
mRNA Therapeutic Modalities Design, Formulation and Manufacturing under Pharma 4.0 Principles

 , , ,
, , ,  and
and 
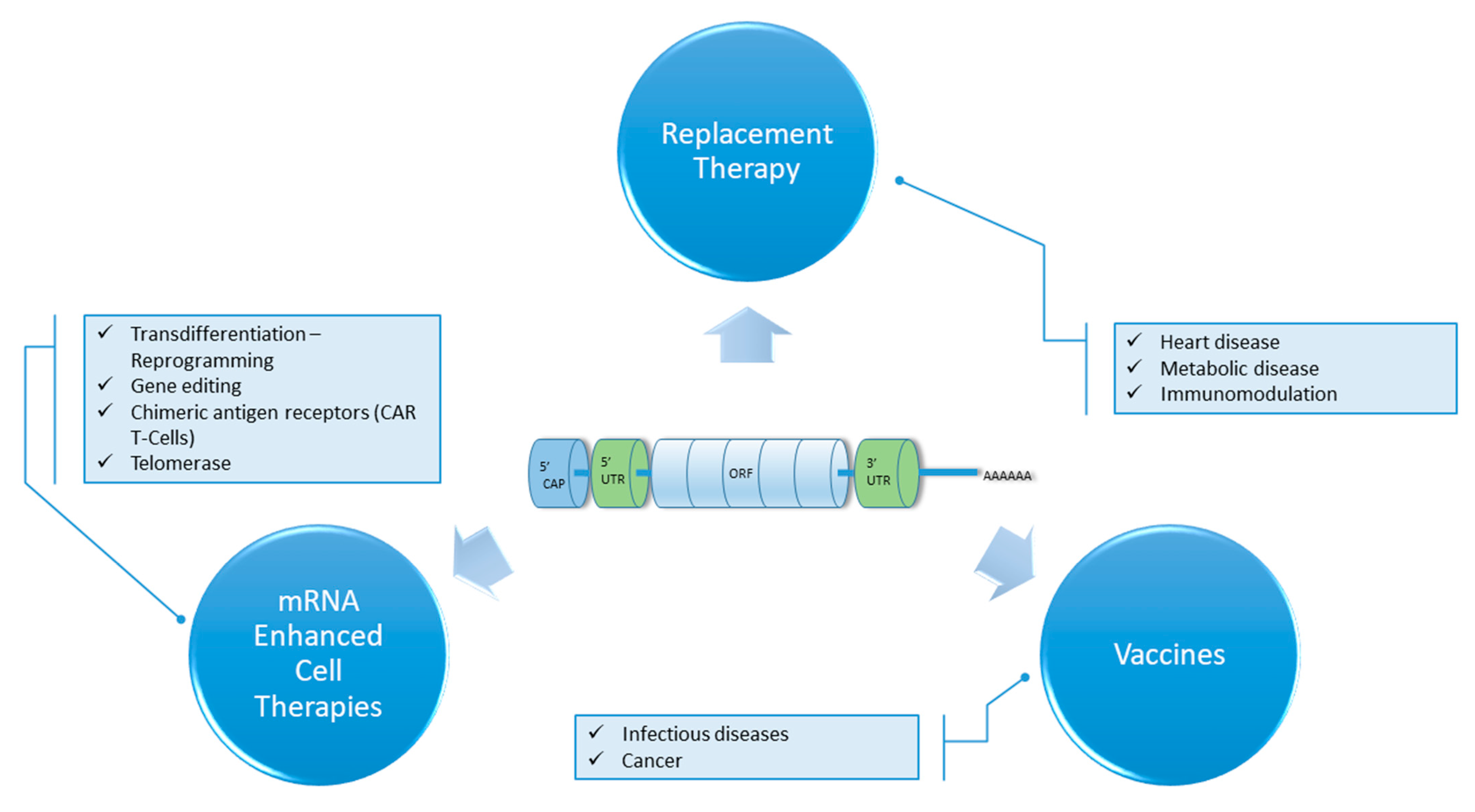
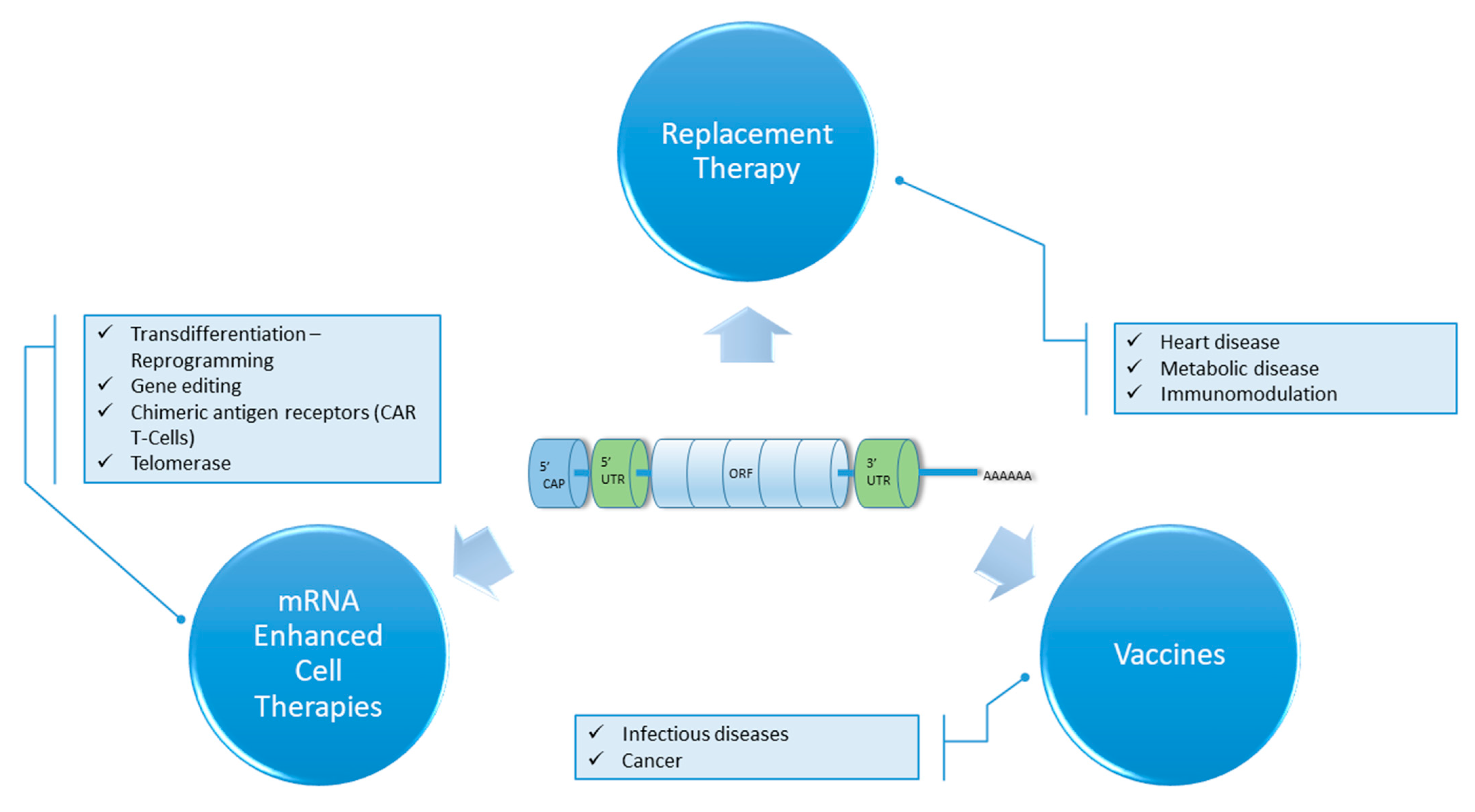{kind=link}
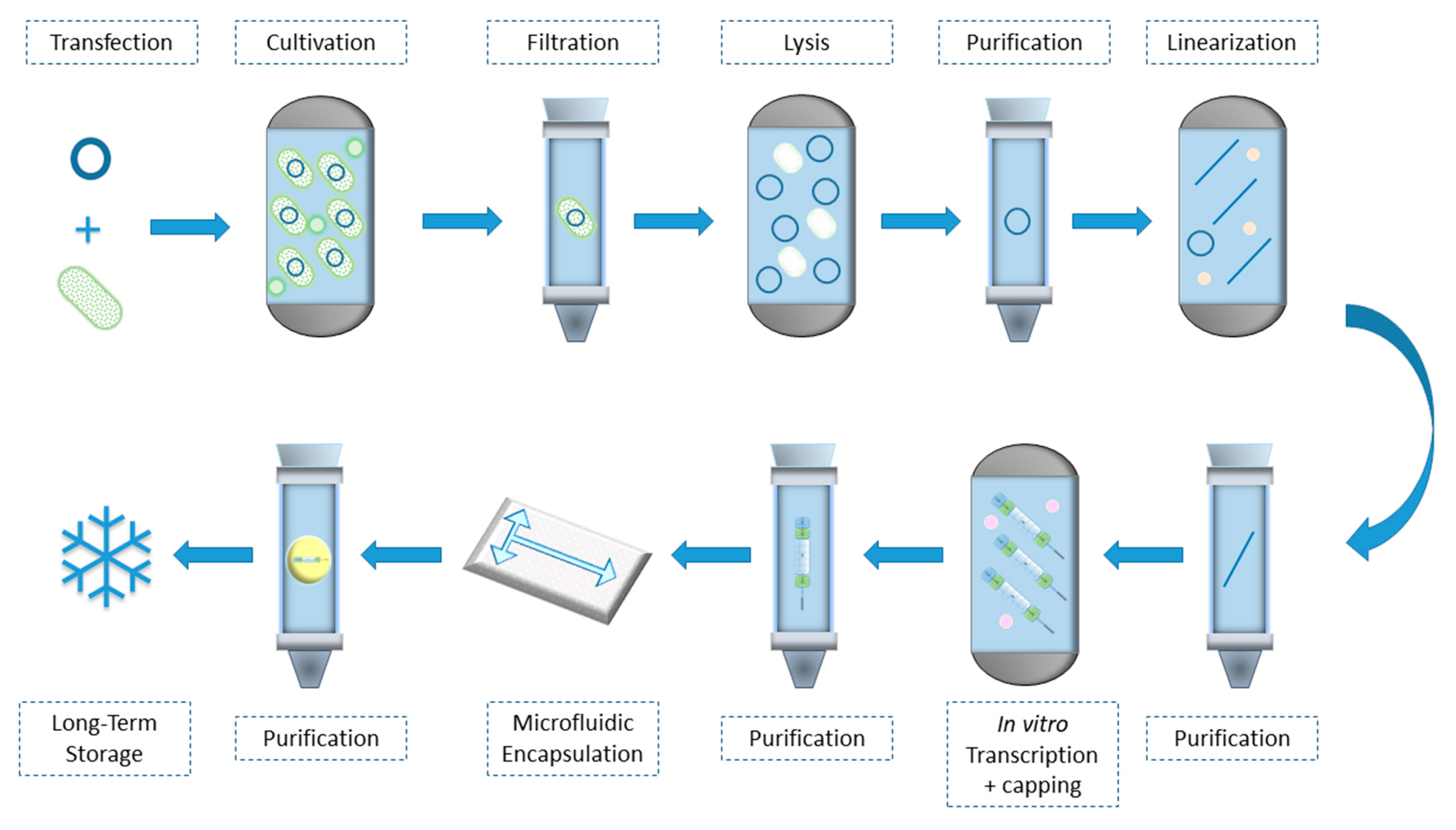{kind=link}
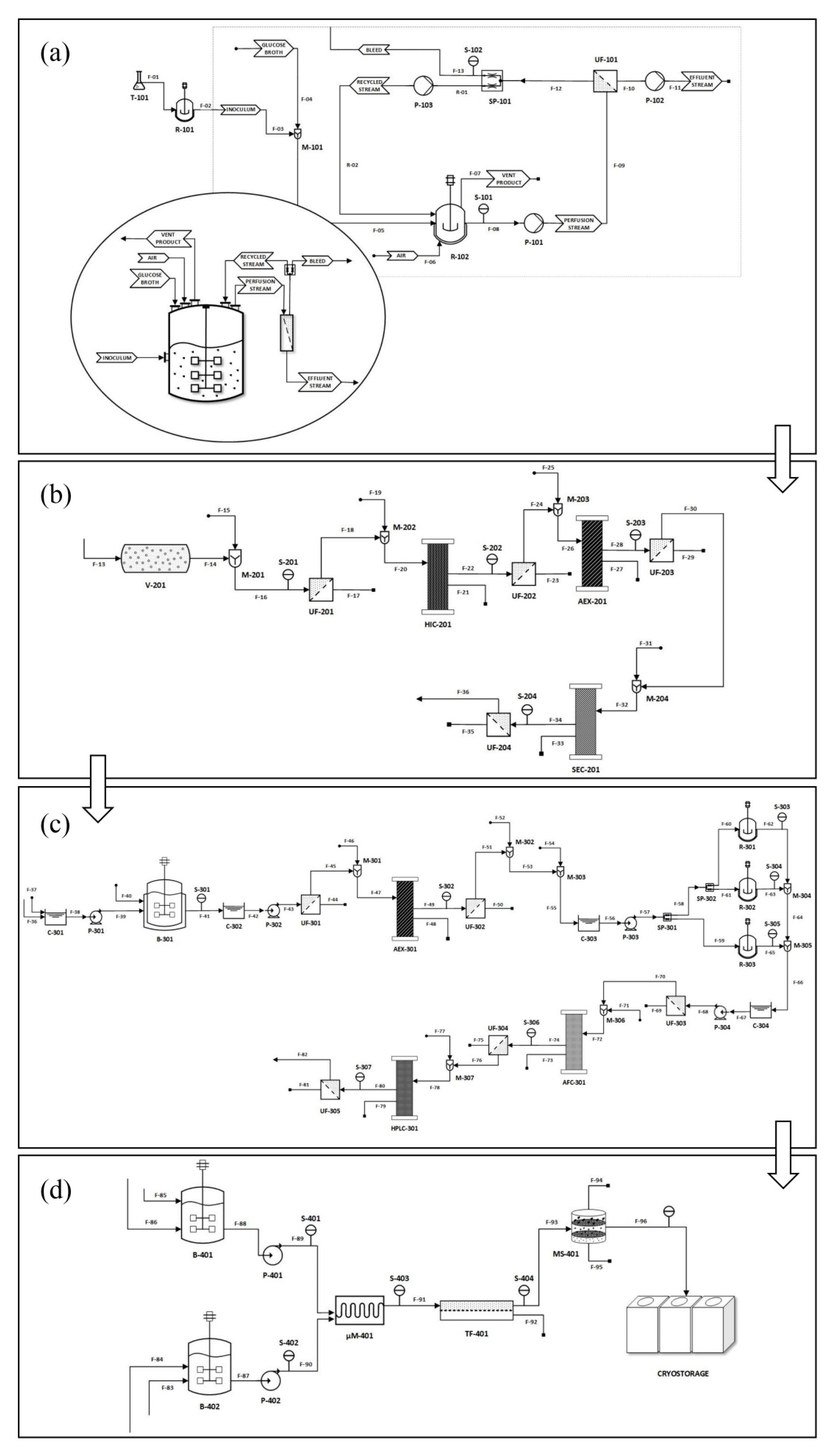{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Basic Features of Moderna mRNA-1273 and Pfizer-BioNTech BNT162b2 COVID-19 mRNA Vaccines
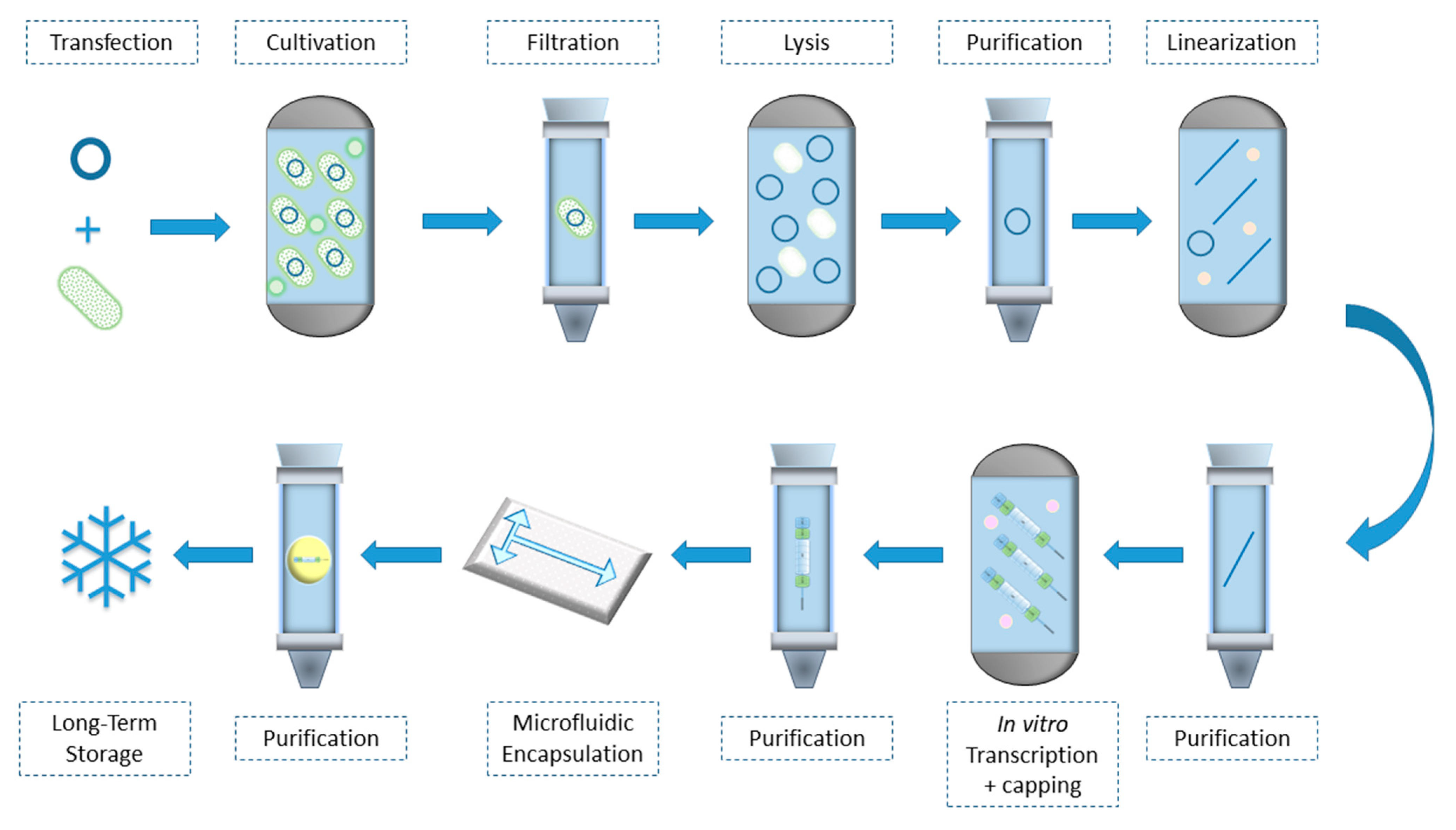
3. mRNA Preparation Process
3.1. Plasmid Linearization
3.2. In-Vitro mRNA Transcription
3.3. mRNA Purification
3.4. mRNA Encapsulation
4. Analytical Approaches
4.1. For mRNA
4.2. For the LPNs of mRNA
4.3. For DNA Plasmid Purification
4.4. For DNA Analysis
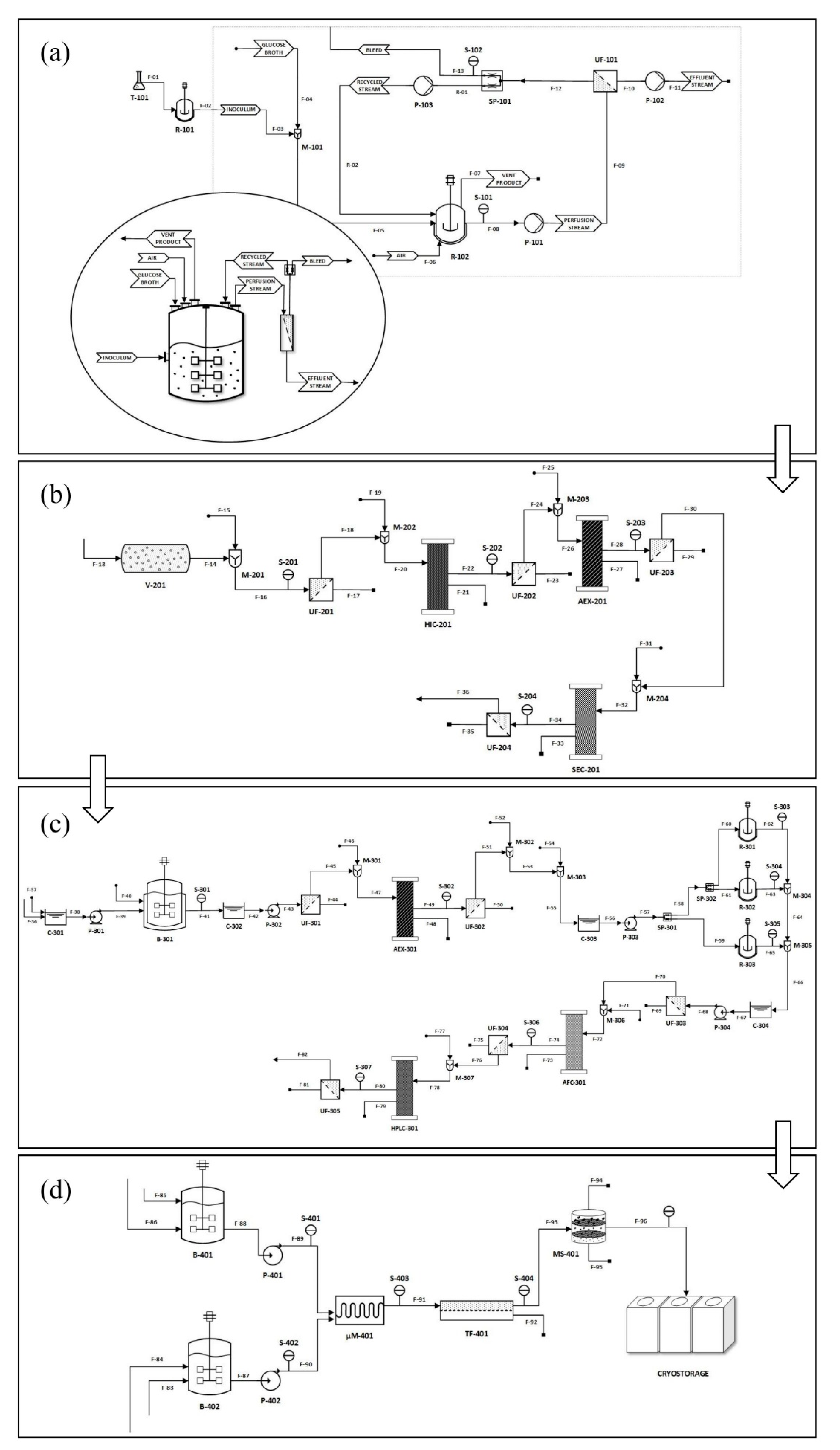
5. Differences between Laboratory and Industrial Scale
6. Formulation Strategies
6.1. Viral Vectors
6.2. Non-Viral Vectors
6.2.1. Polymer-Based Vectors
6.2.2. Lipid Based Vectors
6.2.3. Polymer-Lipid Hybrid Vectors
6.2.4. Other Vectors
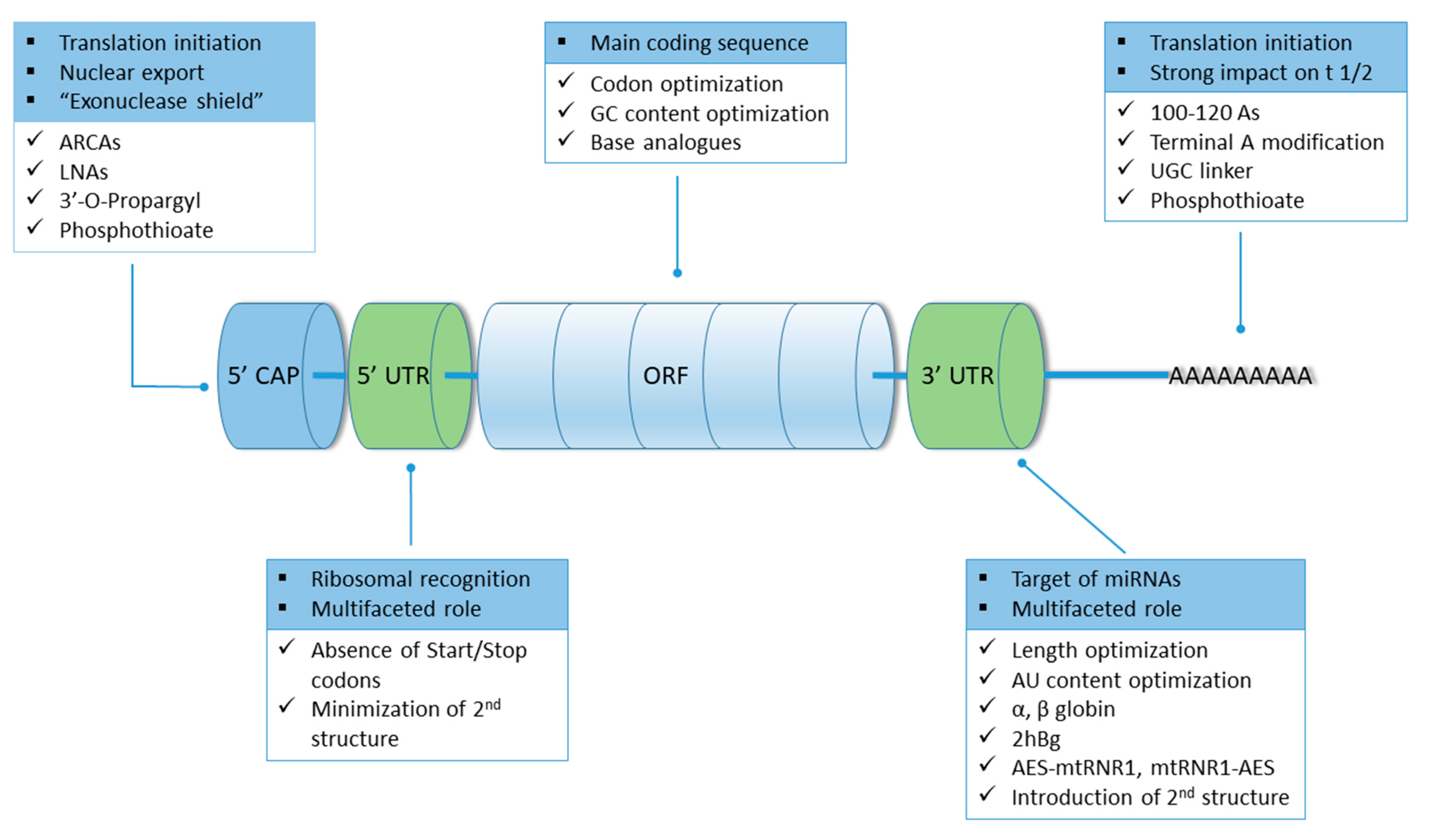
7. mRNA Stability and Modifications
7.1. Structural Modifications
7.1.1. 5′ Cap
7.1.2. 5′ and 3′ UTRs
7.1.3. Open Reading Frame
7.1.4. Poly(A) Tail
8. Storage Considerations
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef]
- Gupta, A.; Andresen, J.L.; Manan, R.S.; Langer, R. Nucleic Acid Delivery for Therapeutic Applications. Adv. Drug Deliv. Rev. 2021, 178, 113834. [Google Scholar] [CrossRef]
- Wayment-Steele, H.K.; Kim, D.S.; Choe, C.A.; Nicol, J.J.; Wellington-Oguri, R.; Watkins, A.M.; Parra Sperberg, R.A.; Huang, P.-S.; Participants, E.; Das, R. Theoretical Basis for Stabilizing Messenger RNA through Secondary Structure Design. Nucleic Acids Res. 2021, 49, 10604–10617. [Google Scholar] [CrossRef]
- Ouranidis, A.; Choli-Papadopoulou, T.; Papachristou, E.T.; Papi, R.; Kostomitsopoulos, N. Biopharmaceutics 4.0, Advanced Pre-Clinical Development of mRNA-Encoded Monoclonal Antibodies to Immunosuppressed Murine Models. Vaccines 2021, 9, 890. [Google Scholar] [CrossRef] [PubMed]
- Soundara Rajan, T.; Gugliandolo, A.; Bramanti, P.; Mazzon, E. In Vitro-Transcribed MRNA Chimeric Antigen Receptor T Cell (IVT MRNA CAR T) Therapy in Hematologic and Solid Tumor Management: A Preclinical Update. Int. J. Mol. Sci. 2020, 21, 6514. [Google Scholar] [CrossRef]
- Parayath, N.N.; Stephan, S.B.; Koehne, A.L.; Nelson, P.S.; Stephan, M.T. In Vitro-Transcribed Antigen Receptor MRNA Nanocarriers for Transient Expression in Circulating T Cells in Vivo. Nat. Commun. 2020, 11, 6080. [Google Scholar] [CrossRef]
