
Cisplatin Induces Senescent Lung Cancer Cell-Mediated Stemness Induction via GRP78/Akt-Dependent Mechanism

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
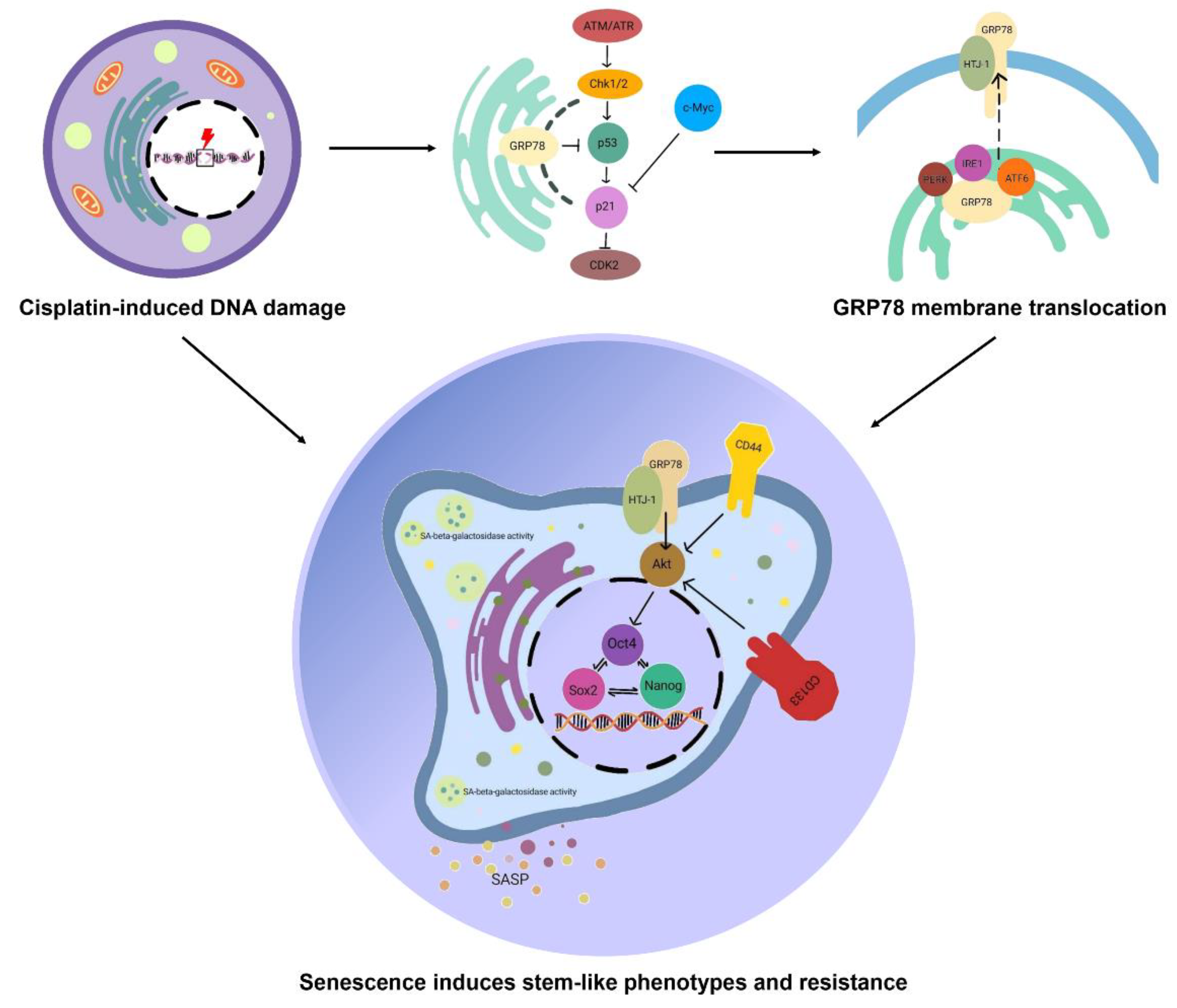{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Preparation of Cisplatin Stock Solution
2.3. Cell Lines and Culture
2.4. SA-β-Galactosidase Staining
2.5. Colony Formation Assay
2.6. Cell Cycle Analysis
2.7. RNA Isolation, Reverse Transcription and Quantitative Real-Time PCR (qRT-PCR)
2.8. Western Blot Analysis
2.9. Immunocytochemistry Assay (ICC)
2.10. Immunoprecipitation (IP)
2.11. Statistical Analysis
3. Results
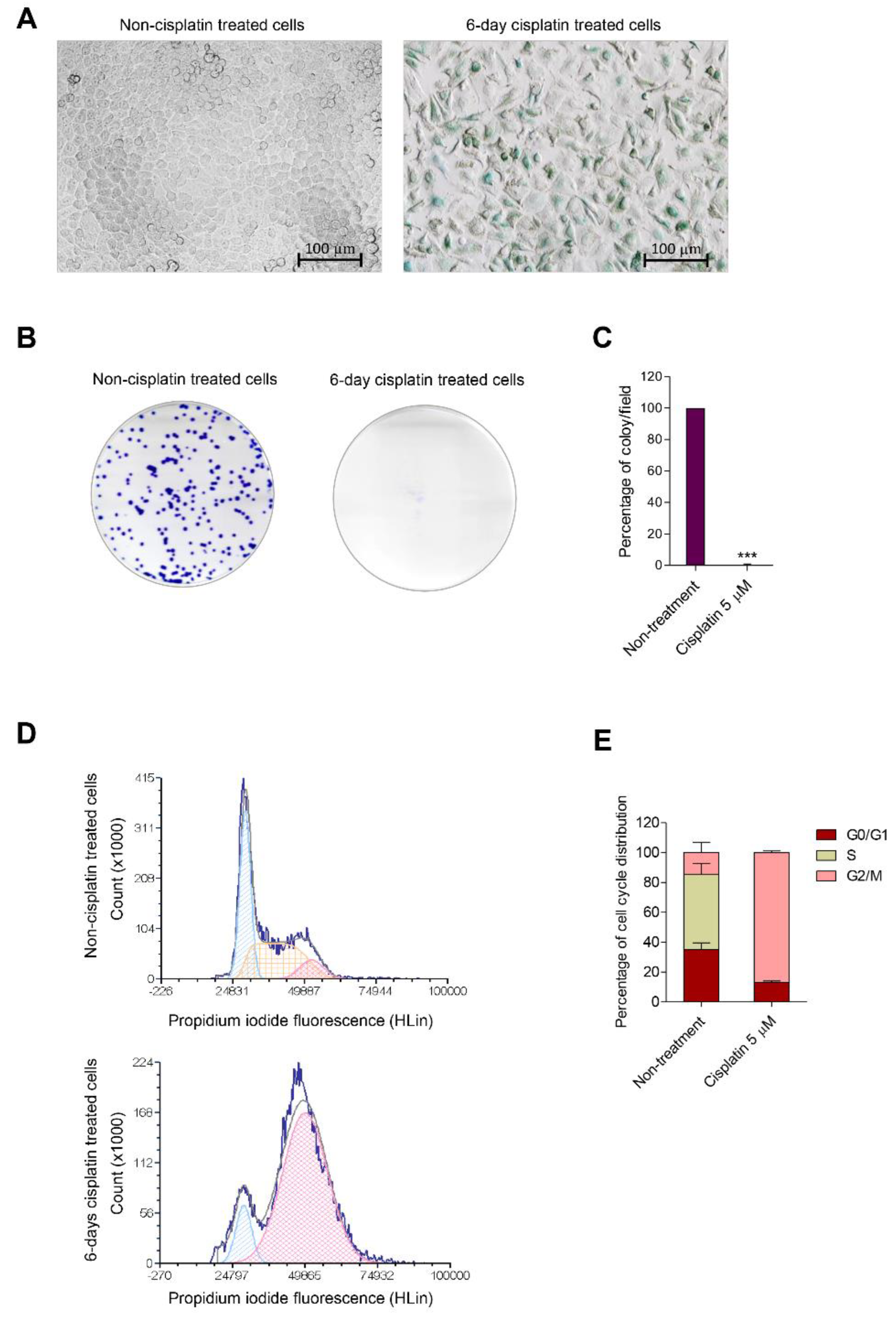
3.1. Cisplatin Induces Senescence in Human Lung Carcinoma Cells
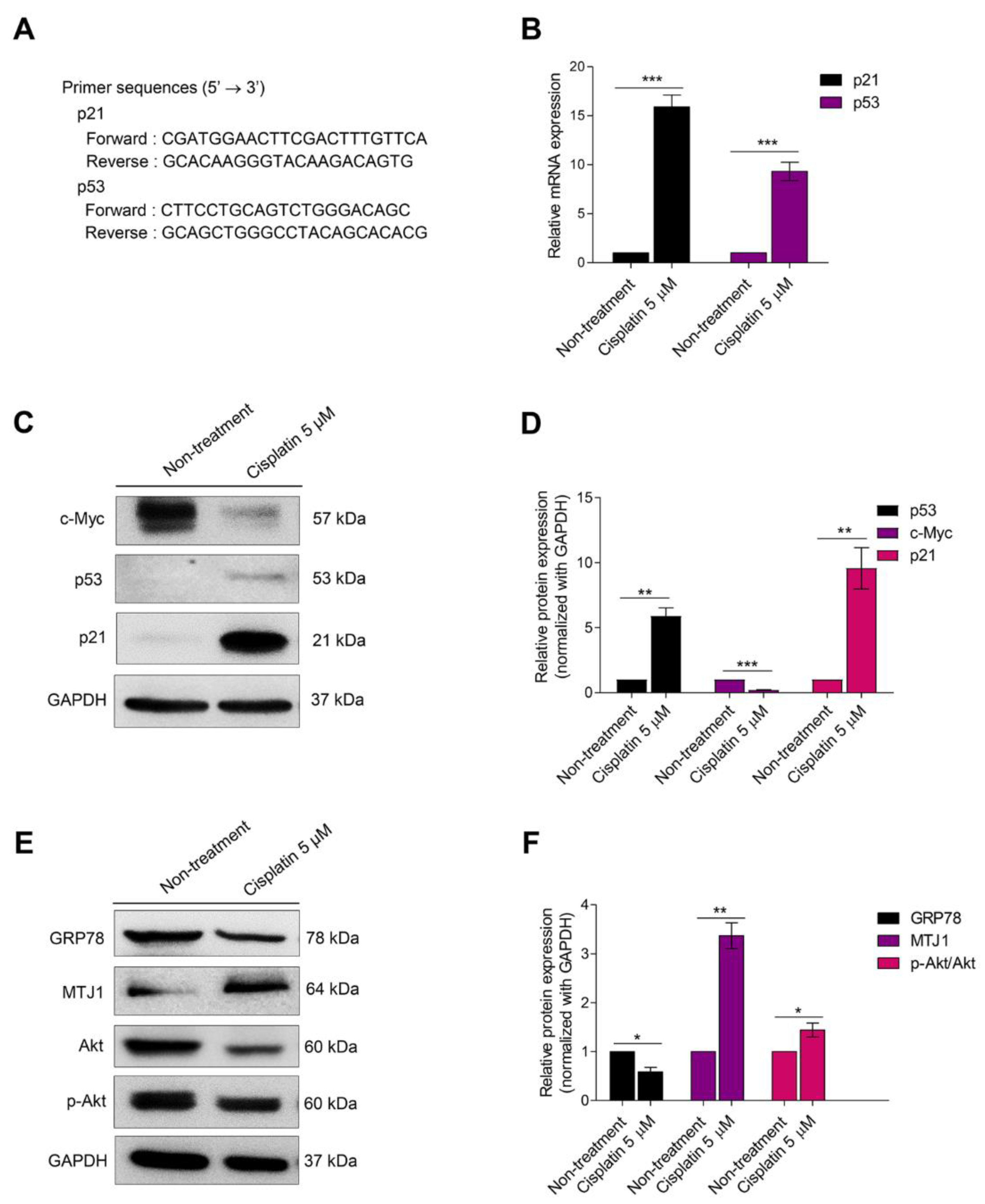
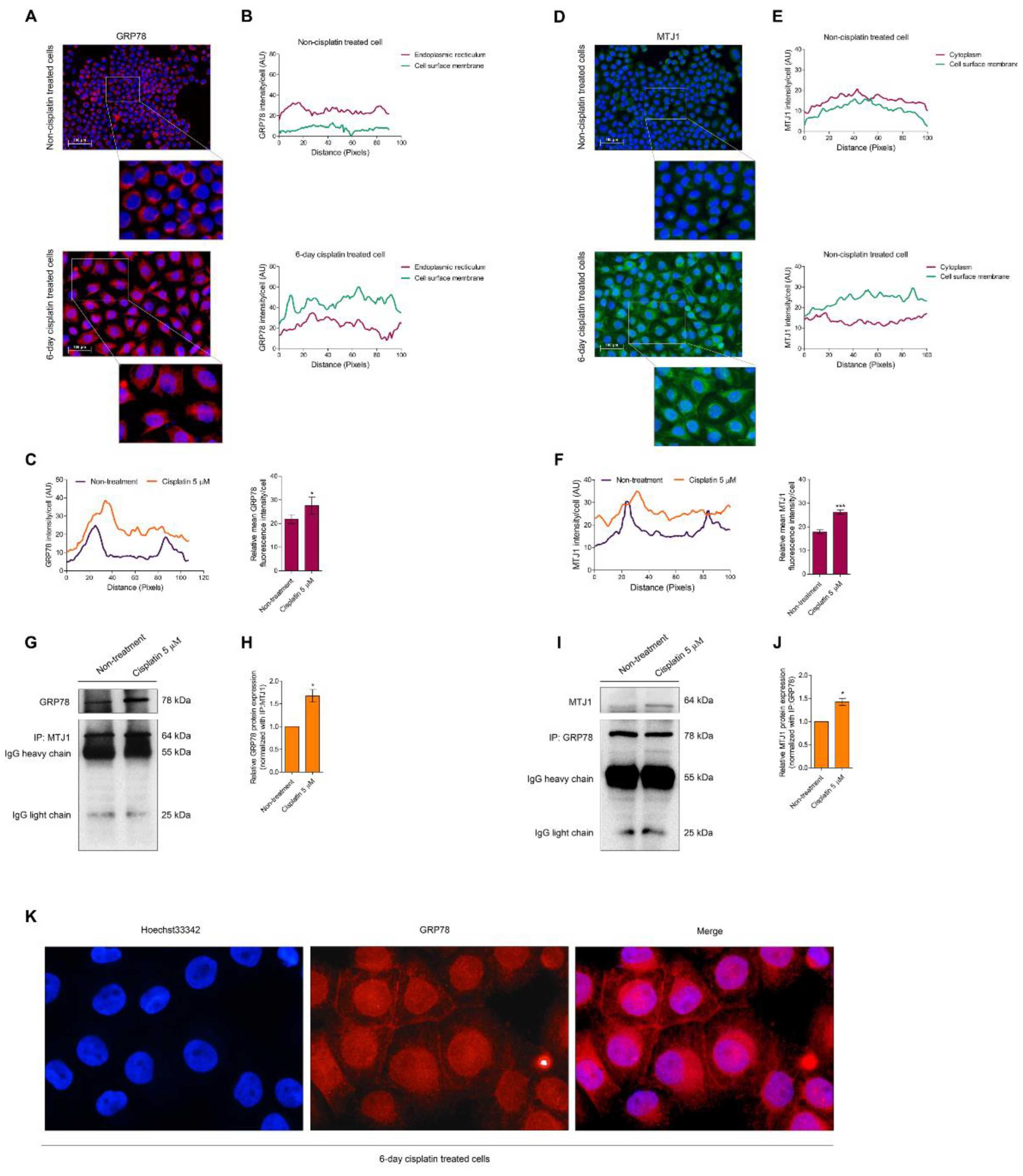
3.2. Senescence Activation Is Related to Mitigating GRP78
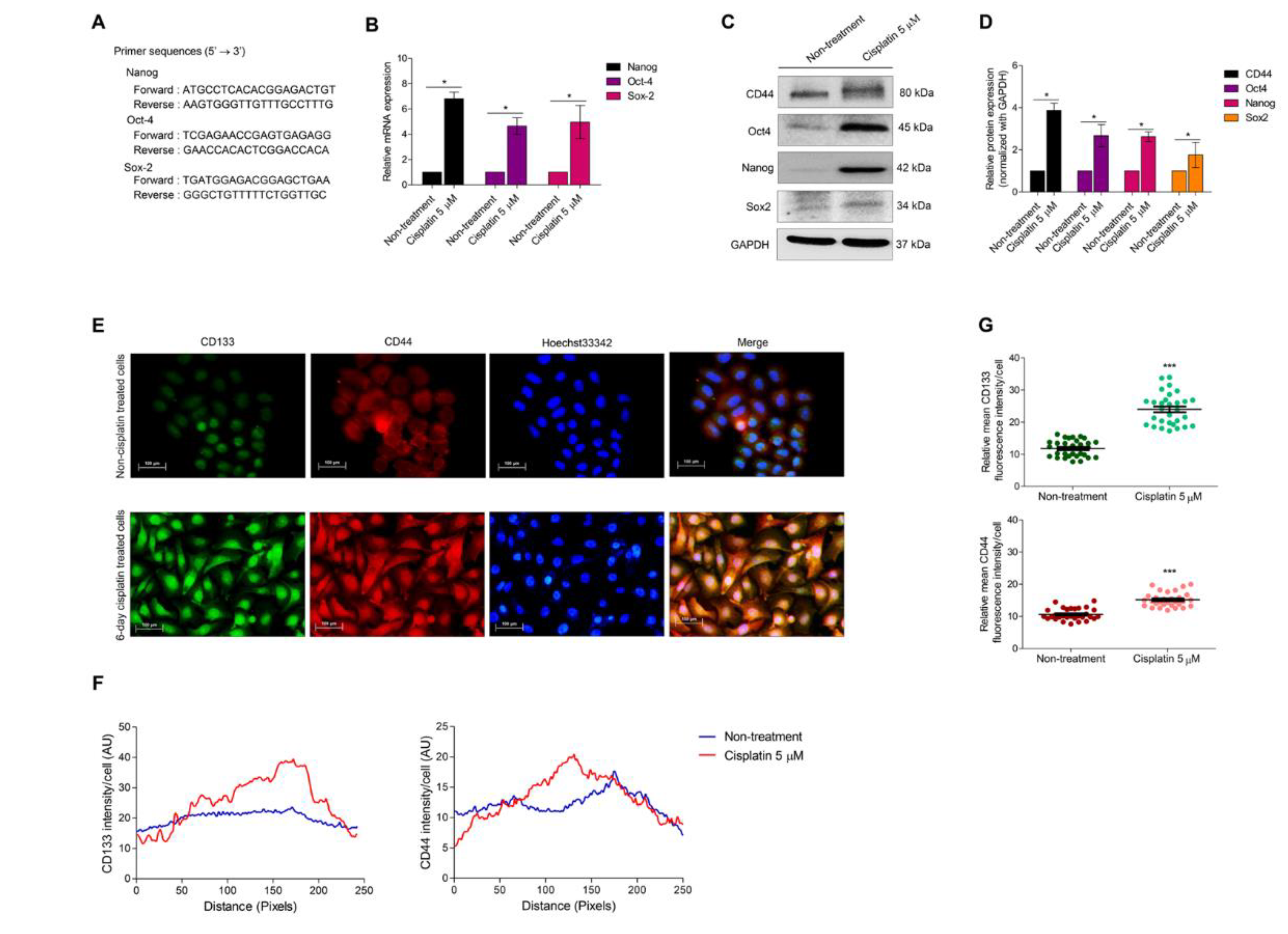
3.3. Cellular Senescence Induces Stem-like Phenotype
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stinchcombe, T.E.; Socinski, M.A. Current Treatments for Advanced Stage Non-Small Cell Lung Cancer. Proc. Am. Thorac. Soc. 2009, 6, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-Induced Senescence in Cancer. JNCI J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef]
- Fang, K.; Chiu, C.C.; Li, C.H.; Chang, Y.T.; Hwang, H.T. Cisplatin-Induced Senescence and Growth Inhibition in Human Non-Small Cell Lung Cancer Cells with Ectopic Transfer of p16I NK4a. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2007, 16, 479–488. [Google Scholar] [CrossRef]
