framingham, QRISK and others: How to choose. Heart 2011, 97, 442–444. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Solomon, S.D.; Claggett, B.; Lewis, E.F.; Desai, A.; Anand, I.; Sweitzer, N.K.; O′Meara, E.; Shah, S.J.; McKinlay, S.; Fleg, J.L.; et al. TOPCAT Investigators. Influence of ejection fraction on outcomes and efficacy of spironolactone in patients with heart failure with preserved ejection fraction. Eur. Heart J. 2015, 37, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A. Defining HFpEF: Where do we draw the line? Eur. Heart J. 2015, 37, 463–465. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Kazutaka, K.; Misakwa, K.; Kei-ichi, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arango, L.F.; Barrett, H.L.; McIntyre, H.D.; Callaway, L.K.; Morrison, M.; Nitert, M.D. Increased Systolic and Diastolic Blood Pressure Is Associated With Altered Gut Microbiota Composition and Butyrate Production in Early Pregnancy. Hypertension 2016, 68, 974–981. [Google Scholar] [CrossRef]
- Koren, O.; Spor, A.; Felin, J.; Fåk, F.; Stombaugh, J.; Tremaroli, V.; Behre, C.J.; Knight, R.; Fagerberg, B.; Ley, R.E.; et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA 2010, 108 (Suppl. 1), 4592–4598. [Google Scholar] [CrossRef]
- Li, H.-B.; Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. Maternal Treatment With Captopril Persistently Alters Gut-Brain Communication and Attenuates Hypertension of Male Offspring. Hypertension 2020, 75, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Luedde, M.; Winkler, T.; Heinsen, F.-A.; Rühlemann, M.C.; Spehlmann, M.E.; Bajrovic, A.; Lieb, W.; Franke, A.; Ott, S.J.; Frey, N. Heart failure is associated with depletion of core intestinal microbiota. ESC Heart Fail. 2017, 4, 282–290. [Google Scholar] [CrossRef]
- Tan, C.; Wu, Q.; Wang, H.; Gao, X.; Xu, R.; Cui, Z.; Zhu, J.; Zeng, X.; Zhou, H.; He, Y.; et al. Dysbiosis of Gut Microbiota and Short-Chain Fatty Acids in Acute Ischemic Stroke and the Subsequent Risk for Poor Functional Outcomes. J. Parenter. Enter. Nutr. 2020, 45, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Fadeeva, M.V.; Kudryavtseva, A.V.; Krasnov, G.S.; Skhirtladze, M.R.; Ivashkin, V.T. Intestinal Microbiota in Patients with Chronic Heart Failure and Systolic Dysfunction. Russ. J. Gastroenterol. Hepatol. Coloproctol. 2020, 30, 35–44. [Google Scholar] [CrossRef]
- Zhang, W.-J.; Su, W.-W.; Li, P.-B.; Rao, H.-Y.; Lin, Q.-W.; Zeng, X.; Chen, T.-B.; Yan, Z.-H.; Liu, H.; Yao, H.-L. Naoxintong Capsule Inhibits the Development of Cardiovascular Pathological Changes in Bama Minipig Through Improving Gut Microbiota. Front. Pharmacol. 2019, 10, 1128. [Google Scholar] [CrossRef]