- Caruso, H.G.; Torikai, H.; Zhang, L.; Maiti, S.; Dai, J.; Do, K.-A.; Singh, H.; Huls, H.; Lee, D.A.; Champlin, R.E.; et al. Redirecting T-Cell Specificity to EGFR Using MRNA to Self-Limit Expression of Chimeric Antigen Receptor. J. Immunother. 2016, 39, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, M.; Lanitis, E.; Migliorini, D.; Ivics, Z.; Guedan, S. Choosing the Right Tool for Genetic Engineering: Clinical Lessons from Chimeric Antigen Receptor-T Cells. Hum. Gene Ther. 2021, 32, 1044–1058. [Google Scholar] [CrossRef]
- Sharp, P.A. The Centrality of RNA. Cell 2009, 136, 577–580. [Google Scholar] [CrossRef] [Green Version]
- Midoux, P.; Pichon, C. Lipid-Based MRNA Vaccine Delivery Systems. Expert Rev Vaccines 2015, 14, 221–234. [Google Scholar] [CrossRef] [Green Version]
- Avci-Adali, M.; Behring, A.; Steinle, H.; Keller, T.; Krajeweski, S.; Schlensak, C.; Wendel, H.P. In Vitro Synthesis of Modified MRNA for Induction of Protein Expression in Human Cells. J. Vis. Exp. 2014, 93, e51943. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Ryals, R.C.; Weller, K.K.; Pennesi, M.E.; Sahay, G. Lipid Nanoparticles for Delivery of Messenger RNA to the Back of the Eye. J. Control. Release 2019, 303, 91–100. [Google Scholar] [CrossRef]
- Matsui, A.; Uchida, S.; Ishii, T.; Itaka, K.; Kataoka, K. Messenger RNA-Based Therapeutics for the Treatment of Apoptosis-Associated Diseases. Sci. Rep. 2015, 5, 15810. [Google Scholar] [CrossRef]
- Poveda, C.; Biter, A.B.; Bottazzi, M.E.; Strych, U. Establishing Preferred Product Characterization for the Evaluation of RNA Vaccine Antigens. Vaccines 2019, 7, 131. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.; Kessler, J.A. Design, Assembly, Production, and Transfection of Synthetic Modified MRNA. Methods 2018, 133, 29–43. [Google Scholar] [CrossRef]
- Yu, A.-M.; Tu, M.-J. Deliver the Promise: RNAs as a New Class of Molecular Entities for Therapy and Vaccination. Pharmacol. Ther. 2021, 107967. [Google Scholar] [CrossRef]
- Yamamoto, A.; Kormann, M.; Rosenecker, J.; Rudolph, C. Current Prospects for MRNA Gene Delivery. Eur. J. Pharm. Biopharm. 2009, 71, 484–489. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Bhattacharya, M.; Lee, S.-S. From COVID-19 to Cancer MRNA Vaccines: Moving from Bench to Clinic in the Vaccine Landscape. Front. Immunol. 2021, 12, 679344. [Google Scholar] [CrossRef]
- Rosa, S.S.; Prazeres, D.M.F.; Azevedo, A.M.; Marques, M.P.C. MRNA Vaccines Manufacturing: Challenges and Bottlenecks. Vaccine 2021, 39, 2190–2200. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. MRNA Vaccines—A New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Milane, L.; Amiji, M. Clinical Approval of Nanotechnology-Based SARS-CoV-2 MRNA Vaccines: Impact on Translational Nanomedicine. Drug Deliv. Transl. Res. 2021, 11, 1309–1315. [Google Scholar] [CrossRef]
- Ho, W.; Gao, M.; Li, F.; Li, Z.; Zhang, X.-Q.; Xu, X. Next-Generation Vaccines: Nanoparticle-Mediated DNA and MRNA Delivery. Adv. Healthc. Mater. 2021, 10, e2001812. [Google Scholar] [CrossRef]
