
A Conserved uORF Regulates APOBEC3G Translation and Is Targeted by HIV-1 Vif Protein to Repress the Antiviral Factor

 , , , , ,
, , , , ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
- Plasmids
- RACE-PCR
- RNA modification by SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension)
- Cell culture
- Immunoblotting
- Real-time qPCR
- FISH and immunofluorescence (IF) assays
- Microscopy and image analysis
- APOBEC3 genotype data
3. Results
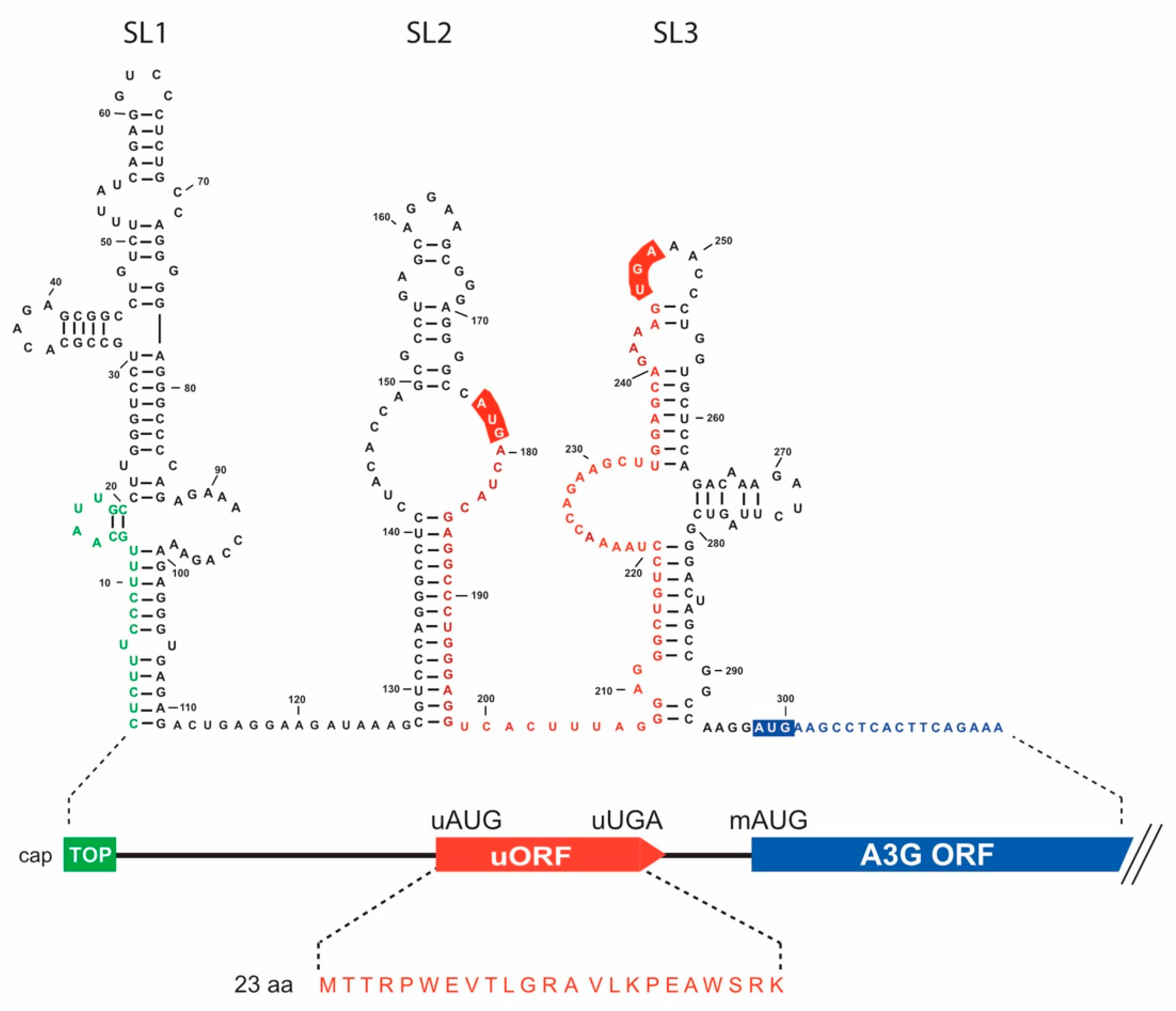
3.1. In Silico Analyses of A3G mRNA 5′-UTR Identify a Conserved uORF
3.2. Vif Specifically Regulates the Translation of A3G and A3F among Members of the A3 Family
3.3. Mechanism of A3G mRNA Translation
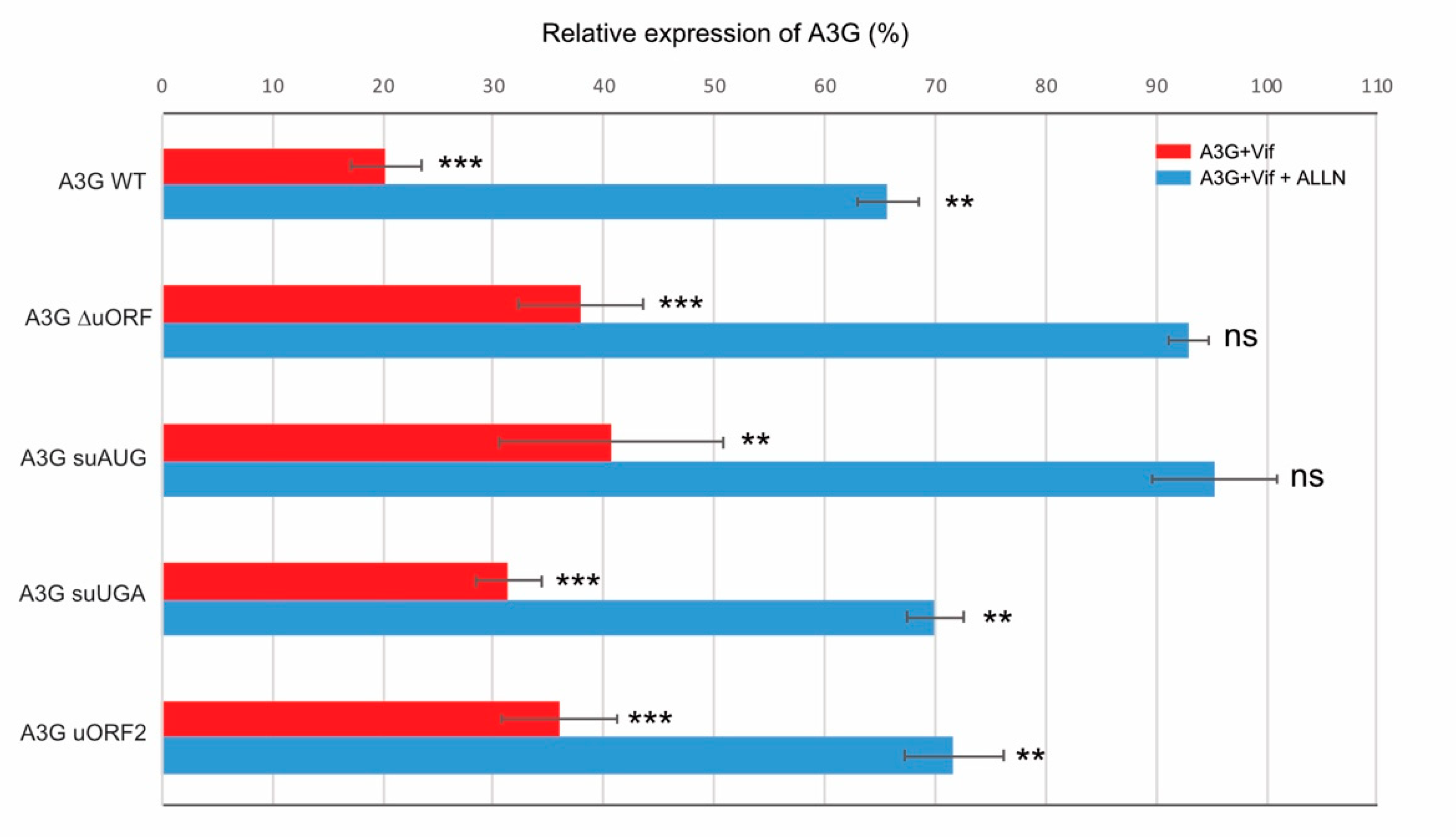
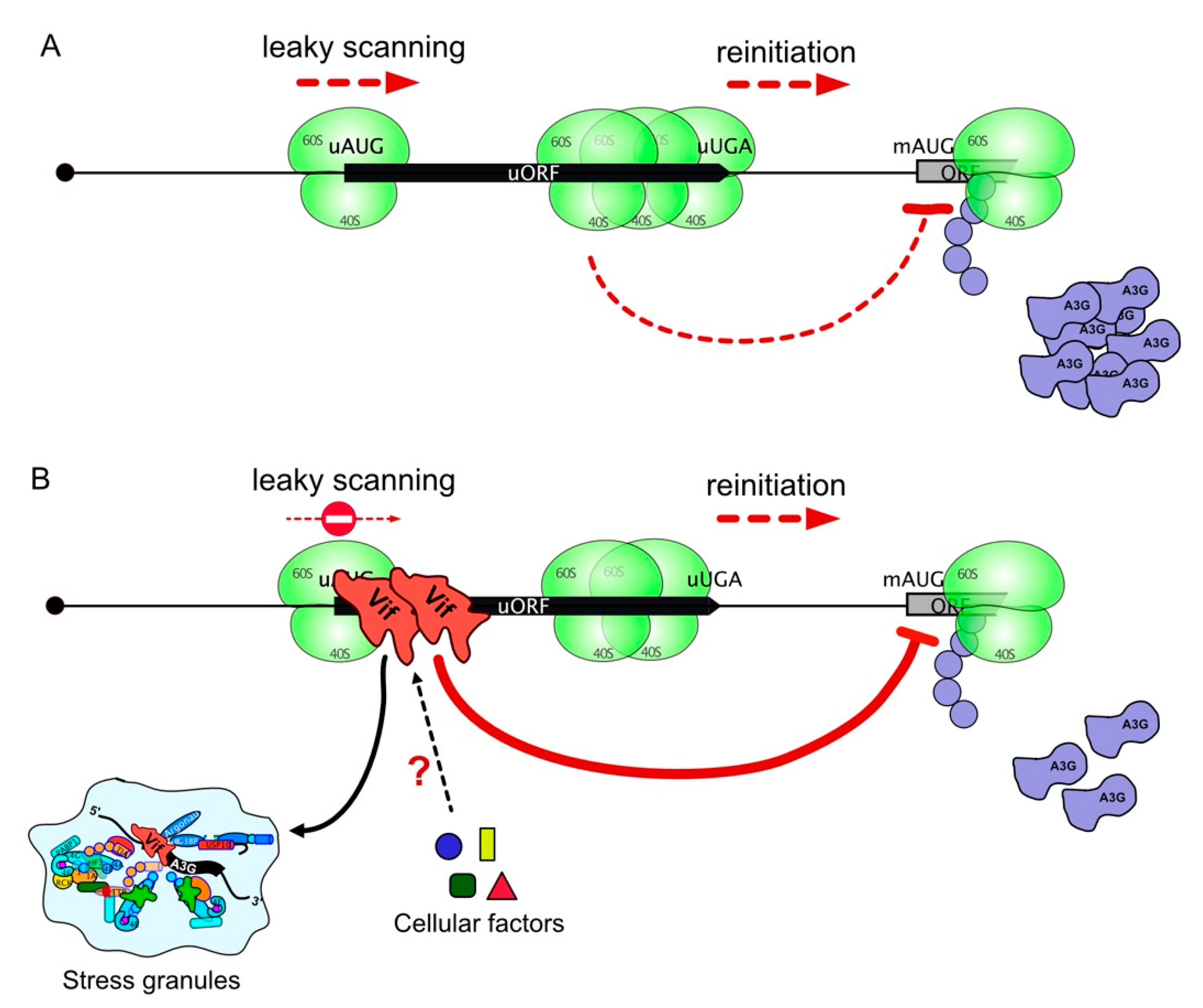
3.4. Initiation at the uORF Is Required in Vif-Mediated A3G Translation Inhibition
3.5. Relocation of A3G mRNA to Stress Granules Is Dependent on the uORF and Vif
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutants | Primers | Sequences (5′ to 3′) |
|---|---|---|
| A3G ∆uORF | pS-∆uORF | GAAGCGGGAGGGGCCAACCCTGGTGCTCCA |
| pAS-∆uORF | TGGAGCACCAGGGTTGGCCCCTCCCGCTTC | |
| A3G suAUG | pS-suAUG | GAAGGGGGAGGGGCCAAGACTACGAGGCCCTGG |
| pAS-suAUG | CCAGGGCCTCGTAGTCTTGGCCCCTCCCCCTTC | |
| A3G suUGA | pS-suUGA | GCCTGGAGCAGAAAGGAAACCCTGGTGCTCCA |
| pAS-suUGA | TGGAGCACCAGGGTTTCCTTTCTGCTCCAGGC | |
| A3G 2aa | pS-A3G2aa | GCCATGACTACGTGATGATGGGAGGTCACT |
| pAS-A3G2aa | AGTGACCTCCCATCATCACGTAGTCATGGC | |
| A3G 5aa | pS-A3G5aa | ACGAGGCCCTGGTGATGAACTTTAGGGAGG |
| pAS-A3G5aa | CCTCCCTAAAGTTCATCACCAGGGCCTCGT | |
| A3G 10aa | pS-A3G10aa | GTCACTTTAGGGTGATGAGTCCTAAAACCA |
| pAS-A3G10aa | TGGTTTTAGGACTCATCACCCTAAAGTGAC | |
| A3G 15aa | pS-A3G15aa | GCTGTCCTAAAATGATGAGCTTGGAGCAGA |
| pAS-A3G15aa | TCTGCTCCAAGCTCATCATTTTAGGACAGC | |
| A3G ∆249–273 | pS-∆249–273 | TGGAGCAGAAAGTGATTAGTCGGGACTAGC |
| pAS-∆249–273 | GCTAGTCCCGACTAATCACTTTCTGCTCCA | |
| A3G ∆249–291 | pS-∆249–291 | TGGAGCAGAAAGTGACCAAGGATGAAGCCT |
| pAS-∆249–291 | AGGCTTCATCCTTGGTCACTTTCTGCTCCA | |
| A3G WK | pS-WK | GAAGCGGGAAAAAAAATGGCTACGAGGCCCT |
| pAS-WK | AGGGCCTCGTAGCCATTTTTTTTCCCGCTTC | |
| A3G uORF2 | pS-uORF2 | GGGAGGGGCCATGGACTACGAGGCCCTGG |
| pAS-uORF2 | CCAGGGCCTCGTAGTCCATGGCCCCTCCC | |
| pS-uORF2 | CTTGGAGCAGAAATGAAACCCTGGTGCTCC | |
| pAS-uORF2 | GGAGCACCAGGGTTTCATTTCTGCTCCAAG | |
| A3G uUGA276 | pS-uUGA276 | CTCCAGACAAAGATCTGATTAGTCGGGACTAGC |
| pAS-uUGA276 | GCTAGTCCCGACTAATCAGATCTTTGTCTGGAG | |
| A3G uUGA289 | pS-uUGA289 | TTAGTCGGGACTAGCTGACGGCCAAGGATGAAG |
| pAS-uUGA289 | CTTCATCCTTGGCCGTCAGCTAGTCCCGACTAA | |
| A3G SSL | pS-SSL | TAGTGAACCGTCAGAAGCTCCACCACGGCCCAAGCTTGGGCCGTGGTGGAGCTCTCTTTCCCTTTGCA |
| pAS-SSL | TGCAAAGGGAAAGAGAGCTCCACCACGGCCCAAGCTTGGGCCGTGGTGGAGCTTCTGACGGTTCACTA | |
| PCR A3G | pST7-A3G1000 | TAATACGACTCACTATAGGGCTCTTTCCCTTTGCAATTGC |
| pAS-A3G1000 | GCAGGACCCAGGTGTCATTG | |
| RTqPCR | A3G-fp | GGATCCACCCACATTCACTT |
| A3G-rp | ATGCGCTCCACCTCATAAC | |
| RTqPCR | β-actin-fp | GGACTTCGAGCAAGAGATGG |
| β-actin-rp | AGCACTGTGTTGGCGTACAG |
| Target Gene | Primer Name | Primer Sequence | Tm |
|---|---|---|---|
| APOBEC3B | A3B SP1 | GCA CAG CCC CAG GAG AAG CA | 62.7 °C |
| A3B SP2 | GAC CCT GTA GAT CTG GGC CG | 59.6 °C | |
| A3B SP3 | GGC GCT CCA CCT CAT AGC AC | 60.7 °C | |
| A3B SP5 | CGG CCC AGA TCT ACA GGG TC | 59.6 °C | |
| A3B SP6 | ACC AGC AAA GCA ATG TGC TC | 56.6 °C | |
| APOBEC3C | A3C SP1 | GAG ACT CTC CCG TAG CCT TC | 56.5 °C |
| A3C SP2 | CAT GAT CTC CAC AGC GAC CC | 57.9 °C | |
