



Novel Expression of Thymine Dimers in Renal Cell Carcinoma, Demonstrated through Immunohistochemistry
 ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Processing, Morphologic Evaluation and Case Selection
2.2. TD-Targeted Immunohistochemistry, Microscopic Evaluation, Data Quantification and Digitalization, Statistical Analysis
3. Results
3.1. Descriptive Analysis
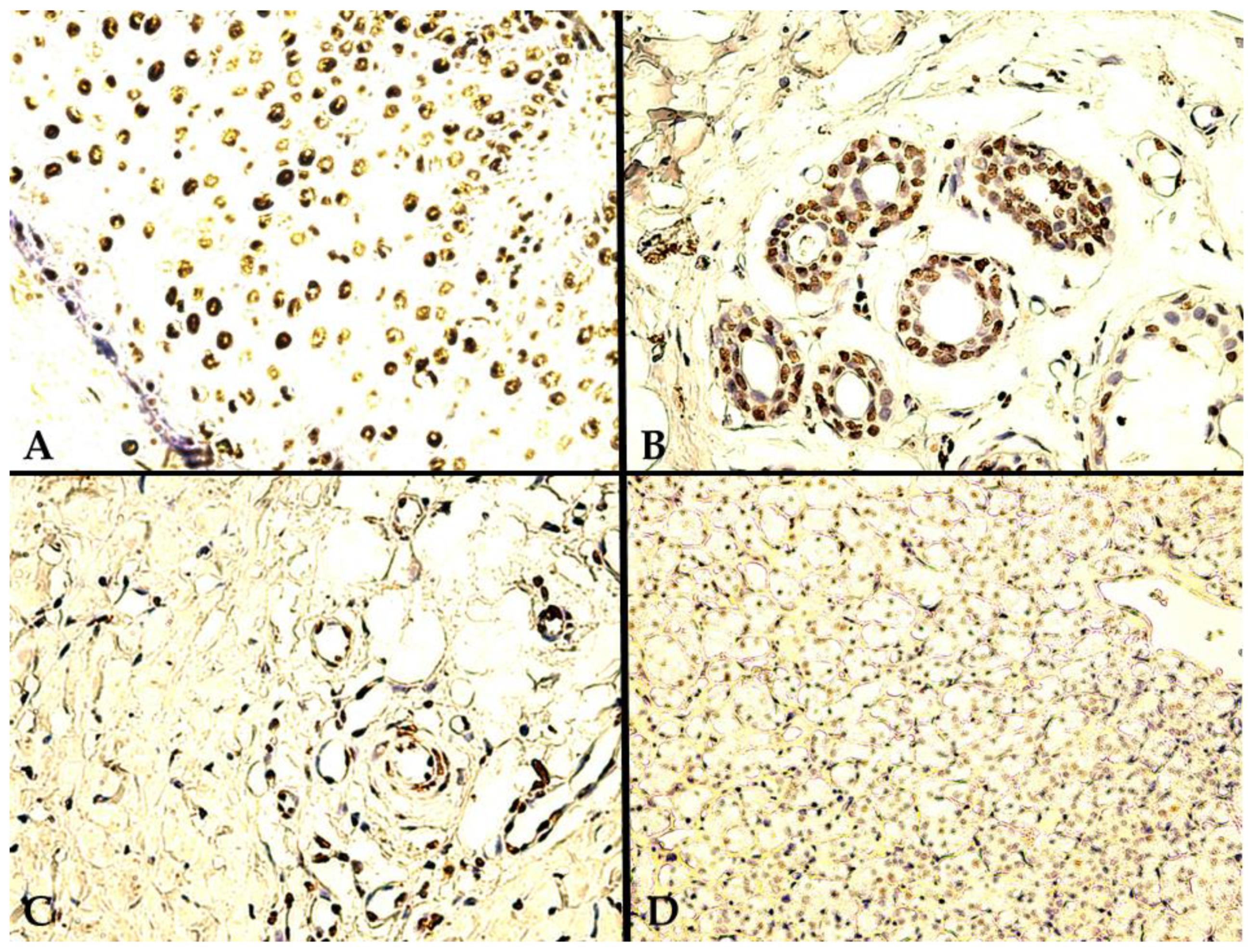
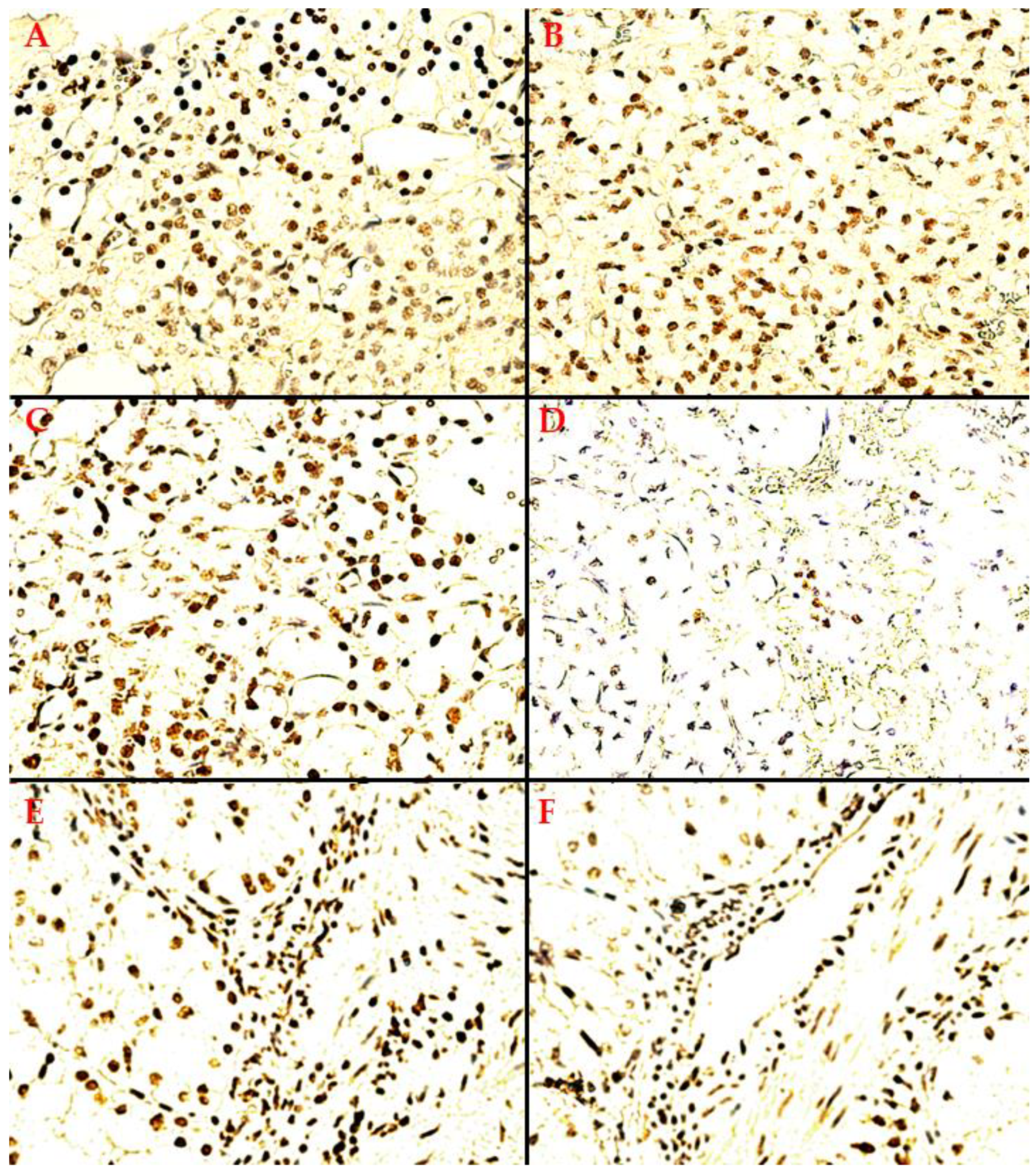
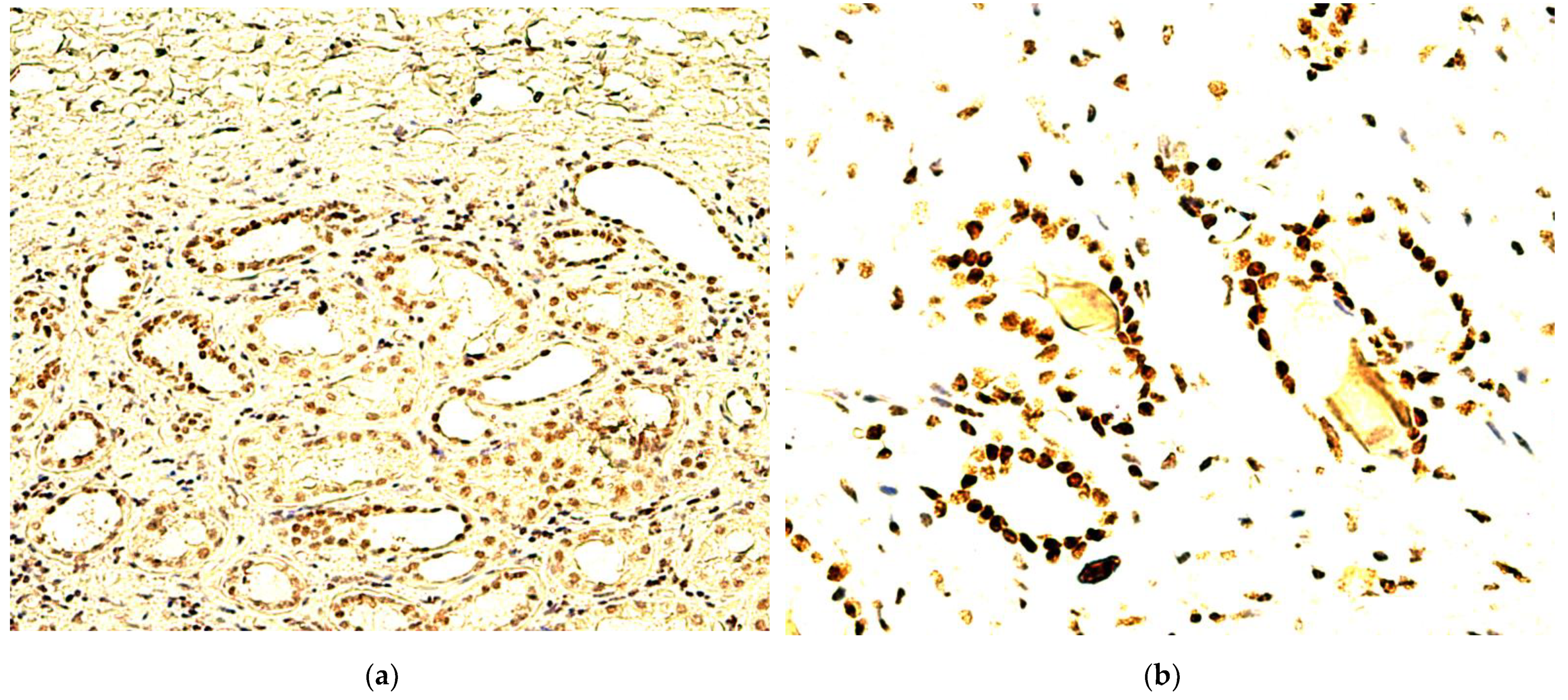
- A heterogeneous expression pattern, concentrated, in moderate to high density (SQ = +2/+3), at the level of the proliferation front, with variable intensity of TD staining (mostly moderate to high as well, i.e., SI = +2/+3), yet inconsistent in more central tumor areas, or along the transitional area, between tumor and healthy renal tissue (Figure 2A);
- A homogenous, diffuse, and high-intensity (SI = +2/+3) staining pattern, with the majority of RCC tumor tissue cellularity being positive for TDs (SQ = +2/+3) (Figure 2B,C).