- Guillon, J.; Petit, C.; Toutain, B.; Guette, C.; Lelievre, E.; Coqueret, O. Chemotherapy-induced senescence, an adaptive mechanism driving resistance and tumor heterogeneity. Cell Cycle 2019, 18, 2385–2397. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Wang, X.; Wong, S.C.; Pan, J.; Tsao, S.W.; Fung, K.H.; Kwong, D.L.; Sham, J.S.; Nicholls, J.M. Evidence of cisplatin-induced senescent-like growth arrest in nasopharyngeal carcinoma cells. Cancer Res. 1998, 58, 5019–5022. [Google Scholar] [PubMed]
- Di Leonardo, A.; Linke, S.P.; Clarkin, K.; Wahl, G.M. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994, 8, 2540–2551. [Google Scholar] [CrossRef]
- Mosieniak, G. Polyploidy: The Link Between Senescence and Cancer. Curr. Pharm. Des. 2010, 16, 734–740. [Google Scholar] [CrossRef]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef]
- Zhang, D.; Monteiro, M.J.; Liu, J.; Gu, W. Mechanisms of cancer stem cell senescence: Current understanding and future perspectives. Clin. Exp. Pharmacol. Physiol. 2021, 48, 1185–1202. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Schosserer, M.; Grillari, J.; Breitenbach, M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front. Oncol. 2017, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Triana-Martínez, F.; Loza, M.I.; Domínguez, E. Beyond Tumor Suppression: Senescence in Cancer Stemness and Tumor Dormancy. Cells 2020, 9, 346. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Cheng, X.; Zhang, J.; Liao, Y.; Jia, Y.; Qing, C. Possibility of inducing tumor cell senescence during therapy (Review). Oncol. Lett. 2021, 22, 496. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef]