| A3C SP3 | AGA GGC GGG CGG TGA AGA TG | 62.3 °C | |
| A3C SP5 | GGG TCG CTG TGG AGA TCA TG | 57.9 °C | |
| A3C SP6 | ATC CAT CCA CCC CCA CAG AC | 59.2 °C | |
| APOBEC3D | A3DE SP1 | CAT TGG GGT GCT CAG CCA AG | 59.1 °C |
| A3DE SP2 | AGG TGA TCT GGA AGC GCC TG | 59.7 °C | |
| A3DE SP3 | CAC ATT TCT GCG TGG TTC TC | 54.2 °C | |
| A3DE SP5 | TGC AGC CTG AGT CAG GAA GG | 59.5 °C | |
| A3DE SP6 | TAG AGT GCA ATG GCT GGA TC | 55.6 °C | |
| APOBEC3H | A3H SP1 | AGC GGT TTC TCG TGG TCC AC | 60 °C |
| A3H SP2 | TCC ACA CAG AAG CCG CAG CC | 63 °C | |
| A3H SP3 | GTC AAC CAG CTC CCA GGC AC | 61 °C | |
| A3H SP5 | GGC TGC GGC TTC TGT GTG GA | 63 °C | |
| A3H SP6 | GGT CCC GGT GGA GGT CAT GG | 62.5 °C | |
| PCR Anchor Primer | GAC CAC GCG TAT CGA TGT CGA C | 59.8 °C | |
| dT Anchor Primer | GAC CAC GCG TAT CGA TGT CGA CTT TTT TTT TTT TTT TTV |
References
- Strebel, K.; Daugherty, D.; Clouse, K.; Cohen, D.; Folks, T.; Martin, M.A. The HIV A (sor) gene product is essential for virus infectivity. Nat. Cell Biol. 1987, 328, 728–730. [Google Scholar] [CrossRef]
- Sharma, B. Effect of omeprazole and domperidone on adult asthmatics with gastroesophageal reflux. World J. Gastroenterol. 2007, 13, 1706–1710. [Google Scholar] [CrossRef] [Green Version]
- Sakai, H.; Shibata, R.; Sakuragi, J.; Kawamura, M.; Adachi, A. Cell-dependent requirement of human immunodeficiency virus type 1 Vif protein for maturation of virus particles. J. Virol. 1993, 67, 1663–1666. [Google Scholar] [CrossRef] [Green Version]
- An, P.; Bleiber, G.; Duggal, P.; Nelson, G.; May, M.; Mangeat, B.; Alobwede, I.; Trono, D.; Vlahov, D.; Donfield, S.; et al. APOBEC3G Genetic Variants and Their Influence on the Progression to AIDS. J. Virol. 2004, 78, 11070–11076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madani, N.; Kabat, D. An Endogenous Inhibitor of Human Immunodeficiency Virus in Human Lymphocytes Is Overcome by the Viral Vif Protein. J. Virol. 1998, 72, 10251–10255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, J.H.M.; Gaddis, N.; Fouchier, R.; Malim, M. Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat. Med. 1998, 4, 1397–1400. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.; Choi, J.D.; Malim, M. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nat. Cell Biol. 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M. DNA Deamination Mediates Innate Immunity to Retroviral Infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Malim, M.H. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos. Trans. R. Soc. B Biol. Sci. 2008, 364, 675–687. [Google Scholar] [CrossRef] [Green Version]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nat. Cell Biol. 2003, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Mbisa, J.L.; Barr, R.; Thomas, J.; Vandegraaff, N.; Dorweiler, I.J.; Svarovskaia, E.S.; Brown, W.L.; Mansky, L.M.; Gorelick, R.J.; Harris, R.S.; et al. Human Immunodeficiency Virus Type 1 cDNAs Produced in the Presence of APOBEC3G Exhibit Defects in Plus-Strand DNA Transfer and Integration. J. Virol. 2007, 81, 7099–7110. [Google Scholar] [CrossRef] [Green Version]
- Ooms, M.; Brayton, B.; Letko, M.; Maio, S.M.; Pilcher, C.D.; Hecht, F.M.; Barbour, J.D.; Simon, V. HIV-1 Vif Adaptation to Human APOBEC3H Haplotypes. Cell Host Microbe 2013, 14, 411–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Izumi, T.; Misawa, N.; Kobayashi, T.; Yamashita, Y.; Ohmichi, M.; Ito, M.; Takaori-Kondo, A.; Koyanagi, Y. Remarkable Lethal G-to-A Mutations in vif- Proficient HIV-1 Provirus by Individual APOBEC3 Proteins in Humanized Mice. J. Virol. 2010, 84, 9546–9556. [Google Scholar] [CrossRef] [Green Version]