- Jackson, M.A.; Goodrich, J.K.; Maxan, M.-E.; Freedberg, D.E.; Abrams, J.A.; Poole, A.C.; Sutter, J.L.; Welter, D.; Ley, R.E.; Bell, J.T.; et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut 2016, 65, 749–756. [Google Scholar] [CrossRef]
- Battson, M.L.; Lee, D.M.; Weir, T.L.; Gentile, C.L. The gut microbiota as a novel regulator of cardiovascular function and disease. J. Nutr. Biochem. 2018, 56, 1–15. [Google Scholar] [CrossRef]
- Byappanahalli, M.; Nevers, M.; Korajkic, A.; Staley, Z.R.; Harwood, V.J. Enterococci in the Environment. Microbiol. Mol. Biol. Rev. 2012, 76, 685–706. [Google Scholar] [CrossRef]
- Gouba, N.; Yimagou, E.; Hassani, Y.; Drancourt, M.; Fellag, M.; Fonkou, M.M. Enterococcus burkinafasonensis sp. nov. isolated from human gut microbiota. New Microbes New Infect. 2020, 36, 100702. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Song, J.; Lu, S.; Liu, Y.; Tang, L.; Wen, S. Influence of Diet on the Effect of the Probiotic Lactobacillus paracasei in Rats Suffering from Allergic Asthma. Front. Microbiol. 2021, 12, 737622. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Du, D.; Fu, T.; Han, Y.; Li, P.; Ju, H. Alterations of the Gut Microbiota in Patients with Severe Chronic Heart Failure. Front. Microbiol. 2022, 12, 813289. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yu, M.; Zhang, R.; Chen, S.; Xi, Y.; Duan, G. A meta-analysis of the association between Helicobacter pylori infection and risk of atherosclerotic cardiovascular disease. Helicobacter 2020, 25, e12761. [Google Scholar] [CrossRef] [PubMed]
- Jie, Z.; Xia, H.; Zhong, S.-L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef]
- Dahl, A.; Rasmussen, R.V.; Bundgaard, H.; Hassager, C.; Bruun, L.E.; Lauridsen, T.K.; Moser, C.; Sogaard, P.; Arpi, M.; Bruun, N.E. Enterococcus faecalis infective endocarditis: A pilot study of the relationship between duration of gentamicin treatment and outcome. Circulation 2013, 127, 1810–1817. [Google Scholar] [CrossRef]
- Knuefermann, P.; Sakata, Y.; Baker, J.S.; Huang, C.-H.; Sekiguchi, K.; Hardarson, H.S.; Takeuchi, O.; Akira, S.; Vallejo, J.G. Toll-Like Receptor 2 Mediates Staphylococcus aureus—Induced Myocardial Dysfunction and Cytokine Production in the Heart. Circulation 2004, 110, 3693–3698. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | HFpEF (n = 59) | HFrEF (n = 50) | ASCVD (n = 100) | Control Group (n = 50) |
|---|---|---|---|---|
| Age, y | 67.0 [61.0; 71.0] | 68.5 [61.0; 73.0] | 66.0 [59.0; 71.0] | 57.5 [48.0; 63.0] |
| Sex, male, n (%) | 30 (50.8%) | 46 (92%) | 66 (66.0%) | 22 (44%) |
| BMI, kg/m2 | 31.2 [27.4; 33.6] | 28.9 [25.8; 32.0] | 29.1 [27.3; 31.2] | 26.3 [22.9; 29.7] |
| NYHA functional class, n (%) | - | - | ||
| I | 42 (71.1%) | 9 (18.0%) | ||