3.2. Statistical Data Analysis
4. Discussion
4.1. Speculative Models and Future Research Perspectives
4.2. Study Limitations and Validation Issues
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs—Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Reuter, V.E.; Argani, P.; Zhou, M.; Delahunt, B.; Members of the ISUP Immunohistochemistry in Diagnostic Urologic Pathology Group. Best Practices Recommendations in the Application of Immunohistochemistry in the Kidney Tumors: Report from the International Society of Urologic Pathology Consensus Conference. Am. J. Surg. Pathol. 2014, 38, e35–e49. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.B.; Epstein, J.I.; Ulbright, T.M.; Humphrey, P.A.; Egevad, L.; Montironi, R.; Grignon, D.; Trpkov, K.; Lopez-Beltran, A.; Zhou, M.; et al. Best Practices Recommendations in the Application of Immunohistochemistry in Urologic Pathology: Report from the International Society of Urological Pathology Consensus Conference. Am. J. Surg. Pathol. 2014, 38, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Joo, J.W.; Lee, S.J.; Cho, Y.A.; Park, C.K.; Cho, N.H. Comprehensive Immunoprofiles of Renal Cell Carcinoma Subtypes. Cancers 2020, 12, 602. [Google Scholar] [CrossRef] [PubMed]
- Kane, C.J.; Mallin, K.; Ritchey, J.; Cooperberg, M.R.; Carroll, P.R. Renal Cell Cancer Stage Migration: Analysis of the National Cancer Data Base. Cancer 2008, 113, 78–83. [Google Scholar] [CrossRef]
- Mancini, M.; Righetto, M.; Baggio, G. Gender-Related Approach to Kidney Cancer Management: Moving Forward. Int. J. Mol. Sci. 2020, 21, 3378. [Google Scholar] [CrossRef]
- Kuzminov, A. Pyrimidine Dimers. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 538–539. [Google Scholar] [CrossRef]
- Cooke, M.S.; Robson, A. Immunochemical Detection of UV-Induced DNA Damage and Repair. Methods Mol. Biol. 2006, 314, 215–228. [Google Scholar] [CrossRef]
- Peak, M.J.; Peak, J.G.; Moehring, M.P.; Webb, R.B. Ultraviolet Action Spectra for DNA Dimer Induction, Lethality, and Mutagenesis in Escherichia Coli with Emphasis on the UVB Region. Photochem. Photobiol. 1984, 40, 613–620. [Google Scholar] [CrossRef]
- Ananthaswamy, H.N.; Pierceall, W.E. Molecular Mechanisms of Ultraviolet Radiation Carcinogenesis. Photochem. Photobiol. 1990, 52, 1119–1136. [Google Scholar] [CrossRef]
- Clingen, P.H.; Arlett, C.F.; Cole, J.; Waugh, A.P.; Lowe, J.E.; Harcourt, S.A.; Hermanova, N.; Roza, L.; Mori, T.; Nikaido, O. Correlation of UVC and UVB Cytotoxicity with the Induction of Specific Photoproducts in T-Lymphocytes and Fibroblasts from Normal Human Donors. Photochem. Photobiol. 1995, 61, 163–170. [Google Scholar] [CrossRef]
- Mitchell, D.L.; Jen, J.; Cleaver, J.E. Relative Induction of Cyclobutane Dimers and Cytosine Photohydrates in DNA Irradiated in Vitro and in Vivo with Ultraviolet-C and Ultraviolet-B Light. Photochem. Photobiol. 1991, 54, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Doetsch, P.W.; Zasatawny, T.H.; Martin, A.M.; Dizdaroglu, M. Monomeric Base Damage Products from Adenine, Guanine, and Thymine Induced by Exposure of DNA to Ultraviolet Radiation. Biochemistry 1995, 34, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Anselmino, C.; Douki, T.; Voituriez, L. Photochemistry of Nucleic Acids in Cells. J. Photochem. Photobiol. B 1992, 15, 277–298. [Google Scholar] [CrossRef]
- Goodsell, D.S. The Molecular Perspective: Ultraviolet Light and Pyrimidine Dimers. Oncologist 2001, 6, 298–299. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D. (Eds.) DNA Repair and Mutagenesis, 2nd ed.; ASM Press: Washington, DC, USA, 2006; pp. 109–110. [Google Scholar]
- Beukers, R.; Eker, A.P.M.; Lohman, P.H.M. 50 Years Thymine Dimer. DNA Repair 2008, 7, 530–543. [Google Scholar] [CrossRef]
- Ramasamy, K.; Shanmugam, M.; Balupillai, A.; Govindhasamy, K.; Gunaseelan, S.; Muthusamy, G.; Robert, B.M.; Nagarajan, R.P. Ultraviolet Radiation-Induced Carcinogenesis: Mechanisms and Experimental Models. J. Radiat. Cancer Res. 2017, 8, 4. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA Damage-Induced Cell Death: From Specific DNA Lesions to the DNA Damage Response and Apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Mallet, J.D.; Dorr, M.M.; Drigeard Desgarnier, M.-C.; Bastien, N.; Gendron, S.P.; Rochette, P.J. Faster DNA Repair of Ultraviolet-Induced Cyclobutane Pyrimidine Dimers and Lower Sensitivity to Apoptosis in Human Corneal Epithelial Cells than in Epidermal Keratinocytes. PLoS ONE 2016, 11, e0162212. [Google Scholar] [CrossRef]
- Brenner, M.; Degitz, K.; Besch, R.; Berking, C. Differential Expression of Melanoma-Associated Growth Factors in Keratinocytes and Fibroblasts by Ultraviolet A and Ultraviolet B Radiation. Br. J. Dermatol. 2005, 153, 733–739. [Google Scholar] [CrossRef]
- Jhappan, C.; Noonan, F.P.; Merlino, G. Ultraviolet Radiation and Cutaneous Malignant Melanoma. Oncogene 2003, 22, 3099–3112. [Google Scholar] [CrossRef]