- Refsland, E.W.; Hultquist, J.; Luengas, E.M.; Ikeda, T.; Shaban, N.M.; Law, E.K.; Brown, W.L.; Reilly, C.; Emerman, M.; Harris, R.S. Natural Polymorphisms in Human APOBEC3H and HIV-1 Vif Combine in Primary T Lymphocytes to Affect Viral G-to-A Mutation Levels and Infectivity. PLoS Genet. 2014, 10, e1004761. [Google Scholar] [CrossRef]
- Sato, K.; Takeuchi, J.S.; Misawa, N.; Izumi, T.; Kobayashi, T.; Kimura, Y.; Iwami, S.; Takaori-Kondo, A.; Hu, W.-S.; Aihara, K.; et al. APOBEC3D and APOBEC3F Potently Promote HIV-1 Diversification and Evolution in Humanized Mouse Model. PLoS Pathog. 2014, 10, e1004453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alce, T.M.; Popik, W. APOBEC3G Is Incorporated into Virus-like Particles by a Direct Interaction with HIV-1 Gag Nucleocapsid Protein. J. Biol. Chem. 2004, 279, 34083–34086. [Google Scholar] [CrossRef] [Green Version]
- Douaisi, M.; Dussart, S.; Courcoul, M.; Bessou, G.; Vigne, R.; Decroly, E. HIV-1 and MLV Gag proteins are sufficient to recruit APOBEC3G into virus-like particles. Biochem. Biophys. Res. Commun. 2004, 321, 566–573. [Google Scholar] [CrossRef]
- Strebel, K.; A Khan, M. APOBEC3G encapsidation into HIV-1 virions: Which RNA is it? Retrovirology 2008, 5, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zennou, V.; Perez-Caballero, D.; Göttlinger, H.; Bieniasz, P.D. APOBEC3G Incorporation into Human Immunodeficiency Virus Type 1 Particles. J. Virol. 2004, 78, 12058–12061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svarovskaia, E.S.; Xu, H.; Mbisa, J.L.; Barr, R.; Gorelick, R.J.; Ono, A.; Freed, E.O.; Hu, W.-S.; Pathak, V.K. Human Apolipoprotein B mRNA-editing Enzyme-catalytic Polypeptide-like 3G (APOBEC3G) Is Incorporated into HIV-1 Virions through Interactions with Viral and Nonviral RNAs. J. Biol. Chem. 2004, 279, 35822–35828. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, X.; Ma, J.; Zhang, L.; Ma, L.; Mi, Z.; Zhou, J.; Guo, F.; Kleiman, L.; Cen, S. Human APOBEC3F incorporation into human immunodeficiency virus type 1 particles. Virus Res. 2014, 191, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, K.; Langlois, M.-A. Comparative analysis of the gene-inactivating potential of retroviral restriction factors APOBEC3F and APOBEC3G. J. Gen. Virol. 2015, 96, 2878–2887. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.N.; Verma, M.; Kim, E.-Y.; Wolinsky, S.; Malim, M.H. APOBEC3G Inhibits Elongation of HIV-1 Reverse Transcripts. PLoS Pathog. 2008, 4, e1000231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwatani, Y.; Chan, D.S.; Wang, F.; Stewart-Maynard, K.; Sugiura, W.; Gronenborn, A.M.; Rouzina, I.; Williams, M.C.; Musier-Forsyth, K.; Levin, J.G. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007, 35, 7096–7108. [Google Scholar] [CrossRef] [PubMed]
- Mbisa, J.L.; Bu, W.; Pathak, V.K. APOBEC3F and APOBEC3G Inhibit HIV-1 DNA Integration by Different Mechanisms. J. Virol. 2010, 84, 5250–5259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillick, K.; Pollpeter, D.; Phalora, P.; Kim, E.-Y.; Wolinsky, S.; Malim, M.H. Suppression of HIV-1 Infection by APOBEC3 Proteins in Primary Human CD4+T Cells Is Associated with Inhibition of Processive Reverse Transcription as Well as Excessive Cytidine Deamination. J. Virol. 2013, 87, 1508–1517. [Google Scholar] [CrossRef] [Green Version]