- Casas, C. GRP78 at the Centre of the Stage in Cancer and Neuroprotection. Front. Neurosci. 2017, 11, 177. [Google Scholar] [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef]
- Xia, P.; Xu, X.Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar]
- Lin, Y.; Yang, Y.; Li, W.; Chen, Q.; Li, J.; Pan, X.; Zhou, L.; Liu, C.; Chen, C.; He, J.; et al. Reciprocal Regulation of Akt and Oct4 Promotes the Self-Renewal and Survival of Embryonal Carcinoma Cells. Mol. Cell 2012, 48, 627–640. [Google Scholar] [CrossRef]
- Schaefer, T.; Steiner, R.; Lengerke, C. SOX2 and p53 Expression Control Converges in PI3K/AKT Signaling with Versatile Implications for Stemness and Cancer. Int. J. Mol. Sci. 2020, 21, 4902. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Michowski, W.; Kolodziejczyk, A.; Sicinski, P. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 2019, 21, 1060–1067. [Google Scholar] [CrossRef]
- Liu, H.; Lv, L.; Yang, K. Chemotherapy targeting cancer stem cells. Am. J. Cancer Res. 2015, 5, 880–893. [Google Scholar] [PubMed]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor p21cip1/waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Ei, Z.Z.; Choochuay, K.; Tubsuwan, A.; Pinkaew, D.; Suksomtip, M.; Vinayanuwattikun, C.; Chanvorachote, P.; Chunhacha, P. GRP78/BiP determines senescence evasion cell fate after cisplatin-based chemotherapy. Sci. Rep. 2021, 11, 22448. [Google Scholar] [CrossRef]