- Takahashi, A.; Ohnishi, T. Does GammaH2AX Foci Formation Depend on the Presence of DNA Double Strand Breaks? Cancer Lett. 2005, 229, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Schul, W.; Jans, J.; Rijksen, Y.M.A.; Klemann, K.H.M.; Eker, A.P.M.; de Wit, J.; Nikaido, O.; Nakajima, S.; Yasui, A.; Hoeijmakers, J.H.J.; et al. Enhanced Repair of Cyclobutane Pyrimidine Dimers and Improved UV Resistance in Photolyase Transgenic Mice. EMBO J. 2002, 21, 4719–4729. [Google Scholar] [CrossRef] [PubMed]
- Jans, J.; Schul, W.; Sert, Y.-G.; Rijksen, Y.; Rebel, H.; Eker, A.P.M.; Nakajima, S.; van Steeg, H.; de Gruijl, F.R.; Yasui, A.; et al. Powerful Skin Cancer Protection by a CPD-Photolyase Transgene. Curr. Biol. 2005, 15, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Jans, J.; Garinis, G.A.; Schul, W.; van Oudenaren, A.; Moorhouse, M.; Smid, M.; Sert, Y.-G.; van der Velde, A.; Rijksen, Y.; de Gruijl, F.R.; et al. Differential Role of Basal Keratinocytes in UV-Induced Immunosuppression and Skin Cancer. Mol. Cell. Biol. 2006, 26, 8515–8526. [Google Scholar] [CrossRef]
- DiGiovanna, J.J.; Kraemer, K.H. Shining a Light on Xeroderma Pigmentosum. J. Investig. Dermatol. 2012, 132, 785–796. [Google Scholar] [CrossRef]
- Lawrence, K.P.; Delinasios, G.J.; Premi, S.; Young, A.R.; Cooke, M.S. Perspectives on Cyclobutane Pyrimidine Dimers—Rise of the Dark Dimers. Photochem. Photobiol. 2022, 98, 609–616. [Google Scholar] [CrossRef]
- Premi, S.; Wallisch, S.; Mano, C.M.; Weiner, A.B.; Bacchiocchi, A.; Wakamatsu, K.; Bechara, E.J.H.; Halaban, R.; Douki, T.; Brash, D.E. Chemiexcitation of Melanin Derivatives Induces DNA Photoproducts Long after UV Exposure. Science 2015, 347, 842–847. [Google Scholar] [CrossRef]
- Ruven, H.J.; Berg, R.J.; Seelen, C.M.; Dekkers, J.A.; Lohman, P.H.; Mullenders, L.H.; van Zeeland, A.A. Ultraviolet-Induced Cyclobutane Pyrimidine Dimers Are Selectively Removed from Transcriptionally Active Genes in the Epidermis of the Hairless Mouse. Cancer Res 1993, 53, 1642–1645. [Google Scholar]
- Snopov, S.A.; de Gruijl, F.R.; Roza, L.; van der Leun, J.C. Immunochemical Study of DNA Modifications in the Nuclei of UV-Damaged Lymphocytes. Photochem. Photobiol. Sci. 2004, 3, 85–90. [Google Scholar] [CrossRef]
- Bykov, V.J.; Sheehan, J.M.; Hemminki, K.; Young, A.R. In Situ Repair of Cyclobutane Pyrimidine Dimers and 6-4 Photoproducts in Human Skin Exposed to Solar Simulating Radiation. J. Investig. Dermatol. 1999, 112, 326–331. [Google Scholar] [CrossRef]
- Strickland, P.T. Distribution of Thymine Dimers Induced in Mouse Skin by Ultraviolet Radiation. Photo-Dermatology 1988, 5, 1–8. [Google Scholar] [PubMed]
- Handoko, H.Y.; Rodero, M.P.; Boyle, G.M.; Ferguson, B.; Engwerda, C.; Hill, G.; Muller, H.K.; Khosrotehrani, K.; Walker, G.J. UVB-Induced Melanocyte Proliferation in Neonatal Mice Driven by CCR2-Independent Recruitment of Ly6cLowMHCIIHi Macrophages. J. Investig. Dermatol. 2013, 133, 1803–1812. [Google Scholar] [CrossRef] [PubMed]
- Redmond, R.W.; Rajadurai, A.; Udayakumar, D.; Sviderskaya, E.V.; Tsao, H. Melanocytes Are Selectively Vulnerable to UVA-Mediated Bystander Oxidative Signaling. J. Investig. Dermatol. 2014, 134, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.-M.; Huang, C.-H.; Li, H.-J.; Hsiao, C.-Y.; Su, C.-C.; Lee, P.-L.; Hung, C.-F. Protective Effects of Resveratrol against UVA-Induced Damage in ARPE19 Cells. Int. J. Mol. Sci. 2015, 16, 5789–5802. [Google Scholar] [CrossRef] [PubMed]
- Niculiţe, C.; Nechifor, M.; Urs, A.; Olariu, L.; Ceafalan, L.; Leabu, M. Keratinocyte Motility Is Affected by UVA Radiation—A Comparison between Normal and Dysplastic Cells. Int. J. Mol. Sci. 2018, 19, 1700. [Google Scholar] [CrossRef]
- Cimpean, A.M.; Sava, M.P.; Raica, M. DNA Damage in Human Pterygium: One-Shot Multiple Targets. Mol. Vis. 2013, 19, 348–356. [Google Scholar]
- Ferguson, B.; Ram, R.; Handoko, H.Y.; Mukhopadhyay, P.; Muller, H.K.; Soyer, H.P.; Morahan, G.; Walker, G.J. Melanoma Susceptibility as a Complex Trait: Genetic Variation Controls All Stages of Tumor Progression. Oncogene 2015, 34, 2879–2886. [Google Scholar] [CrossRef]
- Murray, H.C.; Maltby, V.E.; Smith, D.W.; Bowden, N.A. Nucleotide Excision Repair Deficiency in Melanoma in Response to UVA. Exp. Hematol. Oncol. 2015, 5, 6. [Google Scholar] [CrossRef]
- Tyagi, N.; Bhardwaj, A.; Srivastava, S.K.; Arora, S.; Marimuthu, S.; Deshmukh, S.K.; Singh, A.P.; Carter, J.E.; Singh, S. Development and Characterization of a Novel in Vitro Progression Model for UVB-Induced Skin Carcinogenesis. Sci. Rep. 2015, 5, 13894. [Google Scholar] [CrossRef]
- Protić-Sabljić, M.; Tuteja, N.; Munson, P.J.; Hauser, J.; Kraemer, K.H.; Dixon, K. UV Light-Induced Cyclobutane Pyrimidine Dimers Are Mutagenic in Mammalian Cells. Mol. Cell. Biol. 1986, 6, 3349–3356. [Google Scholar]
- Gibson-D’Ambrosio, R.E.; Leong, Y.; D’Ambrosio, S.M. DNA Repair Following Ultraviolet and N-Ethyl-N-Nitrosourea Treatment of Cells Cultured from Human Fetal Brain, Intestine, Kidney, Liver, and Skin. Cancer Res. 1983, 43 Pt 1, 5846–5850. [Google Scholar] [PubMed]
- Thymine dimers expression in RCCs database. Available online: https://data.mendeley.com/datasets/v42wd7xsxj/1 (accessed on 21 July 2022).
