
HDL Is Not Dead Yet


Abstract
1. Introduction
2. Early Clinical Trials of HDL-Raising Drugs
3. Recent Clinical Trials of HDL-Raising Drugs
4. Genetic Studies of HDL Causality in CHD Prevention
5. Very High HDL-C May Not Be Good
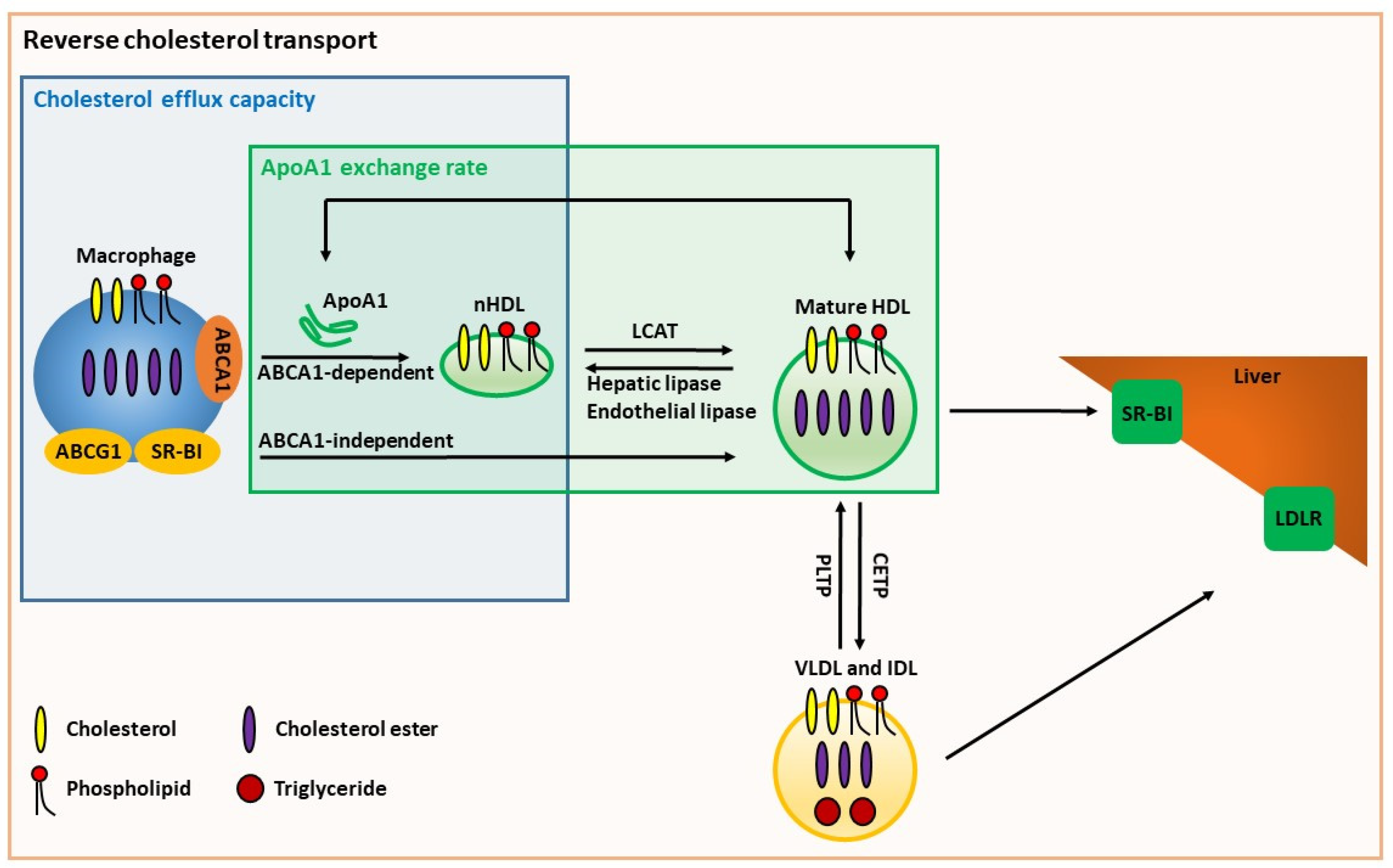
6. Cell-Based HDL Function Assays
6.1. Cholesterol Efflux Capacity
6.2. Endothelial Protective Actions
6.3. Antithrombotic Activity
7. In Vivo Assays for HDL Function
8. HDL Structure and Cell-Free HDL Function Assays
8.1. HDL Subpopulations
8.2. HDL Proteome
8.3. ApoA1 Exchange Assays
8.4. Antioxidant Activity
9. Cholesterol Efflux and Inflammation
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Gordon, T.; Castelli, W.P.; Hjortland, M.C.; Kannel, W.B.; Dawber, T.R. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am. J. Med. 1977, 62, 707–714. [Google Scholar] [CrossRef]
- Gordon, D.J.; Probstfield, J.L.; Garrison, R.J.; Neaton, J.D.; Castelli, W.P.; Knoke, J.D.; Jacobs, D.R., Jr.; Bangdiwala, S.; Tyroler, H.A. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 1989, 79, 8–15. [Google Scholar] [CrossRef]
- Smith, J.D.; Le Goff, W.; Settle, M.; Brubaker, G.; Waelde, C.; Horwitz, A.; Oda, M.N. ABCA1 mediates concurrent cholesterol and phospholipid efflux to apolipoprotein A-I. J. Lipid Res. 2004, 45, 635–644. [Google Scholar] [CrossRef]
- Rothblat, G.H.; Phillips, M.C. High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr. Opin. Lipidol. 2010, 21, 229–238. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Davidson, W.S.; Fayad, Z.A.; Fuster, V.; Goldstein, J.; Hellerstein, M.; Jiang, X.C.; Phillips, M.C.; Rader, D.J.; et al. Cholesterol efflux and atheroprotection: Advancing the concept of reverse cholesterol transport. Circulation 2012, 125, 1905–1919. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A.; Westerterp, M.; von Eckardstein, A.; Remaley, A.; Rye, K.A. HDL in the 21st Century: A Multifunctional Roadmap for Future HDL Research. Circulation 2021, 143, 2293–2309. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, H.; Vincent, V.; Sen, A.; Singh, A.; Roy, A. Changing Perspectives on HDL: From Simple Quantity Measurements to Functional Quality Assessment. J. Lipids 2021, 2021, 5585521. [Google Scholar] [CrossRef]
- Rubin, E.M.; Krauss, R.M.; Spangler, E.A.; Verstuyft, J.G.; Clift, S.M. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature 1991, 353, 265–267. [Google Scholar] [CrossRef]
- Plump, A.S.; Scott, C.J.; Breslow, J.L. Human apolipoprotein A-I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E-deficient mouse. Proc. Natl. Acad. Sci. USA 1994, 91, 9607–9611. [Google Scholar] [CrossRef]
- van Eck, M.; Bos, I.S.; Kaminski, W.E.; Orso, E.; Rothe, G.; Twisk, J.; Bottcher, A.; Van Amersfoort, E.S.; Christiansen-Weber, T.A.; Fung-Leung, W.P.; et al. Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc. Natl. Acad. Sci. USA 2002, 99, 6298–6303. [Google Scholar] [CrossRef]