- Aldrich, C.; Leroux-Roels, I.; Huang, K.B.; Bica, M.A.; Loeliger, E.; Schoenborn-Kellenberger, O.; Walz, L.; Leroux-Roels, G.; von Sonnenburg, F.; Oostvogels, L. Proof-of-Concept of a Low-Dose Unmodified MRNA-Based Rabies Vaccine Formulated with Lipid Nanoparticles in Human Volunteers: A Phase 1 Trial. Vaccine 2021, 39, 1310–1318. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the Delivery of RNA Therapeutics: From Concept to Clinical Reality. Genome Med. 2017, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Jackson, N.A.C.; Kester, K.E.; Casimiro, D.; Gurunathan, S.; DeRosa, F. The Promise of MRNA Vaccines: A Biotech and Industrial Perspective. NPJ Vaccines 2020, 5, 11. [Google Scholar] [CrossRef]
- Chu, L.; McPhee, R.; Huang, W.; Bennett, H.; Pajon, R.; Nestorova, B.; Leav, B.; mRNA-1273 Study Group. A Preliminary Report of a Randomized Controlled Phase 2 Trial of the Safety and Immunogenicity of MRNA-1273 SARS-CoV-2 Vaccine. Vaccine 2021, 39, 2791–2799. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Frenck, R.W.; Walsh, E.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Bailey, R.; Swanson, K.A.; Xu, X.; et al. SARS-CoV-2 Neutralization with BNT162b2 Vaccine Dose 3. N. Engl. J. Med. 2021, 385, 1627–1629. [Google Scholar] [CrossRef]
- Sahin, U.; Muik, A.; Vogler, I.; Derhovanessian, E.; Kranz, L.M.; Vormehr, M.; Quandt, J.; Bidmon, N.; Ulges, A.; Baum, A.; et al. BNT162b2 Vaccine Induces Neutralizing Antibodies and Poly-Specific T Cells in Humans. Nature 2021, 595, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Frenck, R.W.; Klein, N.P.; Kitchin, N.; Gurtman, A.; Absalon, J.; Lockhart, S.; Perez, J.L.; Walter, E.B.; Senders, S.; Bailey, R.; et al. Safety, Immunogenicity, and Efficacy of the BNT162b2 COVID-19 Vaccine in Adolescents. N. Engl. J. Med. 2021, 385, 239–250. [Google Scholar] [CrossRef]
- Choi, A.; Koch, M.; Wu, K.; Dixon, G.; Oestreicher, J.; Legault, H.; Stewart-Jones, G.B.E.; Colpitts, T.; Pajon, R.; Bennett, H.; et al. Serum Neutralizing Activity of MRNA-1273 against SARS-CoV-2 Variants. J. Virol. 2021, 95, e0131321. [Google Scholar] [CrossRef]
- Choi, A.; Koch, M.; Wu, K.; Chu, L.; Ma, L.; Hill, A.; Nunna, N.; Huang, W.; Oestreicher, J.; Colpitts, T.; et al. Safety and Immunogenicity of SARS-CoV-2 Variant MRNA Vaccine Boosters in Healthy Adults: An Interim Analysis. Nat. Med. 2021, 27, 2025–2031. [Google Scholar] [CrossRef]
- El Sahly, H.M.; Baden, L.R.; Essink, B.; Doblecki-Lewis, S.; Martin, J.M.; Anderson, E.J.; Campbell, T.B.; Clark, J.; Jackson, L.A.; Fichtenbaum, C.J.; et al. Efficacy of the MRNA-1273 SARS-CoV-2 Vaccine at Completion of Blinded Phase. N. Engl. J. Med. 2021, 385, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Lonza News. Available online: https://www.lonza.com/news/2020-05-01-04-50 (accessed on 2 November 2021).