- Behrooz, A.B.; Syahir, A.; Ahmad, S. CD133: Beyond a cancer stem cell biomarker. J. Drug Target. 2018, 27, 257–269. [Google Scholar] [CrossRef]
- Tothill, P.; Klys, H.S.; Matheson, L.M.; McKay, K.; Smyth, J.F. The long-term retention of platinum in human tissues following the administration of cisplatin or carboplatin for cancer chemotherapy. Eur. J. Cancer 1992, 28, 1358–1361. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef]
- Ismail, I.H.; Nyström, S.; Nygren, J.; Hammarsten, O. Activation of Ataxia Telangiectasia Mutated by DNA Strand Break-inducing Agents Correlates Closely with the Number of DNA Double Strand Breaks. J. Biol. Chem. 2005, 280, 4649–4655. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Ferguson, D.; Song, H.; Bassing, C.; Eckersdorff, M.; Alt, F.W.; Xu, Y. Functional Interaction of H2AX, NBS1, and p53 in ATM-Dependent DNA Damage Responses and Tumor Suppression. Mol. Cell. Biol. 2005, 25, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Stagni, V.; Ferri, A.; Cirotti, C.; Barilà, D. ATM Kinase-Dependent Regulation of Autophagy: A Key Player in Senescence? Front. Cell Dev. Biol. 2021, 8, 599048. [Google Scholar] [CrossRef]
- Li, W.; Wang, W.; Dong, H.; Li, Y.; Li, L.; Han, L.; Han, Z.; Wang, S.; Ma, D.; Wang, H. Cisplatin-induced senescence in ovarian cancer cells is mediated by GRP78. Oncol. Rep. 2014, 31, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Zu, K.; Bihani, T.; Lin, A.; Park, Y.M.; Mori, K.; Ip, C. Enhanced selenium effect on growth arrest by BiP/GRP78 knockdown in p53-null human prostate cancer cells. Oncogene 2005, 25, 546–554. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Shen, H.; Maki, C.G. Persistent p21 Expression after Nutlin-3a Removal Is Associated with Senescence-like Arrest in 4N Cells. J. Biol. Chem. 2010, 285, 23105–23114. [Google Scholar] [CrossRef]
- Campaner, S.; Doni, M.; Verrecchia, A.; Fagà, G.; Bianchi, L.; Amati, B. Myc, Cdk2 and cellular senescence: Old players, new game. Cell Cycle 2010, 9, 3679–3685. [Google Scholar] [CrossRef]
- Bailly, C.; Waring, M.J. Pharmacological effectors of GRP78 chaperone in cancers. Biochem. Pharmacol. 2019, 163, 269–278. [Google Scholar] [CrossRef]
- Misra, U.K.; Gonzalez-Gronow, M.; Gawdi, G.; Pizzo, S.V. The Role of MTJ-1 in Cell Surface Translocation of GRP78, a Receptor for α2-Macroglobulin-Dependent Signaling. J. Immunol. 2005, 174, 2092–2097. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, M.; Rhee, H.; Elguindi, E.C.; Blond, S.Y. Interaction of Murine BiP/GRP78 with the DnaJ Homologue MTJ1. J. Biol. Chem. 2000, 275, 19620–19627. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Durden, B.; Hadad, R.; Banerjee, S.; Dudeja, V.; Saluja, A.; Banerjee, S. ER stress sensor, glucose regulatory protein 78 (GRP78) regulates redox status in pancreatic cancer thereby maintaining “stemness”. Cell Death Dis. 2019, 10, 132. [Google Scholar] [CrossRef]
- Klauzinska, M.; Castro, N.P.; Rangel, M.C.; Spike, B.T.; Gray, P.C.; Bertolette, D.; Cuttitta, F.; Salomon, D. The multifaceted role of the embryonic gene Cripto-1 in cancer, stem cells and epithelial-mesenchymal transition. Semin. Cancer Biol. 2014, 29, 51–58. [Google Scholar] [CrossRef]