- Pollpeter, D.; Parsons, M.; Sobala, A.E.; Coxhead, S.; Lang, R.D.; Bruns, A.M.; Papaioannou, S.; McDonnell, J.M.; Apolonia, L.; Chowdhury, J.A.; et al. Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nat. Microbiol. 2018, 3, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Seissler, T.; Marquet, R.; Paillart, J.-C. Hijacking of the Ubiquitin/Proteasome Pathway by the HIV Auxiliary Proteins. Viruses 2017, 9, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupfler, B.; Verriez, C.; Gallois-Montbrun, S.; Marquet, R.; Paillart, J.-C. Degradation-Independent Inhibition of APOBEC3G by the HIV-1 Vif Protein. Viruses 2021, 13, 617. [Google Scholar] [CrossRef]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.-F. Induction of APOBEC3G Ubiquitination and Degradation by an HIV-1 Vif-Cul5-SCF Complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Takaori-Kondo, A.; Kobayashi, M.; Tomonaga, M.; Izumi, T.; Fukunaga, K.; Sasada, A.; Abudu, A.; Miyauchi, Y.; Akari, H.; et al. Ubiquitination of APOBEC3 proteins by the Vif–Cullin5–ElonginB–ElonginC complex. Virology 2006, 344, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Mehle, A.; Strack, B.; Ancuta, P.; Zhang, C.; McPike, M.; Gabuzda, D. Vif Overcomes the Innate Antiviral Activity of APOBEC3G by Promoting Its Degradation in the Ubiquitin-Proteasome Pathway. J. Biol. Chem. 2004, 279, 7792–7798. [Google Scholar] [CrossRef] [Green Version]
- Hüttenhain, R.; Xu, J.; Burton, L.A.; Gordon, D.E.; Hultquist, J.; Johnson, J.; Satkamp, L.; Hiatt, J.; Rhee, D.; Baek, K.; et al. ARIH2 Is a Vif-Dependent Regulator of CUL5-Mediated APOBEC3G Degradation in HIV Infection. Cell Host Microbe 2019, 26, 86–99.e7. [Google Scholar] [CrossRef]
- Stanley, B.J.; Ehrlich, E.S.; Short, L.; Yu, Y.; Xiao, Z.; Yu, X.-F.; Xiong, Y. Structural Insight into the Human Immunodeficiency Virus Vif SOCS Box and Its Role in Human E3 Ubiquitin Ligase Assembly. J. Virol. 2008, 82, 8656–8663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergeron, J.R.C.; Huthoff, H.; Veselkov, D.A.; Beavil, R.L.; Simpson, P.; Matthews, S.; Malim, M.H.; Sanderson, M.R. The SOCS-Box of HIV-1 Vif Interacts with ElonginBC by Induced-Folding to Recruit Its Cul5-Containing Ubiquitin Ligase Complex. PLoS Pathog. 2010, 6, e1000925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Dong, L.; Qiu, X.; Wang, Y.; Zhang, B.; Liu, H.; Yu, Y.; Zang, Y.; Yang, M.; Huang, Z. Structural basis for hijacking CBF-β and CUL5 E3 ligase complex by HIV-1 Vif. Nat. Cell Biol. 2014, 505, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Kwon, E.; Hartley, P.D.; Crosby, D.C.; Mann, S.; Krogan, N.J.; Gross, J.D. CBFβ Stabilizes HIV Vif to Counteract APOBEC3 at the Expense of RUNX1 Target Gene Expression. Mol. Cell 2013, 49, 632–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, B.D.; Harris, R.S. Transcriptional regulation of APOBEC3 antiviral immunity through the CBF-β/RUNX axis. Sci. Adv. 2015, 1, e1500296. [Google Scholar] [CrossRef] [Green Version]