- Rele, S. COVID-19 Vaccine Development during Pandemic: Gap Analysis, Opportunities, and Impact on Future Emerging Infectious Disease Development Strategies. Hum. Vaccin. Immunother. 2021, 17, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, I.; Liu, M.A.; Peden, K.; Zhou, T.; Kang, H.-N. Development of MRNA Vaccines: Scientific and Regulatory Issues. Vaccines 2021, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.K.; King, C.R.; Feldman, S.R. Biologics and Biosimilars. J. Dermatol. Treat. 2015, 26, 299–302. [Google Scholar] [CrossRef]
- CureVac Streamlines European Network for MRNA Product Manufacturing. 2021. Available online: https://www.curevac.com/en/2021/09/14/curevac-streamlines-european-network-for-mrna-product-manufacturing/ (accessed on 16 November 2021).
- Sahin, U.; Karikó, K.; Türeci, Ö. MRNA-Based Therapeutics—Developing a New Class of Drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Kallen, K.-J.; Theß, A. A Development That May Evolve into a Revolution in Medicine: MRNA as the Basis for Novel, Nucleotide-Based Vaccines and Drugs. Ther. Adv. Vaccines 2014, 2, 10–31. [Google Scholar] [CrossRef] [Green Version]
- Ouranidis, A.; Gkampelis, N.; Vardaka, E.; Karagianni, A.; Tsiptsios, D.; Nikolakakis, I.; Kachrimanis, K. Overcoming the Solubility Barrier of Ibuprofen by the Rational Process Design of a Nanocrystal Formulation. Pharmaceutics 2020, 12, 969. [Google Scholar] [CrossRef]
- Ouranidis, A.; Gkampelis, N.; Markopoulou, C.; Nikolakakis, I.; Kachrimanis, K. Development of a Nanocrystal Formulation of a Low Melting Point API Following a Quality by Design Approach. Processes 2021, 9, 954. [Google Scholar] [CrossRef]
- Ouranidis, A.; Tsiaxerli, A.; Vardaka, E.; Markopoulou, C.K.; Zacharis, C.K.; Nicolaou, I.; Hatzichristou, D.; Haidich, A.-B.; Kostomitsopoulos, N.; Kachrimanis, K. Sildenafil 4.0—Integrated Synthetic Chemistry, Formulation and Analytical Strategies Effecting Immense Therapeutic and Societal Impact in the Fourth Industrial Era. Pharmaceuticals 2021, 14, 365. [Google Scholar] [CrossRef]
- Ouranidis, A.; Davidopoulou, C.; Tashi, R.K.; Kachrimanis, K. Pharma 4.0 Continuous MRNA Drug Products Manufacturing. Pharmaceutics 2021, 13, 1371. [Google Scholar] [CrossRef] [PubMed]
- Ouranidis, A.; Davidopoulou, C.; Kachrimanis, K. Integrating Elastic Tensor and Pc-Saft Modeling with Systems-Based Pharma 4.0 Simulation, to Predict Process Operations and Product Specifications of Ternary Nanocrystalline Suspensions. Pharmaceutics 2021, 13, 1771. [Google Scholar] [CrossRef] [PubMed]
- Granados-Riveron, J.T.; Aquino-Jarquin, G. Engineering of the Current Nucleoside-Modified MRNA-LNP Vaccines against SARS-CoV-2. Biomed. Pharmacother. 2021, 142, 111953. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. Detailed Dissection and Critical Evaluation of the Pfizer/BioNTech and Moderna MRNA Vaccines. Vaccines 2021, 9, 734. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 MRNA Vaccine Design Enabled by Prototype Pathogen Preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Jeeva, S.; Kim, K.-H.; Shin, C.H.; Wang, B.-Z.; Kang, S.-M. An Update on MRNA-Based Viral Vaccines. Vaccines 2021, 9, 965. [Google Scholar] [CrossRef]
- Huang, E.Y.-C.; TSE, S.-W.; Iacovelli, J.; Mckinney, K.; Valiante, N. Immunomodulatory Therapeutic Mrna Compositions Encoding Activating Oncogene Mutation Peptides. WO2018144775A1, 9 August 2018. [Google Scholar]
- Nance, K.D.; Meier, J.L. Modifications in an Emergency: The Role of N1-Methylpseudouridine in COVID-19 Vaccines. ACS Cent. Sci. 2021, 7, 748–756. [Google Scholar] [CrossRef]
- Trepotec, Z.; Geiger, J.; Plank, C.; Aneja, M.K.; Rudolph, C. Segmented Poly(A) Tails Significantly Reduce Recombination of Plasmid DNA without Affecting MRNA Translation Efficiency or Half-Life. RNA 2019, 25, 507–518. [Google Scholar] [CrossRef]
- Baronti, L.; Karlsson, H.; Marušič, M.; Petzold, K. A Guide to Large-Scale RNA Sample Preparation. Anal. Bioanal. Chem. 2018, 410, 3239–3252. [Google Scholar] [CrossRef] [Green Version]
- Beckert, B.; Masquida, B. Synthesis of RNA by in Vitro Transcription. Methods Mol. Biol. 2011, 703, 29–41. [Google Scholar] [CrossRef]
- Walker, S.C.; Avis, J.M.; Conn, G.L. General Plasmids for Producing RNA in Vitro Transcripts with Homogeneous Ends. Nucleic Acids Res. 2003, 31, e82. [Google Scholar] [CrossRef]
- Hadas, Y.; Sultana, N.; Youssef, E.; Sharkar, M.T.K.; Kaur, K.; Chepurko, E.; Zangi, L. Optimizing Modified MRNA In Vitro Synthesis Protocol for Heart Gene Therapy. Mol. Ther. Methods Clin. Dev. 2019, 14, 300–305. [Google Scholar] [CrossRef]
- Tusup, M.; French, L.E.; De Matos, M.; Gatfield, D.; Kundig, T.; Pascolo, S. Design of in Vitro Transcribed MRNA Vectors for Research and Therapy. Chimia 2019, 73, 391–394. [Google Scholar] [CrossRef]
- McNamara, M.A.; Nair, S.K.; Holl, E.K. RNA-Based Vaccines in Cancer Immunotherapy. J. Immunol. Res. 2015, 2015, 794528. [Google Scholar] [CrossRef] [Green Version]
- Pascolo, S. Vaccination with Messenger RNA. Methods Mol. Med. 2006, 127, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Stadler, J.; Lemmens, R.; Nyhammar, T. Plasmid DNA Purification. J. Gene Med. 2004, 6 (Suppl. 1), S54–S66. [Google Scholar] [CrossRef]
- Sierra, F. A Laboratory Guide to In Vitro Transcription; Birkhäuser: Basel, Switzerland, 2013; ISBN 978-3-0348-6383-4. [Google Scholar]
- Brunelle, J.L.; Green, R. In Vitro Transcription from Plasmid or PCR-Amplified DNA. Methods Enzymol. 2013, 530, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Y.; Wang, Y.; Shi, C.; Li, C.; Li, Q.; Linhardt, R.J. Bacteriophage T7 Transcription System: An Enabling Tool in Synthetic Biology. Biotechnol. Adv. 2018, 36, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Calvopina-Chavez, D.G.; Gardner, M.A.; Griffitts, J.S. Engineering Efficient Termination of Bacteriophage T7 RNA Polymerase Transcription. bioRxiv 2021, in press. [Google Scholar] [CrossRef]
- Mairhofer, J.; Wittwer, A.; Cserjan-Puschmann, M.; Striedner, G. Preventing T7 RNA Polymerase Read-through Transcription-A Synthetic Termination Signal Capable of Improving Bioprocess Stability. ACS Synth. Biol. 2015, 4, 265–273. [Google Scholar] [CrossRef]
- Duffy, K.; Arangundy-Franklin, S.; Holliger, P. Modified Nucleic Acids: Replication, Evolution, and next-Generation Therapeutics. BMC Biol. 2020, 18, 112. [Google Scholar] [CrossRef]
- Esvelt, K.M.; Carlson, J.C.; Liu, D.R. A System for the Continuous Directed Evolution of Biomolecules. Nature 2011, 472, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Mirer, P.L.; Zaldivar Santamaria, E.; Klapperich, C.; Sharon, A.; Sauer-Budge, A.F. RNA Isolation from Mammalian Cells Using Porous Polymer Monoliths: An Approach for High-Throughput Automation. Anal. Chem. 2010, 82, 4344–4356. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.M.; Costa, D.; Simões, A.R.; Queiroz, J.A.; Sousa, F.; Sousa, Â. Enhancement of a Biotechnological Platform for the Purification and Delivery of a Human Papillomavirus Supercoiled Plasmid DNA Vaccine. N. Biotechnol. 2020, 59, 1–9. [Google Scholar] [CrossRef]
- Chromatographic Purification with CIMmultusTM Oligo DT Increases MRNA Stability. Available online: https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/2216/Chromatographic-purification-with-CIMmultus-Oligo-dT-increases-mRNA-stability (accessed on 22 November 2021).