- Novacescu, D.; Cut, T.G.; Cumpanas, A.A.; Latcu, S.C.; Bardan, R.; Ferician, O.; Secasan, C.-C.; Rusmir, A.; Raica, M. Evaluating Established Roles, Future Perspectives and Methodological Heterogeneity for Wilms’ Tumor 1 (WT1) Antigen Detection in Adult Renal Cell Carcinoma, Using a Novel N-Terminus Targeted Antibody (Clone WT49). Biomedicines 2022, 10, 912. [Google Scholar] [CrossRef]
- Brash, D.E. UV Signature Mutations. Photochem. Photobiol. 2015, 91, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome Sequencing Identifies Recurrent Somatic RAC1 Mutations in Melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E.; Seetharam, S.; Kraemer, K.H.; Seidman, M.M.; Bredberg, A. Photoproduct Frequency Is Not the Major Determinant of UV Base Substitution Hot Spots or Cold Spots in Human Cells. Proc. Natl. Acad. Sci. USA 1987, 84, 3782–3786. [Google Scholar] [CrossRef]
- Schreier, W.J.; Schrader, T.E.; Koller, F.O.; Gilch, P.; Crespo-Hernández, C.E.; Swaminathan, V.N.; Carell, T.; Zinth, W.; Kohler, B. Thymine Dimerization in DNA Is an Ultrafast Photoreaction. Science 2007, 315, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T.; Ravanat, J.-L. Oxidatively Generated Damage to Cellular DNA by UVB and UVA Radiation. Photochem. Photobiol. 2015, 91, 140–155. [Google Scholar] [CrossRef]
- Lazovich, D.; Vogel, R.I.; Berwick, M.; Weinstock, M.A.; Anderson, K.E.; Warshaw, E.M. Indoor Tanning and Risk of Melanoma: A Case-Control Study in a Highly Exposed Population. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1557–1568. [Google Scholar] [CrossRef]
- Douki, T.; Reynaud-Angelin, A.; Cadet, J.; Sage, E. Bipyrimidine Photoproducts Rather than Oxidative Lesions Are the Main Type of DNA Damage Involved in the Genotoxic Effect of Solar UVA Radiation. Biochemistry 2003, 42, 9221–9226. [Google Scholar] [CrossRef]
- White, E.H.; Miano, J.D.; Watkins, C.J.; Breaux, E.J. Chemically Produced Excited States. Angew. Chem. Int. Ed. Engl. 1974, 13, 229–243. [Google Scholar] [CrossRef]
- Heidegger, I.; Pircher, A.; Pichler, R. Targeting the Tumor Microenvironment in Renal Cell Cancer Biology and Therapy. Front. Oncol. 2019, 9, 490. [Google Scholar] [CrossRef] [PubMed]
- Mier, J.W. The Tumor Microenvironment in Renal Cell Cancer. Curr. Opin. Oncol. 2019, 31, 194–199. [Google Scholar] [CrossRef]
- Shi, J.; Wang, K.; Xiong, Z.; Yuan, C.; Wang, C.; Cao, Q.; Yu, H.; Meng, X.; Xie, K.; Cheng, Z.; et al. Impact of Inflammation and Immunotherapy in Renal Cell Carcinoma. Oncol. Lett. 2020, 20, 272. [Google Scholar] [CrossRef] [PubMed]
- Delinasios, G.J.; Karbaschi, M.; Cooke, M.S.; Young, A.R. Vitamin E Inhibits the UVAI Induction of “Light” and “Dark” Cyclobutane Pyrimidine Dimers, and Oxidatively Generated DNA Damage, in Keratinocytes. Sci. Rep. 2018, 8, 423. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhang, X.; Wang, Y.; Lebwohl, M. Inhibition of Ultraviolet Light-Induced Oxidative Events in the Skin and Internal Organs of Hairless Mice by Isoflavone Genistein. Cancer Lett. 2002, 185, 21–29. [Google Scholar] [CrossRef]
- Fosså, S.D. Interferon in Metastatic Renal Cell Carcinoma. Semin. Oncol. 2000, 27, 187–193. [Google Scholar]
- Koneru, R.; Hotte, S.J. Role of Cytokine Therapy for Renal Cell Carcinoma in the Era of Targeted Agents. Curr. Oncol. 2009, 16 (Suppl. S1), S40–S44. [Google Scholar] [CrossRef]
- Sherwani, M.A.; Ahmad, I.; Lewis, M.J.; Abdelgawad, A.; Rashid, H.; Yang, K.; Chen, C.-Y.; Raman, C.; Elmets, C.A.; Yusuf, N. Type I Interferons Enhance the Repair of Ultraviolet Radiation-Induced DNA Damage and Regulate Cutaneous Immune Suppression. Int. J. Mol. Sci. 2022, 23, 1822. [Google Scholar] [CrossRef]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326.e5. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Creighton, C.J.; Morgan, M.; Gunaratne, P.H.; Wheeler, D.A.; Gibbs, R.A.; Gordon Robertson, A.; Chu, A.; Beroukhim, R.; Cibulskis, K.; Signoretti, S.; et al. Comprehensive Molecular Characterization of Clear Cell Renal Cell Carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef]