- Song, J.; Liu, W.; Wang, J.; Hao, J.; Wang, Y.; You, X.; Du, X.; Zhou, Y.; Ben, J.; Zhang, X.; et al. GALNT6 promotes invasion and metastasis of human lung adenocarcinoma cells through O-glycosylating chaperone protein GRP78. Cell Death Dis. 2020, 11, 352. [Google Scholar] [CrossRef]
- Liao, C.H.; Tzeng, Y.T.; Lai, G.M.; Chang, C.L.; Hu, M.H.; Tsai, W.L.; Liu, Y.R.; Hsia, S.; Chuang, S.E.; Chiou, T.J.; et al. Omega-3 Fatty Acid-Enriched Fish Oil and Selenium Combination Modulates Endoplasmic Reticulum Stress Response Elements and Reverses Acquired Gefitinib Resistance in HCC827 Lung Adenocarcinoma Cells. Mar. Drugs 2020, 18, 399. [Google Scholar] [CrossRef]
- Raiter, A.; Lipovetsky, J.; Hyman, L.; Mugami, S.; Ben-Zur, T.; Yerushalmi, R. Chemotherapy Controls Metastasis through Stimulatory Effects on GRP78 and Its Transcription Factor CREB3L1. Front. Oncol. 2020, 10, 1500. [Google Scholar] [CrossRef]
- Tomicic, M.; Krämer, F.; Nguyen, A.; Schwarzenbach, C.; Christmann, M. Oxaliplatin-Induced Senescence in Colorectal Cancer Cells Depends on p14ARF-Mediated Sustained p53 Activation. Cancers 2021, 13, 2019. [Google Scholar] [CrossRef]
- Xi, J.; Chen, Y.; Huang, S.; Cui, F.; Wang, X. Suppression of GRP78 sensitizes human colorectal cancer cells to oxaliplatin by downregulation of CD24. Oncol. Lett. 2018, 15, 9861–9867. [Google Scholar] [CrossRef] [PubMed]