- The Coronary Drug Project Research Group. Clofibrate and niacin in coronary heart disease. JAMA 1975, 231, 360–381. [Google Scholar] [CrossRef]
- Frick, M.H.; Elo, O.; Haapa, K.; Heinonen, O.P.; Heinsalmi, P.; Helo, P.; Huttunen, J.K.; Kaitaniemi, P.; Koskinen, P.; Manninen, V.; et al. Helsinki Heart Study: Primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N. Engl. J. Med. 1987, 317, 1237–1245. [Google Scholar] [CrossRef]
- Rubins, H.B.; Robins, S.J.; Collins, D.; Fye, C.L.; Anderson, J.W.; Elam, M.B.; Faas, F.H.; Linares, E.; Schaefer, E.J.; Schectman, G.; et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N. Engl. J. Med. 1999, 341, 410–418. [Google Scholar] [CrossRef]
- Rubins, H.B.; Robins, S.J.; Collins, D.; Nelson, D.B.; Elam, M.B.; Schaefer, E.J.; Faas, F.H.; Anderson, J.W. Diabetes, plasma insulin, and cardiovascular disease: Subgroup analysis from the Department of Veterans Affairs high-density lipoprotein intervention trial (VA-HIT). Arch. Intern. Med. 2002, 162, 2597–2604. [Google Scholar] [CrossRef]
- Investigators, A.-H.; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: Trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur. Heart J. 2013, 34, 1279–1291. [Google Scholar] [CrossRef]
- Tall, A.R.; Rader, D.J. Trials and Tribulations of CETP Inhibitors. Circ. Res. 2018, 122, 106–112. [Google Scholar] [CrossRef]
- Ginsberg, H.N.; Reyes-Soffer, G. Niacin: A long history, but a questionable future. Curr. Opin. Lipidol. 2013, 24, 475–479. [Google Scholar] [CrossRef]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef]
- Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; Sammons, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef]
- Nissen, S.E.; Tsunoda, T.; Tuzcu, E.M.; Schoenhagen, P.; Cooper, C.J.; Yasin, M.; Eaton, G.M.; Lauer, M.A.; Sheldon, W.S.; Grines, C.L.; et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: A randomized controlled trial. JAMA 2003, 290, 2292–2300. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Puri, R.; Ballantyne, C.M.; Jukema, J.W.; Kastelein, J.J.P.; Koenig, W.; Wright, R.S.; Kallend, D.; Wijngaard, P.; Borgman, M.; et al. Effect of Infusion of High-Density Lipoprotein Mimetic Containing Recombinant Apolipoprotein A-I Milano on Coronary Disease in Patients with an Acute Coronary Syndrome in the MILANO-PILOT Trial: A Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 806–814. [Google Scholar] [CrossRef]
- Didichenko, S.A.; Navdaev, A.V.; Cukier, A.M.; Gille, A.; Schuetz, P.; Spycher, M.O.; Therond, P.; Chapman, M.J.; Kontush, A.; Wright, S.D. Enhanced HDL Functionality in Small HDL Species Produced Upon Remodeling of HDL by Reconstituted HDL, CSL112: Effects on Cholesterol Efflux, Anti-Inflammatory and Antioxidative Activity. Circ. Res. 2016, 119, 751–763. [Google Scholar] [CrossRef]