- Williams, A.B.; Schumacher, B. P53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.R.; Fishel, M.L.; Amundson, S.; Kelley, M.R.; Smith, M.L. Implication of P53 in Base Excision DNA Repair: In Vivo Evidence. Oncogene 2002, 21, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Maltzman, W.; Czyzyk, L. UV Irradiation Stimulates Levels of P53 Cellular Tumor Antigen in Nontransformed Mouse Cells. Mol. Cell. Biol. 1984, 4, 1689–1694. [Google Scholar]
- Ko, L.J.; Prives, C. P53: Puzzle and Paradigm. Genes Dev. 1996, 10, 1054–1072. [Google Scholar] [CrossRef] [PubMed]
- Dimas-González, J.; Maldonado-Lagunas, V.; Díaz-Chávez, J.; López-Arellano, M.E.; Muñoz-Camacho, J.; Terán-Porcayo, M.A.; Lagunas-Martínez, A. Overexpression of P53 Protein Is a Marker of Poor Prognosis in Mexican Women with Breast Cancer. Oncol. Rep. 2017, 37, 3026–3036. [Google Scholar] [CrossRef][Green Version]
- Kunizaki, M.; Fukuda, A.; Wakata, K.; Tominaga, T.; Nonaka, T.; Miyazaki, T.; Matsumoto, K.; Sumida, Y.; Hidaka, S.; Yasutake, T.; et al. Clinical Significance of Serum P53 Antibody in the Early Detection and Poor Prognosis of Gastric Cancer. Anticancer Res. 2017, 37, 1979–1984. [Google Scholar] [CrossRef]
- Kunizaki, M.; Sawai, T.; Takeshita, H.; Tominaga, T.; Hidaka, S.; To, K.; Miyazaki, T.; Hamamoto, R.; Nanashima, A.; Nagayasu, T. Clinical Value of Serum P53 Antibody in the Diagnosis and Prognosis of Colorectal Cancer. Anticancer Res. 2016, 36, 4171–4175. [Google Scholar]
- Freier, C.P.; Stiasny, A.; Kuhn, C.; Mayr, D.; Alexiou, C.; Janko, C.; Wiest, I.; Jeschke, U.; Kost, B. Immunohistochemical Evaluation of the Role of P53 Mutation in Cervical Cancer: Ser-20 P53-Mutant Correlates with Better Prognosis. Anticancer Res. 2016, 36, 3131–3137. [Google Scholar]
- Singh, R.D.; Patel, K.R.; Patel, P.S. P53 Mutation Spectrum and Its Role in Prognosis of Oral Cancer Patients: A Study from Gujarat, West India. Mutat. Res. 2016, 783, 15–26. [Google Scholar] [CrossRef]
- Hideshima, T.; Cottini, F.; Nozawa, Y.; Seo, H.-S.; Ohguchi, H.; Samur, M.K.; Cirstea, D.; Mimura, N.; Iwasawa, Y.; Richardson, P.G.; et al. P53-Related Protein Kinase Confers Poor Prognosis and Represents a Novel Therapeutic Target in Multiple Myeloma. Blood 2017, 129, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Rice, K.; Greenblatt, M.S.; Soussi, T.; Fuchs, R.; Sørlie, T.; Hovig, E.; Smith-Sørensen, B.; Montesano, R.; Harris, C.C. Database of P53 Gene Somatic Mutations in Human Tumors and Cell Lines. Nucleic Acids Res. 1994, 22, 3551–3555. [Google Scholar] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. P53 Mutations in Human Cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Peng, S.; Jiang, N.; Wang, A.; Liu, S.; Xie, H.; Guo, L.; Cai, Q.; Niu, Y. Prognostic and Clinicopathological Value of P53 Expression in Renal Cell Carcinoma: A Meta-Analysis. Oncotarget 2017, 8, 102361–102370. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; DeDecker, B.S.; Johnson, C.M.; Nikolova, P.V.; Proctor, M.R.; Lane, D.P.; Fersht, A.R. Thermodynamic Stability of Wild-Type and Mutant P53 Core Domain. Proc. Natl. Acad. Sci. USA 1997, 94, 14338–14342. [Google Scholar] [CrossRef] [PubMed]
- Kanjilal, S.; Pierceall, W.E.; Cummings, K.K.; Kripke, M.L.; Ananthaswamy, H.N. High Frequency of P53 Mutations in Ultraviolet Radiation-Induced Murine Skin Tumors: Evidence for Strand Bias and Tumor Heterogeneity. Cancer Res. 1993, 53, 2961–2964. [Google Scholar]
- Van Kranen, H.J.; de Gruijl, F.R.; de Vries, A.; Sontag, Y.; Wester, P.W.; Senden, H.C.; Rozemuller, E.; van Kreijl, C.F. Frequent P53 Alterations but Low Incidence of Ras Mutations in UV-B-Induced Skin Tumors of Hairless Mice. Carcinogenesis 1995, 16, 1141–1147. [Google Scholar] [CrossRef]