- Stopak, K.; de Noronha, C.; Yonemoto, W.; Greene, W.C. HIV-1 Vif Blocks the Antiviral Activity of APOBEC3G by Impairing Both Its Translation and Intracellular Stability. Mol. Cell 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Mercenne, G.; Bernacchi, S.; Richer, D.; Bec, G.; Henriet, S.; Paillart, J.-C.; Marquet, R. HIV-1 Vif binds to APOBEC3G mRNA and inhibits its translation. Nucleic Acids Res. 2009, 38, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.; Khan, M.A.; Miyagi, E.; Plishka, R.; Buckler-White, A.; Strebel, K. The Human Immunodeficiency Virus Type 1 Vif Protein Reduces Intracellular Expression and Inhibits Packaging of APOBEC3G (CEM15), a Cellular Inhibitor of Virus Infectivity. J. Virol. 2003, 77, 11398–11407. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, S.; Libre, C.; Batisse, J.; Mercenne, G.; Richer, D.; Laumond, G.; Decoville, T.; Moog, C.; Marquet, R.; Paillart, J.-C. Translational regulation of APOBEC3G mRNA by Vif requires its 5′UTR and contributes to restoring HIV-1 infectivity. Sci. Rep. 2016, 6, 39507. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.-L.; Llano, M.; Akari, H.; Miyagi, E.; Poeschla, E.M.; Strebel, K.; Bour, S. Codon optimization of the HIV-1 vpu and vif genes stabilizes their mRNA and allows for highly efficient Rev-independent expression. Virology 2004, 319, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Binka, M.; Ooms, M.; Steward, M.; Simon, V. The Activity Spectrum of Vif from Multiple HIV-1 Subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. J. Virol. 2011, 86, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [Green Version]
- Darty, K.; Denise, A.; Ponty, Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 2009, 25, 1974–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.H.; Huang, H.Y.; Hsu, J.B.; Weng, S.L.; Horng, J.T.; Huang, H.D. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. BMC Bioinform. 2013, 14. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Pagliarini, D.J.; Mootha, V.K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA 2009, 106, 7507–7512. [Google Scholar] [CrossRef] [Green Version]
- Renz, P.F.; Valdivia-Francia, F.; Sendoel, A. Some like it translated: Small ORFs in the 5′UTR. Exp. Cell Res. 2020, 396, 112229. [Google Scholar] [CrossRef]
- Johnstone, T.G.; A Bazzini, A.; Giraldez, A.J. Upstream ORF s are prevalent translational repressors in vertebrates. EMBO J. 2016, 35, 706–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, G.-L.; Pauli, A.; Schier, A.F. Conservation of uORF repressiveness and sequence features in mouse, human and zebrafish. Nat. Commun. 2016, 7, 11663. [Google Scholar] [CrossRef] [PubMed]
- Hernández, G.; Osnaya, V.G.; Pérez-Martínez, X. Conservation and Variability of the AUG Initiation Codon Context in Eukaryotes. Trends Biochem. Sci. 2019, 44, 1009–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caval, V.; Suspène, R.; Khalfi, P.; Gaillard, J.; Caignard, G.; Vitour, D.; Roingeard, P.; Vartanian, J.-P.; Wain-Hobson, S. Frame-shifted APOBEC3A encodes two alternative proapoptotic proteins that target the mitochondrial network. J. Biol. Chem. 2021, 297, 297. [Google Scholar] [CrossRef]
- Marin, M.; Golem, S.; Rose, K.M.; Kozak, S.L.; Kabat, D. Human Immunodeficiency Virus Type 1 Vif Functionally Interacts with Diverse APOBEC3 Cytidine Deaminases and Moves with Them between Cytoplasmic Sites of mRNA Metabolism. J. Virol. 2008, 82, 987–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultquist, J.F.; Lengyel, J.A.; Refsland, E.W.; LaRue, R.S.; Lackey, L.; Brown, W.L.; Harris, R.S. Human and Rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H Demonstrate a Conserved Capacity To Restrict Vif-Deficient HIV-1. J. Virol. 2011, 85, 11220–11234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, C.; Peixeiro, I.; Romão, L. Gene Expression Regulation by Upstream Open Reading Frames and Human Disease. PLoS Genet. 2013, 9, e1003529. [Google Scholar] [CrossRef] [Green Version]