- Grier, A.E.; Burleigh, S.; Sahni, J.; Clough, C.A.; Cardot, V.; Choe, D.C.; Krutein, M.C.; Rawlings, D.J.; Jensen, M.C.; Scharenberg, A.M.; et al. PEVL: A Linear Plasmid for Generating MRNA IVT Templates with Extended Encoded Poly(A) Sequences. Mol. Ther. Nucleic Acids 2016, 5, e306. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.W.; Cho, W.C.S. An Overview of Rational Design of MRNA-Based Therapeutics and Vaccines. Expert Opin. Drug Discov. 2021, 16, 1307–1317. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid Nanoparticles for MRNA Delivery. Nat. Rev. Mater. 2021, in press. [Google Scholar] [CrossRef]
- Roces, C.B.; Lou, G.; Jain, N.; Abraham, S.; Thomas, A.; Halbert, G.W.; Perrie, Y. Manufacturing Considerations for the Development of Lipid Nanoparticles Using Microfluidics. Pharmaceutics 2020, 12, 1095. [Google Scholar] [CrossRef]
- Kraft, J.C.; Freeling, J.P.; Wang, Z.; Ho, R.J.Y. Emerging Research and Clinical Development Trends of Liposome and Lipid Nanoparticle Drug Delivery Systems. J. Pharm. Sci. 2014, 103, 29–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid Nanoparticles—From Liposomes to MRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.; Fadda, A.M.; Sinico, C. Liposomes for Brain Delivery. Expert Opin. Drug Deliv. 2013, 10, 1003–1022. [Google Scholar] [CrossRef] [PubMed]
- MacLachlan, I. Liposomal Formulations for Nucleic Acid Delivery. In Antisense Drug Technology, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007; ISBN 978-0-429-18934-0. [Google Scholar]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New Developments in Liposomal Drug Delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef]
- Yang, S.; Chen, J.; Zhao, D.; Han, D.; Chen, X. Comparative Study on Preparative Methods of DC-Chol/DOPE Liposomes and Formulation Optimization by Determining Encapsulation Efficiency. Int. J. Pharm. 2012, 434, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef] [Green Version]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. MRNA Vaccine Delivery Using Lipid Nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A.; Vorauer-Uhl, K.; Kreismayr, G.; Katinger, H. The Crossflow Injection Technique: An Improvement of the Ethanol Injection Method. J. Liposome Res. 2002, 12, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dong, Y. Preparation and Optimization of Lipid-Like Nanoparticles for mRNA Delivery. Methods Mol. Biol. 2017, 1632, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Guevara, M.L.; Persano, F.; Persano, S. Advances in Lipid Nanoparticles for MRNA-Based Cancer Immunotherapy. Front. Chem. 2020, 8, 589959. [Google Scholar] [CrossRef]
- Crommelin, D.J.A.; Anchordoquy, T.J.; Volkin, D.B.; Jiskoot, W.; Mastrobattista, E. Addressing the Cold Reality of MRNA Vaccine Stability. J. Pharm. Sci. 2020, 110, 997–1001. [Google Scholar] [CrossRef]
- Packer, M.; Gyawali, D.; Yerabolu, R.; Schariter, J.; White, P. A Novel Mechanism for the Loss of MRNA Activity in Lipid Nanoparticle Delivery Systems. Nat. Commun. 2021, 12, 6777. [Google Scholar] [CrossRef] [PubMed]
- Demelenne, A.; Servais, A.-C.; Crommen, J.; Fillet, M. Analytical Techniques Currently Used in the Pharmaceutical Industry for the Quality Control of RNA-Based Therapeutics and Ongoing Developments. J. Chromatogr. A 2021, 1651, 462283. [Google Scholar] [CrossRef]
- Beverly, M.; Dell, A.; Parmar, P.; Houghton, L. Label-Free Analysis of MRNA Capping Efficiency Using RNase H Probes and LC-MS. Anal. Bioanal. Chem. 2016, 408, 5021–5030. [Google Scholar] [CrossRef]
- Beverly, M.; Hagen, C.; Slack, O. Poly A Tail Length Analysis of in Vitro Transcribed MRNA by LC-MS. Anal. Bioanal. Chem. 2018, 410, 1667–1677. [Google Scholar] [CrossRef]
- DeLano, M.; Walter, T.H.; Lauber, M.A.; Gilar, M.; Jung, M.C.; Nguyen, J.M.; Boissel, C.; Patel, A.V.; Bates-Harrison, A.; Wyndham, K.D. Using Hybrid Organic—Inorganic Surface Technology to Mitigate Analyte Interactions with Metal Surfaces in UHPLC. Anal. Chem. 2021, 93, 5773–5781. [Google Scholar] [CrossRef]
- Muthmann, N.; Špaček, P.; Reichert, D.; van Dülmen, M.; Rentmeister, A. Quantification of MRNA Cap-Modifications by Means of LC-QqQ-MS. Methods 2021, in press. [Google Scholar] [CrossRef]
- Nair, L.M.; Werling, J.O. Aerosol Based Detectors for the Investigation of Phospholipid Hydrolysis in a Pharmaceutical Suspension Formulation. J. Pharm. Biomed. Anal. 2009, 49, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Ramos, R.G.; Libong, D.; Rakotomanga, M.; Gaudin, K.; Loiseau, P.M.; Chaminade, P. Comparison between Charged Aerosol Detection and Light Scattering Detection for the Analysis of Leishmania Membrane Phospholipids. J. Chromatogr. A 2008, 1209, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Merle, C.; Laugel, C.; Chaminade, P.; Baillet-Guffroy, A. Quantitative Study of the Stratum Corneum Lipid Classes by Normal Phase Liquid Chromatography: Comparison Between Two Universal Detectors. J. Liq. Chromatogr. Relat. Technol. 2010, 33, 629–644. [Google Scholar] [CrossRef]
- ACQUITY PREMIER LC Technology Significantly Improves Sensitivity, Peak Shape and Recovery for Phosphorylated and Carboxylate Lipids. Available online: https://www.waters.com/nextgen/xg/fr/library/application-notes/2021/acquity-premier-lc-technology-significantly-improves-sensitivity-peak-shape-and-recovery-for-phosphorylated-and-carboxylate-lipids.html (accessed on 22 November 2021).
- Waters. Charged Surface Hybrid (CSH) Technology and Its Use in Liquid Chromatography. Available online: http://www.waters.com/waters/library.htm?lid=10167251 (accessed on 22 November 2021).
- Ghanem, A.; Healey, R.; Adly, F.G. Current Trends in Separation of Plasmid DNA Vaccines: A Review. Anal. Chim. Acta 2013, 760, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zöchling, A.; Hahn, R.; Ahrer, K.; Urthaler, J.; Jungbauer, A. Mass Transfer Characteristics of Plasmids in Monoliths. J. Sep. Sci. 2004, 27, 819–827. [Google Scholar] [CrossRef]
- Mota, É.; Sousa, Â.; Černigoj, U.; Queiroz, J.A.; Tomaz, C.T.; Sousa, F. Rapid Quantification of Supercoiled Plasmid Deoxyribonucleic Acid Using a Monolithic Ion Exchanger. J. Chromatogr. A 2013, 1291, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.; Proença, Z.; Queiroz, J.A.; Tomaz, C.; Cruz, C. Plasmid Purification by Using a New Naphthalene Tripodal Support. Sep. Purif. Technol. 2017, 188, 81–89. [Google Scholar] [CrossRef]
- Silva-Santos, A.R.; Alves, C.P.A.; Prazeres, D.M.F.; Azevedo, A.M. Separation of Plasmid DNA Topoisomers by Multimodal Chromatography. Anal. Biochem. 2016, 503, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Yu, X.; Yin, Y.; Liu, X.; Wu, Y.; Chen, Y.; Zhang, X.; Jiang, C.; Kong, W. Large-Scale Purification of Pharmaceutical-Grade Plasmid DNA Using Tangential Flow Filtration and Multi-Step Chromatography. J. Biosci. Bioeng. 2013, 116, 281–286. [Google Scholar] [CrossRef]
- Griffiths, K.R.; Burke, D.G.; Emslie, K.R. Quantitative Polymerase Chain Reaction: A Framework for Improving the Quality of Results and Estimating Uncertainty of Measurement. Anal. Methods 2011, 3, 2201–2211. [Google Scholar] [CrossRef]
- Bhat, S.; Curach, N.; Mostyn, T.; Bains, G.S.; Griffiths, K.R.; Emslie, K.R. Comparison of Methods for Accurate Quantification of DNA Mass Concentration with Traceability to the International System of Units. Anal. Chem. 2010, 82, 7185–7192. [Google Scholar] [CrossRef]
- O’Connor, G.; Dawson, C.; Woolford, A.; Webb, K.S.; Catterick, T. Quantitation of Oligonucleotides by Phosphodiesterase Digestion Followed by Isotope Dilution Mass Spectrometry: Proof of Concept. Anal. Chem. 2002, 74, 3670–3676. [Google Scholar] [CrossRef]
- Leclerc, O.; Fraisse, P.-O.; Labarraque, G.; Oster, C.; Pichaut, J.-P.; Baume, M.; Jarraud, S.; Fisicaro, P.; Vaslin-Reimann, S. Method Development for Genomic Legionella Pneumophila DNA Quantification by Inductively Coupled Plasma Mass Spectrometry. Anal. Biochem. 2013, 435, 153–158. [Google Scholar] [CrossRef]
- Liang, W.; Xu, L.; Sui, Z.; Li, Y.; Li, L.; Wen, Y.; Li, C.; Ren, S.; Liu, G. Quantification of Plasmid DNA Reference Materials for Shiga Toxin-Producing Escherichia Coli Based on UV, HR-ICP-MS and Digital PCR. Chem. Cent. J. 2016, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.trilinkbiotech.com/cleancap-reagent-ag.html (accessed on 22 November 2021).
- Pascolo, S. Messenger MRNA: The Inexpensive Biopharmaceutical. J. Multidiscip. Eng. Sci. Technol. 2017, 4, 6937–6941. [Google Scholar]
- Kwon, H.; Kim, M.; Seo, Y.; Moon, Y.S.; Lee, H.J.; Lee, K.; Lee, H. Emergence of Synthetic MRNA: In Vitro Synthesis of MRNA and Its Applications in Regenerative Medicine. Biomaterials 2018, 156, 172–193. [Google Scholar] [CrossRef]
- Akash, M.S.H.; Rehman, K. Column Chromatography. In Essentials of Pharmaceutical Analysis; Akash, M.S.H., Rehman, K., Eds.; Springer: Singapore, 2020; pp. 167–174. ISBN 9789811515477. [Google Scholar]
- Issa, W.J.; Barberio, J.L.; Aunins, J.G.; Afeyan, N.B. Ion Exchange Purification of MRNA. U.S. Patent 10,590, 161, 17 March 2020. [Google Scholar]
- Bryant, S.; Manning, D.L. Isolation of mRNA by Affinity Chromatography. In The Nucleic Acid Protocols Handbook; Rapley, R., Ed.; Springer Protocols Handbooks; Humana Press: Totowa, NJ, USA, 2000; pp. 9–11. ISBN 978-1-59259-038-4. [Google Scholar]
- Thakor, N.; Holcik, M. RNA Affinity Chromatography; Magdeldin, S., Ed.; InTech: Rijeka, Croatia, 2012; ISBN 978-953-51-0325-7. [Google Scholar]
- Geiger, J.; Treml, M. MRNA Purification by Tangential Flow Filtration. WO2020165158A1, 20 August 2020. [Google Scholar]
- Heartlein, M.; Derosa, F.; Dias, A.; Karve, S. Methods for Purification of Messenger Rna. WO2014152966A1, 25 September 2014. [Google Scholar]
- Feyrer, H.; Munteanu, R.; Baronti, L.; Petzold, K. One-Pot Production of RNA in High Yield and Purity Through Cleaving Tandem Transcripts. Molecules 2020, 25, 1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic MRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of MRNA-Based Vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.A.; Reesor, E.K.G.; Xu, Y.; Zope, H.R.; Zetter, B.R.; Shi, J. Biomaterials for MRNA Delivery. Biomater. Sci. 2015, 3, 1519–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schott, J.W.; Morgan, M.; Galla, M.; Schambach, A. Viral and Synthetic RNA Vector Technologies and Applications. Mol. Ther. 2016, 24, 1513–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibba, M.L.; Ciccone, G.; Esposito, C.L.; Catuogno, S.; Giangrande, P.H. Advances in MRNA Non-Viral Delivery Approaches. Adv. Drug Deliv. Rev. 2021, 177, 113930. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorth, M.; Narvekar, A. Non Viral Vectors in Gene Therapy—An Overview. J. Clin. Diagn. Res. 2015, 9, GE01-6. [Google Scholar] [CrossRef]