| II | 17 (28.9%) | 36 (72.0%) | ||
| III | 0 | 5 (10.0%) | ||
| IV | 0 | 0 | ||
| Hypertensive, n (%) | 58 (98.3%) | 45 (90.0%) | 92 (92.0%) | 27 (54%) |
| Type 2-diabetes, n (%) | 0 (0%) | 38 (76.0%) | 6 (6.0%) | 0 (0%) |
| Dyslipidemia, n (%) | 34 (57.7%) | 10 (20.0%) | 80 (80.0%) | 21 (42%) |
| AF, n (%) | 29 (49.1%) | 24 (48.0%) | 24 (24.0%) | 0 (0%) |
| CAD, n (%) | 3 (5.1%) | 49 (98.0%) | 92 (92.0%) | 0 (0%) |
| Revascularization, n (%) | 2 (3.3%) | 43 (86.0%) | 84 (84.0%) | 0 (0%) |
| Catheter ablation, n (%) | 10 (16. 9%) | 0 (0%) | 5 (5.0%) | 0(0%) |
| GFR, mL/min/1.73 m2 | 78.0 [68.0; 88.0] | 71.5 [54.0; 85.0] | 79.0 [70.5; 90.5] | 86.2 [77.6; 96.5] |
| NT-proBNP, pg/mL | 178.0 [136.0; 295.0] | - | - | - |
| Echocardiography | ||||
| LVEDD, mm | 49.0 [48.0; 52.0] | 65.0 [60.0; 70.0] | 52.0 [48.0; 53.0] | 47.0 [46.0; 49.0] |
| LV EF, % | 64.0 [59.5; 67.0] | 34.0 [30.0; 40.0] | 62.0 [58.0; 67.0] | 63.0 [60.0; 66.0] |
| LV Mass Index, g/m2 | 115.0 [100.5; 126.0] | 255.5 [210.0; 320.0] | 110.0 [90.0; 124.5] | 87.5 [76.0;97.0] |
| LA Volume Index, mL/m2 | 37.0 [35.0; 43.5] | 58.0 [50; 70] | 39.7 [35.0; 43.0] | 26.0 [22.0; 31.0] |
| E, sm/s | 78.0 [67.0; 86.5] | 71.0 [59.0; 85.0] | 67.5 [52.0; 81.0] | 65.0 [61.0; 71.0] |
| A, sm/s | 78.0 [65.0; 86.5] | 63.5 [50.0; 85.0] | 75.0 [64.0; 89.0] | 54.0 [51.0; 65.0] |
| e’, sm/s | 6.0 [5.0; 6.4] | - | - | 9.1 [8.6; 11.0] |
| E/A | 1.05 [0.82; 1.24] | 0.86 [0.7; 1.59] | 0.79 [0.73; 0.98] | 1.2 [1.1; 1.35] |
| E/e’ | 13.0 [13.0; 14.0] | - | - | 6.9 [6.0; 7.2] |
| Medications | ||||
| RAAS inhibitors, n (%) | 56 (94.9%) | 45 (90.0%) | 85 (85.0%) | 25 (50.0%) |
| Beta-clockers, n (%) | 51 (86.4%) | 45 (90.0%) | 66 (66.0%) | 10 (20.0%) |
| Diuretics, n (%) | 49 (83%) | 42 (84.0%) | 28 (28.0%) | 8 (16.0%) |
| Anticoagulants, n (%) | 22 (37.3%) | 22 (44.0%) | 18 (18.0%) | 0 (0%) |
| Antiplatelet agents, n (%) | 11 (18.6%) | 32 (64.0%) | 88 (88.0%) | 7 (14.0%) |
| Statins, n (%) | 34 (57.6%) | 38 (76.0%) | 86 (86.0%) | 17 (34.0%) |
| Proton pump inhibitors, n (%) | 17 (28.8%) | 16 (32.0%) | 43 (43.0%) | 12 (24.0%) |
| Characteristics | HFpEF (n = 37) | Control Group (n = 36) | HFrEF (n = 41) | Control Group (n = 42) | ASCVD (n = 69) | Control Group (n = 43) |
|---|---|---|---|---|---|---|
| Age, y Average ± SD [min; max] | 66.0 ± 8.9 [41; 79] | 53.4 ± 12.7 [25; 76] | 70.0 ± 8.6 [41; 80] | 52.2 ± 13.8 [25; 76] | 66.5 ± 8.5 [40; 80] | 52.3 ± 13.7 [25; 76] |
| Sex, male, n (%) | 21 (56.8%) | 8 (22.2%) | 37 (90.2%) | 9 (21.4%) | 46 (66.7%) | 9 (20.9%) |
| BMI, kg/m2 Average ± SD [min; max] | 28.6 ± 3.5 [21.5; 32.8] | 25.9 ± 3.7 [20.5; 33.9] | 27.8 ± 3.3 [20.4; 33.1] | 25.0 ± 4.1 [18.3; 33.9] | 27.8 ± 2.5 [20.5; 31.8] | 24.9 ± 4.1 [18.2; 33.9] |