- Law, G.L.; Raney, A.; Heusner, C.; Morris, D.R. Polyamine Regulation of Ribosome Pausing at the Upstream Open Reading Frame of S-Adenosylmethionine Decarboxylase. J. Biol. Chem. 2001, 276, 38036–38043. [Google Scholar] [CrossRef]
- Fang, P.; Spevak, C.C.; Wu, C.; Sachs, M.S. A nascent polypeptide domain that can regulate translation elongation. Proc. Natl. Acad. Sci. USA 2004, 101, 4059–4064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J. Mol. Biol. 1987, 196, 947–950. [Google Scholar] [CrossRef]
- Kozak, M. Circumstances and mechanisms of inhibition of translation by secondary structure in eucaryotic mRNAs. Mol. Cell. Biol. 1989, 9, 5134–5142. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 2005, 361, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G. Translational regulation of gcn4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 2005, 59, 407–450. [Google Scholar] [CrossRef] [PubMed]
- Guzikowski, A.R.; Chen, Y.S.; Zid, B.M. Stress-induced mRNP granules: Form and function of processing bodies and stress granules. Wiley Interdiscip. Rev. RNA 2019, 10, e1524. [Google Scholar] [CrossRef]
- White, J.P.; Lloyd, R.E. Regulation of stress granules in virus systems. Trends Microbiol. 2012, 20, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Wethmar, K.; Schulz, J.; Muro, E.M.; Talyan, S.; A Andrade-Navarro, M.; Leutz, A. Comprehensive translational control of tyrosine kinase expression by upstream open reading frames. Oncogene 2015, 35, 1736–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, A.M.; Weitzman, M.D. The spectrum of APOBEC3 activity: From anti-viral agents to anti-cancer opportunities. DNA Repair 2019, 83, 102700. [Google Scholar] [CrossRef] [PubMed]
- Jais, J.-P.; De L’Adulte, F.T.G.D.D.L.; Haioun, C.; Molina, T.; Rickman, D.S.; De Reynies, A.; Berger, F.; Gisselbrecht, C.; Brière, J.; Reyes, F.; et al. The expression of 16 genes related to the cell of origin and immune response predicts survival in elderly patients with diffuse large B-cell lymphoma treated with CHOP and rituximab. Leukemia 2008, 22, 1917–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Pan, T.-H.; Xu, S.; Jia, L.-T.; Zhu, L.-L.; Mao, J.-S.; Zhu, Y.-L.; Cai, J.-T. The virus-induced protein APOBEC3G inhibits anoikis by activation of Akt kinase in pancreatic cancer cells. Sci. Rep. 2015, 5, 12230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Refsland, E.W.; Harris, R.S. The APOBEC3 Family of Retroelement Restriction Factors. Curr. Top. Microbiol. Immunol. 2013, 371, 1–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, J.; Gifford, R.J.; Sato, K. Retroviruses drive the rapid evolution of mammalianAPOBEC3genes. Proc. Natl. Acad. Sci. USA 2020, 117, 610–618. [Google Scholar] [CrossRef] [Green Version]
- Kazazian, H.H.; Wong, C.; Youssoufian, H.; Scott, A.F.; Phillips, D.G.; Antonarakis, S.E. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nat. Cell Biol. 1988, 332, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Morse, B.; Rotherg, P.G.; South, V.J.; Spandorfer, J.M.; Astrin, S.M. Insertional mutagenesis of the myc locus by a LINE-1 sequence in a human breast carcinoma. Nat. Cell Biol. 1988, 333, 87–90. [Google Scholar] [CrossRef]
- Schumann, G. APOBEC3 proteins: Major players in intracellular defence against LINE-1-mediated retrotransposition. Biochem. Soc. Trans. 2007, 35, 637–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinomoto, M.; Kanno, T.; Shimura, M.; Ishizaka, Y.; Kojima, A.; Kurata, T.; Sata, T.; Tokunaga, K. All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 2007, 35, 2955–2964. [Google Scholar] [CrossRef]