- Yasar, H.; Biehl, A.; De Rossi, C.; Koch, M.; Murgia, X.; Loretz, B.; Lehr, C.-M. Kinetics of MRNA Delivery and Protein Translation in Dendritic Cells Using Lipid-Coated PLGA Nanoparticles. J. Nanobiotechnol. 2018, 16, 72. [Google Scholar] [CrossRef]
- Sainz-Ramos, M.; Gallego, I.; Villate-Beitia, I.; Zarate, J.; Maldonado, I.; Puras, G.; Pedraz, J.L. How Far Are Non-Viral Vectors to Come of Age and Reach Clinical Translation in Gene Therapy? Int. J. Mol. Sci. 2021, 22, 7545. [Google Scholar] [CrossRef]
- Zu, H.; Gao, D. Non-Viral Vectors in Gene Therapy: Recent Development, Challenges, and Prospects. AAPS J. 2021, 23, 78. [Google Scholar] [CrossRef]
- Rai, R.; Alwani, S.; Badea, I. Polymeric Nanoparticles in Gene Therapy: New Avenues of Design and Optimization for Delivery Applications. Polymers 2019, 11, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahane, A.; Waghmode, A.; Kapphahn, A.; Dhuri, K.; Gupta, A.; Bahal, R. Role of Lipid-Based and Polymer-Based Non-Viral Vectors in Nucleic Acid Delivery for Next-Generation Gene Therapy. Molecules 2020, 25, 2866. [Google Scholar] [CrossRef]
- Meng, Z.; O’Keeffe-Ahern, J.; Lyu, J.; Pierucci, L.; Zhou, D.; Wang, W. A New Developing Class of Gene Delivery: Messenger RNA-Based Therapeutics. Biomater. Sci. 2017, 5, 2381–2392. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Chan, J.; Farokhzad, O.C. PH-Responsive Nanoparticles for Drug Delivery. Mol. Pharm. 2010, 7, 1913–1920. [Google Scholar] [CrossRef]
- Jarzębińska, A.; Pasewald, T.; Lambrecht, J.; Mykhaylyk, O.; Kümmerling, L.; Beck, P.; Hasenpusch, G.; Rudolph, C.; Plank, C.; Dohmen, C. A Single Methylene Group in Oligoalkylamine-Based Cationic Polymers and Lipids Promotes Enhanced MRNA Delivery. Angew. Chem. Int. Ed. Engl. 2016, 55, 9591–9595. [Google Scholar] [CrossRef]
- Ulkoski, D.; Bak, A.; Wilson, J.T.; Krishnamurthy, V.R. Recent Advances in Polymeric Materials for the Delivery of RNA Therapeutics. Expert Opin. Drug Deliv. 2019, 16, 1149–1167. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of Cationic Lipids and Cationic Polymers in Gene Delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Chiper, M.; Niederreither, K.; Zuber, G. Transduction Methods for Cytosolic Delivery of Proteins and Bioconjugates into Living Cells. Adv. Healthc. Mater. 2018, 7, e1701040. [Google Scholar] [CrossRef]
- Zhao, H.; Song, X.; Aslan, H.; Liu, B.; Wang, J.; Wang, L.; Besenbacher, F.; Dong, M. Self-Assembly of Hydrogen-Bonded Supramolecular Complexes of Nucleic-Acid-Base and Fatty-Acid at the Liquid-Solid Interface. Phys. Chem. Chem. Phys. 2016, 18, 14168–14171. [Google Scholar] [CrossRef]
- Monnery, B.D. Polycation-Mediated Transfection: Mechanisms of Internalization and Intracellular Trafficking. Biomacromolecules 2021, 22, 4060–4083. [Google Scholar] [CrossRef] [PubMed]
- Read, M.L.; Singh, S.; Ahmed, Z.; Stevenson, M.; Briggs, S.S.; Oupicky, D.; Barrett, L.B.; Spice, R.; Kendall, M.; Berry, M.; et al. A Versatile Reducible Polycation-Based System for Efficient Delivery of a Broad Range of Nucleic Acids. Nucleic Acids Res. 2005, 33, e86. [Google Scholar] [CrossRef] [Green Version]
- Piperno, A.; Sciortino, M.T.; Giusto, E.; Montesi, M.; Panseri, S.; Scala, A. Recent Advances and Challenges in Gene Delivery Mediated by Polyester-Based Nanoparticles. Int. J. Nanomed. 2021, 16, 5981–6002. [Google Scholar] [CrossRef]
- Yin, J.; Luan, S. Opportunities and Challenges for the Development of Polymer-Based Biomaterials and Medical Devices. Regen. Biomater. 2016, 3, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Men, K.; Gao, Y.; Wu, J.; Lei, S.; Yang, Y.; Pan, H. Single Micelle Vectors Based on Lipid/Block Copolymer Compositions as MRNA Formulations for Efficient Cancer Immunogene Therapy. Mol. Pharm. 2021, 18, 4029–4045. [Google Scholar] [CrossRef]
- Nuhn, L.; Kaps, L.; Diken, M.; Schuppan, D.; Zentel, R. Reductive Decationizable Block Copolymers for Stimuli-Responsive MRNA Delivery. Macromol. Rapid Commun. 2016, 37, 924–933. [Google Scholar] [CrossRef]
- Bettinger, T.; Carlisle, R.C.; Read, M.L.; Ogris, M.; Seymour, L.W. Peptide-Mediated RNA Delivery: A Novel Approach for Enhanced Transfection of Primary and Post-Mitotic Cells. Nucleic Acids Res. 2001, 29, 3882–3891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huth, S.; Hoffmann, F.; von Gersdorff, K.; Laner, A.; Reinhardt, D.; Rosenecker, J.; Rudolph, C. Interaction of Polyamine Gene Vectors with RNA Leads to the Dissociation of Plasmid DNA-Carrier Complexes. J. Gene Med. 2006, 8, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, A.C.; Piotrowski-Daspit, A.S.; Nakazawa, K.H.; Jiang, Y.; Datye, A.; Saltzman, W.M. Tunability of Biodegradable Poly (Amine-Co-Ester) Polymers for Customized Nucleic Acid Delivery and Other Biomedical Applications. Biomacromolecules 2018, 19, 3861–3873. [Google Scholar] [CrossRef]
- Jiang, Y.; Gaudin, A.; Zhang, J.; Agarwal, T.; Song, E.; Kauffman, A.C.; Tietjen, G.T.; Wang, Y.; Jiang, Z.; Cheng, C.J.; et al. A “Top-down” Approach to Actuate Poly (Amine-Co-Ester) Terpolymers for Potent and Safe MRNA Delivery. Biomaterials 2018, 176, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lu, Q.; Wang, Y.; Xu, E.; Ho, A.; Singh, P.; Wang, Y.; Jiang, Z.; Yang, F.; Tietjen, G.T.; et al. Quantitating Endosomal Escape of a Library of Polymers for MRNA Delivery. Nano Lett. 2020, 20, 1117–1123. [Google Scholar] [CrossRef]
- Kurimoto, S.; Yoshinaga, N.; Igarashi, K.; Matsumoto, Y.; Cabral, H.; Uchida, S. PEG-OligoRNA Hybridization of MRNA for Developing Sterically Stable Lipid Nanoparticles toward In Vivo Administration. Molecules 2019, 24, 1303. [Google Scholar] [CrossRef] [Green Version]
- Felgner, P.L.; Ringold, G.M. Cationic Liposome-Mediated Transfection. Nature 1989, 337, 387–388. [Google Scholar] [CrossRef]
- Aldosari, B.N.; Alfagih, I.M.; Almurshedi, A.S. Lipid Nanoparticles as Delivery Systems for RNA-Based Vaccines. Pharmaceutics 2021, 13, 206. [Google Scholar] [CrossRef]
- Hirsch-Lerner, D.; Zhang, M.; Eliyahu, H.; Ferrari, M.E.; Wheeler, C.J.; Barenholz, Y. Effect of “Helper Lipid” on Lipoplex Electrostatics. Biochim. Biophys. Acta 2005, 1714, 71–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granot, Y.; Peer, D. Delivering the Right Message: Challenges and Opportunities in Lipid Nanoparticles-Mediated Modified MRNA Therapeutics-An Innate Immune System Standpoint. Semin. Immunol. 2017, 34, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.; Luft, D.; Abraham, M.-K.; Reinhardt, S.; Salinas Medina, M.L.; Kurz, J.; Schaller, M.; Avci-Adali, M.; Schlensak, C.; Peter, K.; et al. Cationic Nanoliposomes Meet MRNA: Efficient Delivery of Modified MRNA Using Hemocompatible and Stable Vectors for Therapeutic Applications. Mol. Ther. Nucleic Acids 2017, 8, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Hattori, Y.; Hara, E.; Shingu, Y.; Minamiguchi, D.; Nakamura, A.; Arai, S.; Ohno, H.; Kawano, K.; Fujii, N.; Yonemochi, E. SiRNA Delivery into Tumor Cells by Cationic Cholesterol Derivative-Based Nanoparticles and Liposomes. Biol. Pharm. Bull. 2015, 38, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA Delivery to Dendritic Cells Exploits Antiviral Defence for Cancer Immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, Preparation, and Applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Corrias, F.; Lai, F. New Methods for Lipid Nanoparticles Preparation. Recent Pat. Drug Deliv. Formul. 2011, 5, 201–213. [Google Scholar] [CrossRef]
- Michel, T.; Link, A.; Abraham, M.-K.; Schlensak, C.; Peter, K.; Wendel, H.-P.; Wang, X.; Krajewski, S. Generation of Cationic Nanoliposomes for the Efficient Delivery of In Vitro Transcribed Messenger RNA. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Pollard, C.; Rejman, J.; De Haes, W.; Verrier, B.; Van Gulck, E.; Naessens, T.; De Smedt, S.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN Counteracts the Induction of Antigen-Specific Immune Responses by Lipid-Based Delivery of MRNA Vaccines. Mol. Ther. 2013, 21, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Chabanovska, O.; Galow, A.-M.; David, R.; Lemcke, H. mRNA—A Game Changer in Regenerative Medicine, Cell-Based Therapy and Reprogramming Strategies. Adv. Drug Deliv. Rev. 2021, 179, 114002. [Google Scholar] [CrossRef]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid Nanoparticles for Nucleic Acid Delivery: Current Perspectives. Adv. Drug Deliv. Rev. 2020, 154–155, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Schlich, M.; Longhena, F.; Faustini, G.; O′Driscoll, C.M.; Sinico, C.; Fadda, A.M.; Bellucci, A.; Lai, F. Anionic Liposomes for Small Interfering Ribonucleic Acid (SiRNA) Delivery to Primary Neuronal Cells: Evaluation of Alpha-Synuclein Knockdown Efficacy. Nano Res. 2017, 10, 3496–3508. [Google Scholar] [CrossRef]
- Guevara, M.L.; Persano, S.; Persano, F. Lipid-Based Vectors for Therapeutic MRNA-Based Anti-Cancer Vaccines. Curr. Pharm. Des. 2019, 25, 1443–1454. [Google Scholar] [CrossRef]
- Hafez, I.M.; Maurer, N.; Cullis, P.R. On the Mechanism Whereby Cationic Lipids Promote Intracellular Delivery of Polynucleic Acids. Gene Ther. 2001, 8, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Lee, R.J. The Role of Helper Lipids in Lipid Nanoparticles (LNPs) Designed for Oligonucleotide Delivery. Adv. Drug Deliv. Rev. 2016, 99, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Ryals, R.C.; Patel, S.; Acosta, C.; McKinney, M.; Pennesi, M.E.; Sahay, G. The Effects of PEGylation on LNP Based MRNA Delivery to the Eye. PLoS ONE 2020, 15, e0241006. [Google Scholar] [CrossRef]