- You, Y.H.; Szabó, P.E.; Pfeifer, G.P. Cyclobutane Pyrimidine Dimers Form Preferentially at the Major P53 Mutational Hotspot in UVB-Induced Mouse Skin Tumors. Carcinogenesis 2000, 21, 2113–2117. [Google Scholar] [CrossRef]
- Katiyar, S.K.; Matsui, M.S.; Mukhtar, H. Kinetics of UV Light-Induced Cyclobutane Pyrimidine Dimers in Human Skin in Vivo: An Immunohistochemical Analysis of Both Epidermis and Dermis. Photochem. Photobiol. 2000, 72, 788–793. [Google Scholar] [CrossRef]
- Berardesca, E.; Bertona, M.; Altabas, K.; Altabas, V.; Emanuele, E. Reduced Ultraviolet-Induced DNA Damage and Apoptosis in Human Skin with Topical Application of a Photolyase-Containing DNA Repair Enzyme Cream: Clues to Skin Cancer Prevention. Mol. Med. Rep. 2012, 5, 570–574. [Google Scholar] [CrossRef]
- Hu, J.; Adar, S.; Selby, C.P.; Lieb, J.D.; Sancar, A. Genome-Wide Analysis of Human Global and Transcription-Coupled Excision Repair of UV Damage at Single-Nucleotide Resolution. Genes Dev. 2015, 29, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Razzaque, M.S.; Koji, T.; Taguchi, T.; Harada, T.; Nakane, P.K. In Situ Localization of Type III and Type IV Collagen-Expressing Cells in Human Diabetic Nephropathy. J. Pathol. 1994, 174, 131–138. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Grade | SQ Definition | SI Definition | ST Definition (SQ + SI) |
|---|---|---|---|
| 0 | Negative for TD. | Negative for TD. | Negative for TD. |
| +1 | Rare positive nuclei (1–10% of total tumor cells), in a 400× field. | Weak immunoreaction, visibly less intense than external control | Cannot occur. |
| +2 | Positive nuclei in 11% to 25% of the total tumor cell population, in a 400× field. | Moderate immunoreaction, similar or lesser intensity to external control skin TD staining | 1 + 1 |
| +3 | Positive nuclei in >26% of the total tumor cell population, in a 400× field. | Strong immunoreaction, visibly as or more intense than external control skin TD staining | 1 + 2 or 2 + 1 |
| +4 | 1 + 3 or 2 + 2 or 3 + 1 | ||
| +5 | 2 + 3 or 3 + 2 | ||
| +6 | 3 + 3 | ||
| Variables | TD Positive Tumor Tissue (n = 42) | TD Negative Tumor Tissue (n = 12) | p-Value |
|---|---|---|---|
| Age, years (mean ± SD) | 65.74 ± 7.201 | 67.50 ± 10.113 | 0.499 |
| Sex | 0.402 | ||
| Men | 26 (48.15%) | 9 (16.65%) | |
| Women | 16 (29.65%) | 3 (5.55%) | |
| Histology (HP) | 0.703 | ||
| ccRCC | 30 (55.55%) | 9 (16.65%) | |
| pRCC | 6 (11.1%) | 2 (3.7%) | |
| chRCC | 4 (7.4%) | 0 (0%) | |
| svRCC | 2 (3.7%) | 1 (1.85%) | |
| Local extension (pT) | 0.189 | ||
| 1A | 15 (27.8%) | 2 (3.7%) | |
| 1B | 12 (22.2%) | 1 (1.85%) | |
| 2A | 7 (12.95%) | 5 (9.25%) | |
| 2B | 4 (7.4%) | 2 (3.7%) | |
| 3A | 4 (7.4%) | 2 (3.7%) | |
| Fuhrman score | 0.793 | ||
| 1 | 12 (22.2%) | 3 (5.55%) | |
| 2 | 19 (35.2%) | 7 (12.95%) | |
| 3 | 8 (14.8%) | 1 (1.85%) | |
| 4 | 3 (5.55%) | 1 (1.85%) | |
| Lymph nodes (pN) | 0.665 | ||
| Yes | 5 (9.25%) | 2 (3.7%) | |
| No | 37 (68.5%) | 10 (18.5%) | |
| Distant metastasis | 0.634 | ||
| Yes | 2 (3.7%) | 1 (1.85%) | |
| No | 40 (74%) | 11 (20.35%) | |
| Stage at diagnosis | 0.055 | ||
| 1 | 29 (53.7%) | 3 (5.55%) | |
| 2 | 7 (12.95%) | 5 (9.25%) | |
| 3 | 4 (7.4%) | 3 (5.55%) | |
| 4 | 2 (3.7%) | 1 (1.85%) | |
| TD positive adjacent healthy renal tissue | 0.098 | ||
| Yes | 18 (33.35%) | 2 (3.7%) | |
| No | 24 (44.4%) | 10 (18.5%) |
| Variables | TD-Positive Tumor Tissue (n = 42) |
|---|---|
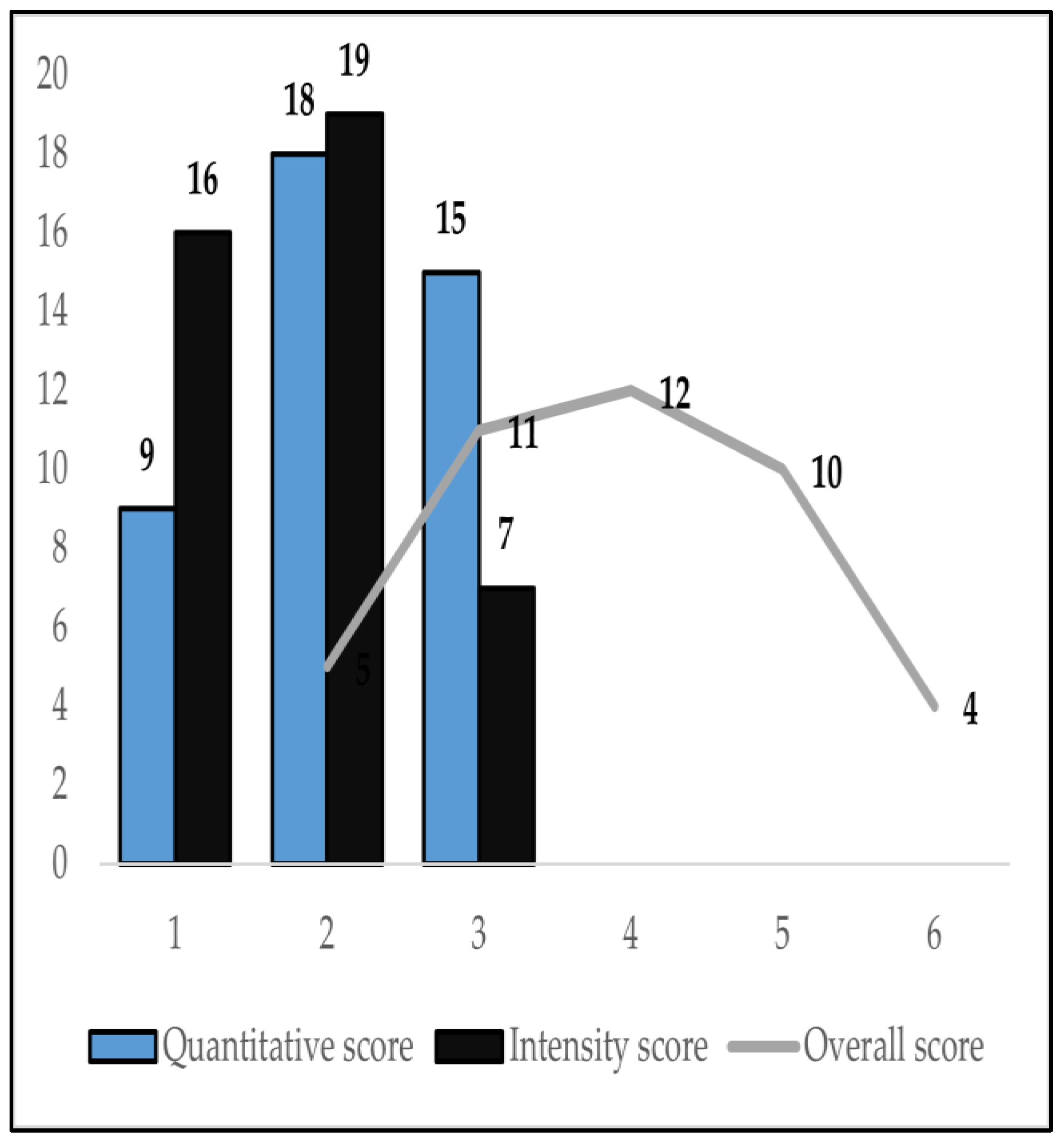
| Quantitative score | |
| 1 | 9 (21.4%) |
| 2 | 18 (42.8%) |
| 3 | 15 (35.7%) |
| Intensity score | |
| 1 | 16 (38.1%) |
| 2 | 19 (45.2%) |
| 3 | 7 (16.7%) |
| Overall expression score | |
| 2 | 5 (11.9%) |
| 3 | 11 (26.2%) |
| 4 | 12 (28.6%) |
| 5 | 10 (23.8%) |
| 6 | 4 (9.5%) |