- Bogerd, H.P. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 2006, 34, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.-L.; Witkowska, H.E.; Hall, S.C.; Santiago, M.; Soros, V.B.; Esnault, C.; Heidmann, T.; Greene, W.C. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. USA 2006, 103, 15588–15593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulliard, Y.; Turelli, P.; Röhrig, U.F.; Zoete, V.; Mangeat, B.; Michielin, O.; Trono, D. Functional Analysis and Structural Modeling of Human APOBEC3G Reveal the Role of Evolutionarily Conserved Elements in the Inhibition of Human Immunodeficiency Virus Type 1 Infection and Alu Transposition. J. Virol. 2009, 83, 12611–12621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulme, A.E.; Bogerd, H.P.; Cullen, B.R.; Moran, J.V. Selective inhibition of Alu retrotransposition by APOBEC3G. Gene 2007, 390, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Koyama, T.; Arias, J.F.; Iwabu, Y.; Yokoyama, M.; Fujita, H.; Sato, H.; Tokunaga, K. APOBEC3G Oligomerization Is Associated with the Inhibition of Both Alu and LINE-1 Retrotransposition. PLoS ONE 2013, 8, e84228. [Google Scholar] [CrossRef]
- McLaughlin, R.N., Jr.; Gable, J.T.; Wittkopp, C.J.; Emerman, M.; Malik, H.S. Conservation and Innovation of APOBEC3A Restriction Functions during Primate Evolution. Mol. Biol. Evol. 2016, 33, 1889–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, D.R.; Geballe, A.P. Upstream Open Reading Frames as Regulators of mRNA Translation. Mol. Cell. Biol. 2000, 20, 8635–8642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, S.K.; Wek, R.C. Upstream open reading frames differentially regulate genespecific translation in the integrated stress response. J. Biol. Chem. 2016, 291, 16927–16935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreev, D.; O’Connor, P.B.F.; Fahey, C.; Kenny, E.M.; Terenin, I.; E Dmitriev, S.; Cormican, P.; Morris, D.; Shatsky, I.N.; Baranov, P.V. Translation of 5′ leaders is pervasive in genes resistant to eIF2 repression. eLife 2015, 4, e03971. [Google Scholar] [CrossRef]
- Terenin, I.; Akulich, K.A.; Andreev, D.E.; Polyanskaya, S.; Shatsky, I.N.; Dmitriev, S.E. Sliding of a 43S ribosomal complex from the recognized AUG codon triggered by a delay in eIF2-bound GTP hydrolysis. Nucleic Acids Res. 2015, 44, 1882–1893. [Google Scholar] [CrossRef] [Green Version]
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene 2002, 299, 1–34. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Lu, P.D.; Harding, H.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Weingarten-Gabbay, S.; Elias-Kirma, S.; Nir, R.; Gritsenko, A.A.; Stern-Ginossar, N.; Yakhini, Z.; Weinberger, A.; Segal, E. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science 2016, 351, 240. [Google Scholar] [CrossRef] [PubMed]
- Wethmar, K. The regulatory potential of upstream open reading frames in eukaryotic gene expression. Wiley Interdiscip. Rev. RNA 2014, 5, 765–768. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Li, X.; Spatrick, P.; Casillo, R.; Dong, S.; Jacobson, A. Genome-Wide Analysis of mRNAs Regulated by the Nonsense-Mediated and 5′ to 3′ mRNA Decay Pathways in Yeast. Mol. Cell 2003, 12, 1439–1452. [Google Scholar] [CrossRef]