- Xiong, H.; Liu, S.; Wei, T.; Cheng, Q.; Siegwart, D.J. Theranostic Dendrimer-Based Lipid Nanoparticles Containing PEGylated BODIPY Dyes for Tumor Imaging and Systemic MRNA Delivery in Vivo. J. Control. Release 2020, 325, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Thanki, K.; Zeng, X.; Justesen, S.; Tejlmann, S.; Falkenberg, E.; Van Driessche, E.; Mørck Nielsen, H.; Franzyk, H.; Foged, C. Engineering of Small Interfering RNA-Loaded Lipidoid-Poly (DL-Lactic-Co-Glycolic Acid) Hybrid Nanoparticles for Highly Efficient and Safe Gene Silencing: A Quality by Design-Based Approach. Eur. J. Pharm. Biopharm. 2017, 120, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.A.A.; Klausen, L.H.; Thanki, K.; Lyngsø, J.; Skov Pedersen, J.; Franzyk, H.; Nielsen, H.M.; van Eden, W.; Dong, M.; Broere, F.; et al. Lipidoid-Polymer Hybrid Nanoparticles Loaded with TNF SiRNA Suppress Inflammation after Intra-Articular Administration in a Murine Experimental Arthritis Model. Eur. J. Pharm. Biopharm. 2019, 142, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Waters, A.K.; Kalyan, P.; Achrol, A.S.; Kesari, S.; Yenugonda, V.M. Lipid-Polymer Hybrid Nanoparticles as a next-Generation Drug Delivery Platform: State of the Art, Emerging Technologies, and Perspectives. Int. J. Nanomed. 2019, 14, 1937–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Zhang, C.; Li, B.; Zhang, X.; Luo, X.; Zeng, C.; Li, W.; Gao, M.; Dong, Y. Lipid Polymer Hybrid Nanomaterials for MRNA Delivery. Cell Mol. Bioeng. 2018, 11, 397–406. [Google Scholar] [CrossRef]
- Ball, R.L.; Hajj, K.A.; Vizelman, J.; Bajaj, P.; Whitehead, K.A. Lipid Nanoparticle Formulations for Enhanced Co-Delivery of SiRNA and MRNA. Nano Lett. 2018, 18, 3814–3822. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Patel, A.K.; Kauffman, K.J.; Fenton, O.S.; Webber, M.J.; Heartlein, M.W.; DeRosa, F.; Anderson, D.G. Polymer-Lipid Nanoparticles for Systemic Delivery of MRNA to the Lungs. Angew. Chem. Int. Ed. Engl. 2016, 55, 13808–13812. [Google Scholar] [CrossRef] [Green Version]
- Udhayakumar, V.K.; De Beuckelaer, A.; McCaffrey, J.; McCrudden, C.M.; Kirschman, J.L.; Vanover, D.; Van Hoecke, L.; Roose, K.; Deswarte, K.; De Geest, B.G.; et al. Arginine-Rich Peptide-Based MRNA Nanocomplexes Efficiently Instigate Cytotoxic T Cell Immunity Dependent on the Amphipathic Organization of the Peptide. Adv. Healthc. Mater. 2017, 6, 1601412. [Google Scholar] [CrossRef] [PubMed]
- van den Brand, D.; Gorris, M.A.J.; van Asbeck, A.H.; Palmen, E.; Ebisch, I.; Dolstra, H.; Hällbrink, M.; Massuger, L.F.A.G.; Brock, R. Peptide-Mediated Delivery of Therapeutic MRNA in Ovarian Cancer. Eur. J. Pharm. Biopharm. 2019, 141, 180–190. [Google Scholar] [CrossRef]
- Coolen, A.-L.; Lacroix, C.; Mercier-Gouy, P.; Delaune, E.; Monge, C.; Exposito, J.-Y.; Verrier, B. Poly (Lactic Acid) Nanoparticles and Cell-Penetrating Peptide Potentiate MRNA-Based Vaccine Expression in Dendritic Cells Triggering Their Activation. Biomaterials 2019, 195, 23–37. [Google Scholar] [CrossRef]
- Lacroix, C.; Humanes, A.; Coiffier, C.; Gigmes, D.; Verrier, B.; Trimaille, T. Polylactide-Based Reactive Micelles as a Robust Platform for MRNA Delivery. Pharm. Res. 2020, 37, 30. [Google Scholar] [CrossRef]
- Aslan, C.; Kiaie, S.H.; Zolbanin, N.M.; Lotfinejad, P.; Ramezani, R.; Kashanchi, F.; Jafari, R. Exosomes for MRNA Delivery: A Novel Biotherapeutic Strategy with Hurdles and Hope. BMC Biotechnol. 2021, 21, 20. [Google Scholar] [CrossRef]
- Kojima, R.; Bojar, D.; Rizzi, G.; Hamri, G.C.-E.; El-Baba, M.D.; Saxena, P.; Ausländer, S.; Tan, K.R.; Fussenegger, M. Designer Exosomes Produced by Implanted Cells Intracerebrally Deliver Therapeutic Cargo for Parkinson’s Disease Treatment. Nat. Commun. 2018, 9, 1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.K.; Nocera, A.L.; Bleier, B.S. Exosome Function in Aerodigestive Mucosa. Nanomedicine 2018, 14, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Shi, J.; Xie, J.; Wang, Y.; Sun, J.; Liu, T.; Zhao, Y.; Zhao, X.; Wang, X.; Ma, Y.; et al. Large-Scale Generation of Functional MRNA-Encapsulating Exosomes via Cellular Nanoporation. Nat. Biomed. Eng. 2020, 4, 69–83. [Google Scholar] [CrossRef]
- Pascolo, S. Synthetic Messenger RNA-Based Vaccines: From Scorn to Hype. Viruses 2021, 13, 270. [Google Scholar] [CrossRef]
- Houseley, J.; Tollervey, D. The Many Pathways of RNA Degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blakney, A.K.; Ip, S.; Geall, A.J. An Update on Self-Amplifying MRNA Vaccine Development. Vaccines 2021, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Kaukinen, U.; Lyytikäinen, S.; Mikkola, S.; Lönnberg, H. The Reactivity of Phosphodiester Bonds within Linear Single-Stranded Oligoribonucleotides Is Strongly Dependent on the Base Sequence. Nucleic Acids Res. 2002, 30, 468–474. [Google Scholar] [CrossRef] [Green Version]
- Clear, K.J.; Virga, K.; Gray, L.; Smith, B.D. Using Membrane Composition to Fine-Tune the PKa of an Optical Liposome PH Sensor. J. Mater. Chem. C Mater. 2016, 4, 2925–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mchedlov-Petrossyan, N.O.; Vodolazkaya, N.A.; Yakubovskaya, A.G.; Grigorovich, A.V.; Alekseeva, V.I.; Savvina, L.P. A Novel Probe for Determination of Electrical Surface Potential of Surfactant Micelles: N,N′-Di-n-Octadecylrhodamine. J. Phys. Org. Chem. 2007, 20, 332–344. [Google Scholar] [CrossRef]
- Temperley, R.J.; Wydro, M.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Human Mitochondrial MRNAs-like Members of All Families, Similar but Different. Biochim. Biophys. Acta 2010, 1797, 1081–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monde, R.A.; Schuster, G.; Stern, D.B. Processing and Degradation of Chloroplast MRNA. Biochimie 2000, 82, 573–582. [Google Scholar] [CrossRef]
- Kim, S.C.; Sekhon, S.S.; Shin, W.-R.; Ahn, G.; Cho, B.-K.; Ahn, J.-Y.; Kim, Y.-H. Modifications of MRNA Vaccine Structural Elements for Improving MRNA Stability and Translation Efficiency. Mol. Cell Toxicol. 2021, 1–8. [Google Scholar] [CrossRef]
- Izaurralde, E.; Lewis, J.; McGuigan, C.; Jankowska, M.; Darzynkiewicz, E.; Mattaj, I.W. A Nuclear Cap Binding Protein Complex Involved in Pre-MRNA Splicing. Cell 1994, 78, 657–668. [Google Scholar] [CrossRef]
- Charenton, C.; Graille, M. MRNA Decapping: Finding the Right Structures. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20180164. [Google Scholar] [CrossRef] [Green Version]
- Pasquinelli, A.E.; Dahlberg, J.E.; Lund, E. Reverse 5′ Caps in RNAs Made in Vitro by Phage RNA Polymerases. RNA 1995, 1, 957–967. [Google Scholar]
- Grudzien-Nogalska, E.; Stepinski, J.; Jemielity, J.; Zuberek, J.; Stolarski, R.; Rhoads, R.E.; Darzynkiewicz, E. Synthesis of Anti-Reverse Cap Analogs (ARCAs) and Their Applications in MRNA Translation and Stability. Methods Enzymol. 2007, 431, 203–227. [Google Scholar] [CrossRef]
- Grudzien, E.; Kalek, M.; Jemielity, J.; Darzynkiewicz, E.; Rhoads, R.E. Differential Inhibition of MRNA Degradation Pathways by Novel Cap Analogs. J. Biol. Chem. 2006, 281, 1857–1867. [Google Scholar] [CrossRef] [Green Version]
- Zohra, F.T.; Chowdhury, E.H.; Tada, S.; Hoshiba, T.; Akaike, T. Effective Delivery with Enhanced Translational Activity Synergistically Accelerates MRNA-Based Transfection. Biochem. Biophys. Res. Commun. 2007, 358, 373–378. [Google Scholar] [CrossRef]
- Stepinski, J.; Waddell, C.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Synthesis and Properties of MRNAs Containing the Novel “Anti-Reverse” Cap Analogs 7-Methyl (3′-O-Methyl) GpppG and 7-Methyl (3′-Deoxy) GpppG. RNA 2001, 7, 1486–1495. [Google Scholar]
- Shanmugasundaram, M.; Charles, I.; Kore, A.R. Design, Synthesis and Biological Evaluation of Dinucleotide MRNA Cap Analog Containing Propargyl Moiety. Bioorg. Med. Chem. 2016, 24, 1204–1208. [Google Scholar] [CrossRef] [PubMed]
- Grudzien, E.; Stepinski, J.; Jankowska-Anyszka, M.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Novel Cap Analogs for in Vitro Synthesis of MRNAs with High Translational Efficiency. RNA 2004, 10, 1479–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grudzien-Nogalska, E.; Jemielity, J.; Kowalska, J.; Darzynkiewicz, E.; Rhoads, R.E. Phosphorothioate Cap Analogs Stabilize MRNA and Increase Translational Efficiency in Mammalian Cells. RNA 2007, 13, 1745–1755. [Google Scholar] [CrossRef] [Green Version]
- Muttach, F.; Muthmann, N.; Rentmeister, A. Synthetic MRNA Capping. Beilstein J. Org. Chem. 2017, 13, 2819–2832. [Google Scholar] [CrossRef]
- Senthilvelan, A.; Vonderfecht, T.; Shanmugasundaram, M.; Pal, I.; Potter, J.; Kore, A.R. Trinucleotide Cap Analogue Bearing a Locked Nucleic Acid Moiety: Synthesis, MRNA Modification, and Translation for Therapeutic Applications. Org. Lett. 2021, 23, 4133–4136. [Google Scholar] [CrossRef] [PubMed]
- Boo, S.H.; Kim, Y.K. The Emerging Role of RNA Modifications in the Regulation of MRNA Stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Boulias, K.; Toczydłowska-Socha, D.; Hawley, B.R.; Liberman, N.; Takashima, K.; Zaccara, S.; Guez, T.; Vasseur, J.-J.; Debart, F.; Aravind, L.; et al. Identification of the M6Am Methyltransferase PCIF1 Reveals the Location and Functions of M6Am in the Transcriptome. Mol. Cell 2019, 75, 631–643. [Google Scholar] [CrossRef]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.-J.; Chen, Q.; et al. Reversible Methylation of M6Am in the 5′ Cap Controls MRNA Stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [Green Version]
- Mugridge, J.S.; Coller, J.; Gross, J.D. Structural and Molecular Mechanisms for the Control of Eukaryotic 5′-3′ MRNA Decay. Nat. Struct. Mol. Biol. 2018, 25, 1077–1085. [Google Scholar] [CrossRef]
- Henderson, J.M.; Ujita, A.; Hill, E.; Yousif-Rosales, S.; Smith, C.; Ko, N.; McReynolds, T.; Cabral, C.R.; Escamilla-Powers, J.R.; Houston, M.E. Cap 1 Messenger RNA Synthesis with Co-Transcriptional CleanCap® Analog by In Vitro Transcription. Curr. Protoc. 2021, 1, e39. [Google Scholar] [CrossRef]
- Tan, X.; Wan, Y. Enhanced Protein Expression by Internal Ribosomal Entry Site-Driven MRNA Translation as a Novel Approach for in Vitro Loading of Dendritic Cells with Antigens. Hum. Immunol. 2008, 69, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A.; Hatzoglou, M. Cellular IRES-Mediated Translation. Cell Cycle 2011, 10, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenik, C.; Chua, H.N.; Zhang, H.; Tarnawsky, S.P.; Akef, A.; Derti, A.; Tasan, M.; Moore, M.J.; Palazzo, A.F.; Roth, F.P. Genome Analysis Reveals Interplay between 5′ UTR Introns and Nuclear MRNA Export for Secretory and Mitochondrial Genes. PLoS Genet. 2011, 7, e1001366. [Google Scholar] [CrossRef] [Green Version]
- Mignone, F.; Gissi, C.; Liuni, S.; Pesole, G. Untranslated Regions of MRNAs. Genome Biol. 2002, 3, reviews0004. [Google Scholar] [CrossRef] [PubMed]