- Gille, A.; D’Andrea, D.; Tortorici, M.A.; Hartel, G.; Wright, S.D. CSL112 (Apolipoprotein A-I [Human]) Enhances Cholesterol Efflux Similarly in Healthy Individuals and Stable Atherosclerotic Disease Patients. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 953–963. [Google Scholar] [CrossRef]
- He, H.; Hong, K.; Liu, L.; Schwendeman, A. Artificial high-density lipoprotein-mimicking nanotherapeutics for the treatment of cardiovascular diseases. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2021, 13, e1737. [Google Scholar] [CrossRef]
- Frikke-Schmidt, R.; Nordestgaard, B.G.; Stene, M.C.; Sethi, A.A.; Remaley, A.T.; Schnohr, P.; Grande, P.; Tybjaerg-Hansen, A. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA 2008, 299, 2524–2532. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, P.; Khetarpal, S.A.; Larach, D.B.; Hancock-Cerutti, W.F.; Millar, J.S.; Cuchel, M.; DerOhannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 2016, 351, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Haase, C.L.; Tybjaerg-Hansen, A.; Qayyum, A.A.; Schou, J.; Nordestgaard, B.G.; Frikke-Schmidt, R. LCAT, HDL cholesterol and ischemic cardiovascular disease: A Mendelian randomization study of HDL cholesterol in 54,500 individuals. J. Clin. Endocrinol. Metab. 2012, 97, E248–E256. [Google Scholar] [CrossRef]
- Johannsen, T.H.; Frikke-Schmidt, R.; Schou, J.; Nordestgaard, B.G.; Tybjaerg-Hansen, A. Genetic inhibition of CETP, ischemic vascular disease and mortality, and possible adverse effects. J. Am. Coll. Cardiol. 2012, 60, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Klarin, D.; Damrauer, S.M.; Cho, K.; Sun, Y.V.; Teslovich, T.M.; Honerlaw, J.; Gagnon, D.R.; DuVall, S.L.; Li, J.; Peloso, G.M.; et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet. 2018, 50, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Holm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef]
- Thomas, D.G.; Wei, Y.; Tall, A.R. Lipid and metabolic syndrome traits in coronary artery disease: A Mendelian randomization study. J. Lipid Res. 2021, 62, 100044. [Google Scholar] [CrossRef]
- Prats-Uribe, A.; Sayols-Baixeras, S.; Fernandez-Sanles, A.; Subirana, I.; Carreras-Torres, R.; Vilahur, G.; Civeira, F.; Marrugat, J.; Fito, M.; Hernaez, A.; et al. High-density lipoprotein characteristics and coronary artery disease: A Mendelian randomization study. Metabolism 2020, 112, 154351. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, J.; Miao, Z.; Zhang, N.R.; Hennessy, S.; Small, D.S.; Rader, D.J. A Mendelian randomization study of the role of lipoprotein subfractions in coronary artery disease. Elife 2021, 10, e58361. [Google Scholar] [CrossRef] [PubMed]