| Characteristics | HFpEF (n = 16) | HFrEF (n = 18) | ASCVD (n = 77) | Control Group (n = 17) |
|---|---|---|---|---|
| Age, y Average ± SD [min; max] | 65.1 ± 11.1 [41; 79] | 65.6 ± 6.6 [51; 74] | 65.7 ± 9.1 [40; 80] | 50.9 ± 13.5 [31; 76] |
| Sex, male, n (%) | 12 (75.0%) | 17 (94.4%) | 51 (66.2%) | 4 (23.5%) |
| BMI, kg/m2 Average ± SD [min; max] | 28.4 ± 4.5 [21.5; 34.6] | 27.8 ± 2.0 [24.2; 31.1] | 28.6 ± 2.9 [20.5; 33.4] | 25.6 ± 4.0 [19.8; 33.9] |
| Characteristics | HFpEF (n = 13) | HFrEF (n = 20) | ASCVD (n = 37) | Control Group (n = 28) |
|---|---|---|---|---|
| Age, years Average ± SD [min; max] | 68.0 ± 8.4 [54; 80] | 68.7 ± 6.5 [56; 80] | 66.2 ± 9.4 [40; 80] | 54.3.2 ± 9.3 [41; 74] |
| Sex, male, n (%) | 6 (46.1%) | 17 (85%) | 26 (70.3%) | 19 (67.8%) |
| BMI, kg/m2 Average ± SD [min; max] | 30.5 ± 3.5 [23.9; 34.6] | 28.9 ± 3.9 [21.8; 34.7] | 28.6 ± 3.7 [19.8; 36.3] | 28.3 ± 3.5 [21.9; 34.5] |
| Characteristics | HFpEF (n = 16) | HFrEF (n = 25) | ASCVD (n = 37) | Control Group (n = 21) |
|---|---|---|---|---|
| Age, y Average ± SD [min; max] | 68.0 ± 9.1 [49; 80] | 70.0 ± 6.6 [55; 80] | 63.7 ± 8.8 [55; 80] | 56.5 ± 8.6 [44; 74] |
| Sex, male, n (%) | 8 (50%) | 22 (88%) | 26 (70.3%) | 13 (61.9%) |
| BMI, kg/m2 Average ± SD [min; max] | 29.5 ± 3.8 [22.1; 34.6] | 28.3 ± 3.9 [20.4; 34.7] | 29.8 ± 2.7 [25.8; 34.7] | 28.2 ± 4.3 [21.1; 34.9] |
| List of Bacterial Species | Relative Abundance | Effect Size | Adjusted p-Value |
|---|---|---|---|
| 1. Atherosclerotic cardiovascular diseases | |||
| Enterococcus faecium | ↓ | 0.23 | <0.001 |
| Enterobacter cloacae | ↓ | 0.2 | <0.001 |
| Bifidobacterium bifidum | ↓ | 0.18 | <0.001 |
| Enterococcus faecalis | ↓ | 0.17 | <0.001 |
| Escherichia coli | ↓ | 0.12 | <0.01 |
| Citrobacter freundii | ↓ | 0.1 | <0.05 |
| Pediococcus acidilactici | ↓ | 0.1 | <0.05 |
| 2. Heart failure with preserved ejection fraction | |||
| Enterococcus faecium | ↓ | 0.45 | <0.001 |
| Enterococcus faecalis | ↓ | 0.19 | <0.01 |
| Bacteroides fragilis | ↓ | 0.12 | <0.05 |
| 3. Heart failure with reduced ejection fraction | |||
| Streptococcus parasanguinis | ↑ | 0.24 | <0.001 |
| Streptococcus sanguinus | ↑ | 0.21 | <0.001 |
| Candida albicans | ↑ | 0.11 | <0.05 |
| Enterococcus faecalis | ↓ | 0.37 | <0.001 |
| Bifidobacterium bifidum | ↓ | 0.18 | <0.01 |
| Bacteroides fragilis | ↓ | 0.14 | <0.05 |
| Enterococcus faecium | ↓ | 0.13 | <0.05 |
| Families | Relative Abundance | Effect Size, RDP/SILVA | Adjusted p-Value |
|---|---|---|---|
| 1. Atherosclerotic cardiovascular diseases | |||
| Erysipelotrichaceae | ↑ | 0.28/0.18 | <0.001 |
| Streptococcaceae | ↑ | 0.2/0.2 | <0.001 |
| Clostridiaceae | ↑ | 0.14/0.14 | <0.01 |
| Peptostreptococcaceae | ↑ | 0.14/0.14 | <0.01 |