- Araujo, P.R.; Yoon, K.; Ko, D.; Smith, A.D.; Qiao, M.; Suresh, U.; Burns, S.C.; Penalva, L.O.F. Before It Gets Started: Regulating Translation at the 5′ UTR. Comp. Funct. Genom. 2012, 2012, 475731. [Google Scholar] [CrossRef] [Green Version]
- Rubio, C.A.; Weisburd, B.; Holderfield, M.; Arias, C.; Fang, E.; DeRisi, J.L.; Fanidi, A. Transcriptome-Wide Characterization of the EIF4A Signature Highlights Plasticity in Translation Regulation. Genome Biol. 2014, 15, 476. [Google Scholar] [CrossRef]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR MRNA Structures in Eukaryotic Translation Regulation and How to Find Them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef]
- Mauger, D.M.; Cabral, B.J.; Presnyak, V.; Su, S.V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J.; et al. MRNA Structure Regulates Protein Expression through Changes in Functional Half-Life. Proc. Natl. Acad. Sci. USA 2019, 116, 24075–24083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppek, K.; Byeon, G.W.; Kladwang, W.; Wayment-Steele, H.K.; Kerr, C.H.; Xu, A.F.; Kim, D.S.; Topkar, V.V.; Choe, C.; Rothschild, D.; et al. Combinatorial Optimization of MRNA Structure, Stability, and Translation for RNA-Based Therapeutics. bioRxiv 2021, in press. [Google Scholar] [CrossRef]
- Mayr, C. What Are 3′ UTRs Doing? Cold Spring Harb. Perspect. Biol. 2019, 11, a034728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Blagev, D.; Pollack, J.L.; Erle, D.J. Toward a Systematic Understanding of MRNA 3′ Untranslated Regions. Proc. Am. Thorac. Soc. 2011, 8, 163–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlandini von Niessen, A.G.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Löwer, M.; Vallazza, B.; Beissert, T.; et al. Improving MRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019, 27, 824–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.Y.; Shyu, A.B. AU-Rich Elements: Characterization and Importance in MRNA Degradation. Trends Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef]
- Otsuka, H.; Fukao, A.; Funakami, Y.; Duncan, K.E.; Fujiwara, T. Emerging Evidence of Translational Control by AU-Rich Element-Binding Proteins. Front. Genet. 2019, 10, 332. [Google Scholar] [CrossRef] [Green Version]
- Asrani, K.H.; Farelli, J.D.; Stahley, M.R.; Miller, R.L.; Cheng, C.J.; Subramanian, R.R.; Brown, J.M. Optimization of MRNA Untranslated Regions for Improved Expression of Therapeutic MRNA. RNA Biol. 2018, 15, 756–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hia, F.; Takeuchi, O. The Effects of Codon Bias and Optimality on MRNA and Protein Regulation. Cell Mol. Life Sci. 2021, 78, 1909–1928. [Google Scholar] [CrossRef]
- Cannarozzi, G.; Cannarrozzi, G.; Schraudolph, N.N.; Faty, M.; von Rohr, P.; Friberg, M.T.; Roth, A.C.; Gonnet, P.; Gonnet, G.; Barral, Y. A Role for Codon Order in Translation Dynamics. Cell 2010, 141, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Presnyak, V.; Alhusaini, N.; Chen, Y.-H.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon Optimality Is a Major Determinant of MRNA Stability. Cell 2015, 160, 1111–1124. [Google Scholar] [CrossRef] [Green Version]
- Courel, M.; Clément, Y.; Bossevain, C.; Foretek, D.; Vidal Cruchez, O.; Yi, Z.; Bénard, M.; Benassy, M.-N.; Kress, M.; Vindry, C.; et al. GC Content Shapes MRNA Storage and Decay in Human Cells. eLife 2019, 8, e49708. [Google Scholar] [CrossRef]
- Komar, A.A. A Code Within a Code: How Codons Fine-Tune Protein Folding in the Cell. Biochemistry 2021, 86, 976–991. [Google Scholar] [CrossRef] [PubMed]
- Minnaert, A.-K.; Vanluchene, H.; Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Raemdonck, K.; Sanders, N.N.; Remaut, K. Strategies for Controlling the Innate Immune Activity of Conventional and Self-Amplifying MRNA Therapeutics: Getting the Message Across. Adv. Drug Deliv. Rev. 2021, 176, 113900. [Google Scholar] [CrossRef] [PubMed]
- Freund, I.; Eigenbrod, T.; Helm, M.; Dalpke, A.H. RNA Modifications Modulate Activation of Innate Toll-Like Receptors. Genes 2019, 10, 92. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Muramatsu, H.; Keller, J.M.; Weissman, D. Increased Erythropoiesis in Mice Injected with Submicrogram Quantities of Pseudouridine-Containing MRNA Encoding Erythropoietin. Mol. Ther. 2012, 20, 948–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diebold, S.S.; Massacrier, C.; Akira, S.; Paturel, C.; Morel, Y.; Reis e Sousa, C. Nucleic Acid Agonists for Toll-like Receptor 7 Are Defined by the Presence of Uridine Ribonucleotides. Eur. J. Immunol. 2006, 36, 3256–3267. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine into MRNA Yields Superior Nonimmunogenic Vector with Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N (1)-Methylpseudouridine-Incorporated MRNA Outperforms Pseudouridine-Incorporated MRNA by Providing Enhanced Protein Expression and Reduced Immunogenicity in Mammalian Cell Lines and Mice. J. Control. Release 2015, 217, 337–344. [Google Scholar] [CrossRef]
- Kormann, M.S.D.; Hasenpusch, G.; Aneja, M.K.; Nica, G.; Flemmer, A.W.; Herber-Jonat, S.; Huppmann, M.; Mays, L.E.; Illenyi, M.; Schams, A.; et al. Expression of Therapeutic Proteins after Delivery of Chemically Modified MRNA in Mice. Nat. Biotechnol. 2011, 29, 154–157. [Google Scholar] [CrossRef]
- Arango, D.; Sturgill, D.; Alhusaini, N.; Dillman, A.A.; Sweet, T.J.; Hanson, G.; Hosogane, M.; Sinclair, W.R.; Nanan, K.K.; Mandler, M.D.; et al. Acetylation of Cytidine in MRNA Promotes Translation Efficiency. Cell 2018, 175, 1872–1886. [Google Scholar] [CrossRef] [Green Version]
- Hoernes, T.P.; Hüttenhofer, A.; Erlacher, M.D. MRNA Modifications: Dynamic Regulators of Gene Expression? RNA Biol. 2016, 13, 760–765. [Google Scholar] [CrossRef] [Green Version]
- Hoernes, T.P.; Clementi, N.; Faserl, K.; Glasner, H.; Breuker, K.; Lindner, H.; Hüttenhofer, A.; Erlacher, M.D. Nucleotide Modifications within Bacterial Messenger RNAs Regulate Their Translation and Are Able to Rewire the Genetic Code. Nucleic Acids Res. 2016, 44, 852–862. [Google Scholar] [CrossRef] [Green Version]
- Hoernes, T.P.; Heimdörfer, D.; Köstner, D.; Faserl, K.; Nußbaumer, F.; Plangger, R.; Kreutz, C.; Lindner, H.; Erlacher, M.D. Eukaryotic Translation Elongation Is Modulated by Single Natural Nucleotide Derivatives in the Coding Sequences of MRNAs. Genes 2019, 10, 84. [Google Scholar] [CrossRef] [Green Version]
- Hoernes, T.P.; Erlacher, M.D. Methylated MRNA Nucleotides as Regulators for Ribosomal Translation. Methods Mol. Biol. 2017, 1562, 283–294. [Google Scholar] [CrossRef]
- Nicholson, A.L.; Pasquinelli, A.E. Tales of Detailed Poly(A) Tails. Trends Cell Biol. 2019, 29, 191–200. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.-J. Developing MRNA-Vaccine Technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Zhang, Q.; Feng, X.-H.; Liu, J. Synthetic Modified Messenger RNA for Therapeutic Applications. Acta Biomater. 2021, 131, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Strzelecka, D.; Smietanski, M.; Sikorski, P.J.; Warminski, M.; Kowalska, J.; Jemielity, J. Phosphodiester Modifications in MRNA Poly(A) Tail Prevent Deadenylation without Compromising Protein Expression. RNA 2020, 26, 1815–1837. [Google Scholar] [CrossRef]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. MRNA Vaccines for Infectious Diseases: Principles, Delivery and Clinical Translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar] [CrossRef]
- Eberle, F.; Sahin, U.; Kuhn, A.; Vallazza, B.; Diken, M. Stabilization of Poly(a) Sequence Encoding Dna Sequences. U.S. Patent US20170166905, 15 June 2017. [Google Scholar]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J.A. MRNA-Lipid Nanoparticle COVID-19 Vaccines: Structure and Stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef] [PubMed]
- Crommelin, D.J.A.; Volkin, D.B.; Hoogendoorn, K.H.; Lubiniecki, A.S.; Jiskoot, W. The Science Is There: Key Considerations for Stabilizing Viral Vector-Based COVID-19 Vaccines. J. Pharm. Sci. 2021, 110, 627–634. [Google Scholar] [CrossRef]
- Arteta, M.Y.; Kjellman, T.; Bartesaghi, S.; Wallin, S.; Wu, X.; Kvist, A.J.; Dabkowska, A.; Székely, N.; Radulescu, A.; Bergenholtz, J.; et al. Successful Reprogramming of Cellular Protein Production through MRNA Delivered by Functionalized Lipid Nanoparticles. Proc. Natl. Acad. Sci. USA 2018, 115, E3351–E3360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Hou, X.; Yan, J.; Du, S.; Xue, Y.; Li, W.; Xiang, G.; Dong, Y. Long-Term Storage of Lipid-like Nanoparticles for MRNA Delivery. Bioact. Mater. 2020, 5, 358–363. [Google Scholar] [CrossRef]
- Kon, E.; Elia, U.; Peer, D. Principles for Designing an Optimal MRNA Lipid Nanoparticle Vaccine. Curr. Opin. Biotechnol. 2022, 73, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jiang, D.; Pardridge, W.M. Lyoprotectant Optimization for the Freeze-Drying of Receptor-Targeted Trojan Horse Liposomes for Plasmid DNA Delivery. Mol. Pharm. 2020, 17, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Amorij, J.-P.; Huckriede, A.; Wilschut, J.; Frijlink, H.W.; Hinrichs, W.L.J. Development of Stable Influenza Vaccine Powder Formulations: Challenges and Possibilities. Pharm. Res. 2008, 25, 1256–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouranidis, A.; Vavilis, T.; Mandala, E.; Davidopoulou, C.; Stamoula, E.; Markopoulou, C.K.; Karagianni, A.; Kachrimanis, K. mRNA Therapeutic Modalities Design, Formulation and Manufacturing under Pharma 4.0 Principles. Biomedicines 2022, 10, 50. https://doi.org/10.3390/biomedicines10010050
Ouranidis A, Vavilis T, Mandala E, Davidopoulou C, Stamoula E, Markopoulou CK, Karagianni A, Kachrimanis K. mRNA Therapeutic Modalities Design, Formulation and Manufacturing under Pharma 4.0 Principles. Biomedicines. 2022; 10(1):50. https://doi.org/10.3390/biomedicines10010050
Chicago/Turabian StyleOuranidis, Andreas, Theofanis Vavilis, Evdokia Mandala, Christina Davidopoulou, Eleni Stamoula, Catherine K. Markopoulou, Anna Karagianni, and Kyriakos Kachrimanis. 2022. "mRNA Therapeutic Modalities Design, Formulation and Manufacturing under Pharma 4.0 Principles" Biomedicines 10, no. 1: 50. https://doi.org/10.3390/biomedicines10010050
APA StyleOuranidis, A., Vavilis, T., Mandala, E., Davidopoulou, C., Stamoula, E., Markopoulou, C. K., Karagianni, A., & Kachrimanis, K. (2022). mRNA Therapeutic Modalities Design, Formulation and Manufacturing under Pharma 4.0 Principles. Biomedicines, 10(1), 50. https://doi.org/10.3390/biomedicines10010050