- Misra, U.K.; Deedwania, R.; Pizzo, S.V. Binding of Activated α2-Macroglobulin to Its Cell Surface Receptor GRP78 in 1-LN Prostate Cancer Cells Regulates PAK-2-dependent Activation of LIMK. J. Biol. Chem. 2005, 280, 26278–26286. [Google Scholar] [CrossRef] [PubMed]
- Conner, C.; Lager, T.W.; Guldner, I.H.; Wu, M.Z.; Hishida, Y.; Hishida, T.; Ruiz, S.; Yamasaki, A.E.; Gilson, R.C.; Belmonte, J.C.I.; et al. Cell surface GRP78 promotes stemness in normal and neoplastic cells. Sci. Rep. 2020, 10, 3474. [Google Scholar] [CrossRef]
- Gopal, U.; Mowery, Y.; Young, K.; Pizzo, S.V. Targeting cell surface GRP78 enhances pancreatic cancer radiosensitivity through YAP/TAZ protein signaling. J. Biol. Chem. 2019, 294, 13939–13952. [Google Scholar] [CrossRef]
- Fu, Y.; Wey, S.; Wang, M.; Ye, R.; Liao, C.-P.; Roy-Burman, P.; Lee, A.S. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc. Natl. Acad. Sci. USA 2008, 105, 19444–19449. [Google Scholar] [CrossRef]
- Yu, J.S.L.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [PubMed]
- Hongwiangchan, N.; Sriratanasak, N.; Wichadakul, D.; Aksorn, N.; Chamni, S.; Chanvorachote, P. Hydroquinone 5-O-Cinnamoyl Ester of Renieramycin M Suppresses Lung Cancer Stem Cells by Targeting Akt and Destabilizes c-Myc. Pharmaceuticals 2021, 14, 1112. [Google Scholar] [CrossRef] [PubMed]
- Suksamai, D.; Racha, S.; Sriratanasak, N.; Chaotham, C.; Aphicho, K.; Lin, A.C.K.; Chansriniyom, C.; Suwanborirux, K.; Chamni, S.; Chanvorachote, P. 5-O-(N-Boc-l-Alanine)-Renieramycin T Induces Cancer Stem Cell Apoptosis via Targeting Akt Signaling. Mar. Drugs 2022, 20, 235. [Google Scholar] [CrossRef]