| Age | Sex | RS | cN | Stage | FG | HKE | HSE | TD+/− | SQ | SI | ST | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | Rho | 1 | −0.234 | −0.420 ** | −0.149 | −0.159 | −0.061 | −0.235 | 0.101 | −0.136 | 0.007 | −0.114 | −0.040 |
| p−value | - | 0.088 | 0.002 | 0.283 | 0.251 | 0.662 | 0.087 | 0.465 | 0.327 | 0.961 | 0.410 | 0.773 | |
| Sex | Rho | −0.234 | 1 | 0.242 | 0.177 | 0.104 | 0.046 | 0.238 | 0.186 | 0.114 | −0.043 | 0.248 | 0.100 |
| p−value | 0.088 | - | 0.078 | 0.199 | 0.453 | 0.744 | 0.083 | 0.177 | 0.412 | 0.759 | 0.071 | 0.471 | |
| RS | Rho | −0.420 ** | 0.242 | 1 | 0.420 ** | 0.261 | 0.159 | 0.245 | −0.084 | 0.036 | −0.041 | −0.115 | −0.102 |
| p−value | 0.002 | 0.078 | - | 0.002 | 0.056 | 0.250 | 0.074 | 0.546 | 0.794 | 0.769 | 0.407 | 0.461 | |
| cN | Rho | −0.149 | 0.177 | 0.420 ** | 1 | 0.615 ** | 0.356 ** | 0.161 | 0.356 ** | −0.059 | −0.088 | 0.022 | −0.031 |
| p−value | 0.283 | 0.199 | 0.002 | - | 0.000 | 0.008 | 0.246 | 0.008 | 0.672 | 0.526 | 0.874 | 0.826 | |
| Stage | Rho | 0.251 | 0.104 | 0.261 | 0.615 ** | 1 | 0.295 * | 0.099 | 0.205 | −0.353 ** | −0.212 | −0.195 | −0.170 |
| p−value | 0.540 | 0.453 | 0.056 | 0.000 | - | 0.031 | 0.476 | 0.138 | 0.009 | 0.123 | 0.157 | 0.220 | |
| FG | Rho | −0.061 | 0.046 | 0.159 | 0.356 ** | 0.295 * | 1 | 0.111 | 0.176 | 0.025 | 0.064 | 0.109 | 0.068 |
| p−value | 0.662 | 0.744 | 0.250 | 0.008 | 0.031 | - | 0.423 | 0.204 | 0.860 | 0.644 | 0.433 | 0.625 | |
| HKE | Rho | −0.235 | 0.238 | 0.245 | 0.161 | 0.099 | 0.111 | 1 | 0.179 | 0.225 | 0.341 * | 0.353 ** | 0.394 ** |
| p−value | 0.087 | 0.083 | 0.074 | 0.246 | 0.476 | 0.423 | - | 0.195 | 0.101 | 0.012 | 0.009 | 0.003 | |
| HSE | Rho | 0.101 | 0.186 | −0.084 | 0.356 ** | 0.205 | 0.176 | 0.179 | 1 | 0.073 | 0.179 | 0.216 | 0.225 |
| p−value | 0.465 | 0.177 | 0.546 | 0.008 | 0.138 | 0.204 | 0.195 | - | 0.598 | 0.196 | 0.116 | 0.103 | |
| TD+/− | Rho | −0.136 | 0.114 | 0.036 | −0.059 | −0.353 ** | 0.025 | 0.225 | 0.073 | 1 | 0.748 ** | 0.752 ** | 0.734 ** |
| p−value | 0.327 | 0.412 | 0.794 | 0.672 | 0.009 | 0.860 | 0.101 | 0.598 | - | 0.000 | 0.000 | 0.000 | |
| SQ | Rho | 0.007 | −0.043 | −0.041 | −0.088 | −0.212 | 0.064 | 0.341 * | 0.179 | 0.748 ** | 1 | 0.692 ** | 0.917 ** |
| p−value | 0.961 | 0.759 | 0.769 | 0.526 | 0.123 | 0.644 | 0.012 | 0.196 | 0.000 | - | 0.000 | 0.000 | |
| SI | Rho | −0.114 | 0.248 | −0.115 | 0.022 | −0.195 | 0.109 | 0.353 ** | 0.216 | 0.752 ** | 0.692 ** | 1 | 0.905 ** |
| p−value | 0.410 | 0.071 | 0.407 | 0.874 | 0.157 | 0.433 | 0.009 | 0.116 | 0.000 | 0.000 | - | 0.000 | |
| ST | Rho | −0.040 | 0.100 | −0.102 | −0.031 | −0.170 | 0.112 | 0.394 ** | 0.225 | 0.734 ** | 0.917 ** | 0.905 ** | 1 |
| p−value | 0.773 | 0.471 | 0.461 | 0.826 | 0.220 | 0.421 | 0.003 | 0.103 | 0.000 | 0.000 | 0.000 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novacescu, D.; Cut, T.G.; Cumpanas, A.A.; Bratosin, F.; Ceausu, R.A.; Raica, M. Novel Expression of Thymine Dimers in Renal Cell Carcinoma, Demonstrated through Immunohistochemistry. Biomedicines 2022, 10, 2673. https://doi.org/10.3390/biomedicines10112673
Novacescu D, Cut TG, Cumpanas AA, Bratosin F, Ceausu RA, Raica M. Novel Expression of Thymine Dimers in Renal Cell Carcinoma, Demonstrated through Immunohistochemistry. Biomedicines. 2022; 10(11):2673. https://doi.org/10.3390/biomedicines10112673
Chicago/Turabian StyleNovacescu, Dorin, Talida Georgiana Cut, Alin Adrian Cumpanas, Felix Bratosin, Raluca Amalia Ceausu, and Marius Raica. 2022. "Novel Expression of Thymine Dimers in Renal Cell Carcinoma, Demonstrated through Immunohistochemistry" Biomedicines 10, no. 11: 2673. https://doi.org/10.3390/biomedicines10112673
APA StyleNovacescu, D., Cut, T. G., Cumpanas, A. A., Bratosin, F., Ceausu, R. A., & Raica, M. (2022). Novel Expression of Thymine Dimers in Renal Cell Carcinoma, Demonstrated through Immunohistochemistry. Biomedicines, 10(11), 2673. https://doi.org/10.3390/biomedicines10112673