- Mendell, J.T.; A Sharifi, N.; Meyers, J.L.; Martinez-Murillo, F.; Dietz, H.C. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 2004, 36, 1073–1078. [Google Scholar] [CrossRef] [Green Version]
- Medenbach, J.; Seiler, M.; Hentze, M.W. Translational Control via Protein-Regulated Upstream Open Reading Frames. Cell 2011, 145, 902–913. [Google Scholar] [CrossRef] [Green Version]
- Gebauer, F.; Merendino, L.; Hentze, M.; Valcarcel, J. The Drosophila splicing regulator sex-lethal directly inhibits translation of male-specific-lethal 2 mRNA. RNA 1998, 4, 142–150. [Google Scholar]
- Beckmann, K.; Grskovic, M.; Gebauer, F.; Hentze, M.W. A Dual Inhibitory Mechanism Restricts msl-2 mRNA Translation for Dosage Compensation in Drosophila. Cell 2005, 122, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Graindorge, A.; Militti, C.; Gebauer, F. Posttranscriptional control of X-chromosome dosage compensation. Wiley Interdiscip. Rev. RNA 2011, 2, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.E.; Strein, C.; Hentze, M.W. The SXL-UNR Corepressor Complex Uses a PABP-Mediated Mechanism to Inhibit Ribosome Recruitment to msl-2 mRNA. Mol. Cell 2009, 36, 571–582. [Google Scholar] [CrossRef]
- Gallois-Montbrun, S.; Kramer, B.; Swanson, C.; Byers, H.; Lynham, S.; Ward, M.; Malim, M.H. Antiviral Protein APOBEC3G Localizes to Ribonucleoprotein Complexes Found in P Bodies and Stress Granules. J. Virol. 2007, 81, 2165–2178. [Google Scholar] [CrossRef] [Green Version]
- Wichroski, M.J.; Robb, G.B.; Rana, T.M. Human Retroviral Host Restriction Factors APOBEC3G and APOBEC3F Localize to mRNA Processing Bodies. PLoS Pathog. 2006, 2, e41. [Google Scholar] [CrossRef] [PubMed]
- Kozak, S.L.; Marin, M.; Rose, K.M.; Bystrom, C.; Kabat, D. The Anti-HIV-1 Editing Enzyme APOBEC3G Binds HIV-1 RNA and Messenger RNAs That Shuttle between Polysomes and Stress Granules. J. Biol. Chem. 2006, 281, 29105–29119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30, A117. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Libre, C.; Seissler, T.; Guerrero, S.; Batisse, J.; Verriez, C.; Stupfler, B.; Gilmer, O.; Cabrera-Rodriguez, R.; Weber, M.M.; Valenzuela-Fernandez, A.; et al. A Conserved uORF Regulates APOBEC3G Translation and Is Targeted by HIV-1 Vif Protein to Repress the Antiviral Factor. Biomedicines 2022, 10, 13. https://doi.org/10.3390/biomedicines10010013
Libre C, Seissler T, Guerrero S, Batisse J, Verriez C, Stupfler B, Gilmer O, Cabrera-Rodriguez R, Weber MM, Valenzuela-Fernandez A, et al. A Conserved uORF Regulates APOBEC3G Translation and Is Targeted by HIV-1 Vif Protein to Repress the Antiviral Factor. Biomedicines. 2022; 10(1):13. https://doi.org/10.3390/biomedicines10010013
Chicago/Turabian StyleLibre, Camille, Tanja Seissler, Santiago Guerrero, Julien Batisse, Cédric Verriez, Benjamin Stupfler, Orian Gilmer, Romina Cabrera-Rodriguez, Melanie M. Weber, Agustin Valenzuela-Fernandez, and et al. 2022. "A Conserved uORF Regulates APOBEC3G Translation and Is Targeted by HIV-1 Vif Protein to Repress the Antiviral Factor" Biomedicines 10, no. 1: 13. https://doi.org/10.3390/biomedicines10010013
APA StyleLibre, C., Seissler, T., Guerrero, S., Batisse, J., Verriez, C., Stupfler, B., Gilmer, O., Cabrera-Rodriguez, R., Weber, M. M., Valenzuela-Fernandez, A., Cimarelli, A., Etienne, L., Marquet, R., & Paillart, J.-C. (2022). A Conserved uORF Regulates APOBEC3G Translation and Is Targeted by HIV-1 Vif Protein to Repress the Antiviral Factor. Biomedicines, 10(1), 13. https://doi.org/10.3390/biomedicines10010013