- Nör, C.; Zhang, Z.; Warner, K.A.; Bernardi, L.; Visioli, F.; Helman, J.I.; Roesler, R.; Nör, J.E. Cisplatin Induces Bmi-1 and Enhances the Stem Cell Fraction in Head and Neck Cancer. Neoplasia 2014, 16, 137–146. [Google Scholar] [CrossRef]
- Deschênes-Simard, X.; Parisotto, M.; Rowell, M.C.; Le Calvé, B.; Igelmann, S.; Moineau-Vallée, K.; Saint-Germain, E.; Kalegari, P.; Bourdeau, V.; Kottakis, F.; et al. Circumventing senescence is associated with stem cell properties and metformin sensitivity. Aging Cell 2019, 18, e12889. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sriratanasak, N.; Chunhacha, P.; Ei, Z.Z.; Chanvorachote, P. Cisplatin Induces Senescent Lung Cancer Cell-Mediated Stemness Induction via GRP78/Akt-Dependent Mechanism. Biomedicines 2022, 10, 2703. https://doi.org/10.3390/biomedicines10112703
Sriratanasak N, Chunhacha P, Ei ZZ, Chanvorachote P. Cisplatin Induces Senescent Lung Cancer Cell-Mediated Stemness Induction via GRP78/Akt-Dependent Mechanism. Biomedicines. 2022; 10(11):2703. https://doi.org/10.3390/biomedicines10112703
Chicago/Turabian StyleSriratanasak, Nicharat, Preedakorn Chunhacha, Zin Zin Ei, and Pithi Chanvorachote. 2022. "Cisplatin Induces Senescent Lung Cancer Cell-Mediated Stemness Induction via GRP78/Akt-Dependent Mechanism" Biomedicines 10, no. 11: 2703. https://doi.org/10.3390/biomedicines10112703
APA StyleSriratanasak, N., Chunhacha, P., Ei, Z. Z., & Chanvorachote, P. (2022). Cisplatin Induces Senescent Lung Cancer Cell-Mediated Stemness Induction via GRP78/Akt-Dependent Mechanism. Biomedicines, 10(11), 2703. https://doi.org/10.3390/biomedicines10112703