| Lactobacillaceae | ↑ | 0.12/0.12 | <0.01 |
| Bifidobacteriaceae | ↑ | 0.11/0.11 | <0.01 |
| Sutterellaceae | ↓ | 0.28/0.35 | <0.001 |
| Acidaminococcaceae | ↓ | 0.17/0.17 | <0.005 |
| Odoribacteraceae | ↓ | 0.13/0.13 | <0.001 |
| 2. Heart failure with preserved ejection fraction | |||
| Erysipelotrichaceae | ↑ | 0.28/0.11 | <0.05 |
| 3. Heart failure with reduced ejection fraction | |||
| Streptococcaceae | ↑ | 0.48/0.48 | <0.01 |
| Erysipelotrichaceae | ↑ | 0.5/0.34 | <0.05 |
| Lactobacillaceae | ↑ | 0.45/0.39 | <0.01 |
| Clostridiaceae | ↑ | 0.38/0.38 | <0.01 |
| Methanobacteriaceae | ↑ | 0.28/0.28 | <0.05 |
| Bifidobacteriaceae | ↑ | 0.28/0.28 | <0.05 |
| Eggerthellaceae | ↑ | 0.25/0.36 | <0.05 |
| Enterococcaceae | ↑ | 0.25/0.25 | <0.05 |
| Acidaminococcaceae | ↓ | 0.43/0.43 | <0.01 |
| Barnesiellaceae | ↓ | 0.25/0.36 | <0.05 |
| Odoribacteraceae | ↓ | 0.27/.027 | <0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drapkina, O.M.; Ashniev, G.A.; Zlobovskaya, O.A.; Yafarova, A.A.; Dementeva, E.V.; Kaburova, A.N.; Meshkov, I.O.; Sheptulina, A.F.; Kiselev, A.R.; Kontsevaya, A.V.; et al. Diversities in the Gut Microbial Patterns in Patients with Atherosclerotic Cardiovascular Diseases and Certain Heart Failure Phenotypes. Biomedicines 2022, 10, 2762. https://doi.org/10.3390/biomedicines10112762
Drapkina OM, Ashniev GA, Zlobovskaya OA, Yafarova AA, Dementeva EV, Kaburova AN, Meshkov IO, Sheptulina AF, Kiselev AR, Kontsevaya AV, et al. Diversities in the Gut Microbial Patterns in Patients with Atherosclerotic Cardiovascular Diseases and Certain Heart Failure Phenotypes. Biomedicines. 2022; 10(11):2762. https://doi.org/10.3390/biomedicines10112762
Chicago/Turabian StyleDrapkina, Oxana M., German A. Ashniev, Olga A. Zlobovskaya, Adel A. Yafarova, Elena V. Dementeva, Anastasia N. Kaburova, Ivan O. Meshkov, Anna F. Sheptulina, Anton R. Kiselev, Anna V. Kontsevaya, and et al. 2022. "Diversities in the Gut Microbial Patterns in Patients with Atherosclerotic Cardiovascular Diseases and Certain Heart Failure Phenotypes" Biomedicines 10, no. 11: 2762. https://doi.org/10.3390/biomedicines10112762
APA StyleDrapkina, O. M., Ashniev, G. A., Zlobovskaya, O. A., Yafarova, A. A., Dementeva, E. V., Kaburova, A. N., Meshkov, I. O., Sheptulina, A. F., Kiselev, A. R., Kontsevaya, A. V., Zhamalov, L. M., Koretskiy, S. N., Pokrovskaya, M. S., Akinshina, A. I., Zagaynova, A. V., Lukashina, M. V., Kirillov, A. V., Abramov, I. A., Tolkacheva, L. R., ... Yudin, S. M. (2022). Diversities in the Gut Microbial Patterns in Patients with Atherosclerotic Cardiovascular Diseases and Certain Heart Failure Phenotypes. Biomedicines, 10(11), 2762. https://doi.org/10.3390/biomedicines10112762