- Stensvold, I.; Urdal, P.; Thurmer, H.; Tverdal, A.; Lund-Larsen, P.G.; Foss, O.P. High-density lipoprotein cholesterol and coronary, cardiovascular and all cause mortality among middle-aged Norwegian men and women. Eur. Heart J. 1992, 13, 1155–1163. [Google Scholar] [CrossRef]
- van der Steeg, W.A.; Holme, I.; Boekholdt, S.M.; Larsen, M.L.; Lindahl, C.; Stroes, E.S.; Tikkanen, M.J.; Wareham, N.J.; Faergeman, O.; Olsson, A.G.; et al. High-density lipoprotein cholesterol, high-density lipoprotein particle size, and apolipoprotein A-I: Significance for cardiovascular risk: The IDEAL and EPIC-Norfolk studies. J. Am. Coll. Cardiol. 2008, 51, 634–642. [Google Scholar] [CrossRef]
- Wilkins, J.T.; Ning, H.; Stone, N.J.; Criqui, M.H.; Zhao, L.; Greenland, P.; Lloyd-Jones, D.M. Coronary heart disease risks associated with high levels of HDL cholesterol. J. Am. Heart Assoc. 2014, 3, e000519. [Google Scholar] [CrossRef]
- Ko, D.T.; Alter, D.A.; Guo, H.; Koh, M.; Lau, G.; Austin, P.C.; Booth, G.L.; Hogg, W.; Jackevicius, C.A.; Lee, D.S.; et al. High-Density Lipoprotein Cholesterol and Cause-Specific Mortality in Individuals Without Previous Cardiovascular Conditions: The CANHEART Study. J. Am. Coll. Cardiol. 2016, 68, 2073–2083. [Google Scholar] [CrossRef]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: Two prospective cohort studies. Eur. Heart J. 2017, 38, 2478–2486. [Google Scholar] [CrossRef] [PubMed]
- Bowe, B.; Xie, Y.; Xian, H.; Balasubramanian, S.; Zayed, M.A.; Al-Aly, Z. High Density Lipoprotein Cholesterol and the Risk of All-Cause Mortality among U.S. Veterans. Clin. J. Am. Soc. Nephrol. 2016, 11, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Hamer, M.; O’Donovan, G.; Stamatakis, E. High-Density Lipoprotein Cholesterol and Mortality: Too Much of a Good Thing? Arterioscler. Thromb. Vasc. Biol. 2018, 38, 669–672. [Google Scholar] [CrossRef]
- Hirata, A.; Sugiyama, D.; Watanabe, M.; Tamakoshi, A.; Iso, H.; Kotani, K.; Kiyama, M.; Yamada, M.; Ishikawa, S.; Murakami, Y.; et al. Association of extremely high levels of high-density lipoprotein cholesterol with cardiovascular mortality in a pooled analysis of 9 cohort studies including 43,407 individuals: The EPOCH-JAPAN study. J. Clin. Lipidol. 2018, 12, 674–684.e5. [Google Scholar] [CrossRef]
- Li, Z.H.; Lv, Y.B.; Zhong, W.F.; Gao, X.; Kraus, V.B.; Zou, M.C.; Zhang, X.R.; Li, F.R.; Yuan, J.Q.; Shi, X.M.; et al. High-Density Lipoprotein Cholesterol and All-Cause and Cause-Specific Mortality Among the Elderly. J. Clin. Endocrinol. Metab. 2019, 104, 3370–3378. [Google Scholar] [CrossRef]
- Zhong, G.C.; Huang, S.Q.; Peng, Y.; Wan, L.; Wu, Y.Q.; Hu, T.Y.; Hu, J.J.; Hao, F.B. HDL-C is associated with mortality from all causes, cardiovascular disease and cancer in a J-shaped dose-response fashion: A pooled analysis of 37 prospective cohort studies. Eur. J. Prev. Cardiol. 2020, 27, 1187–1203. [Google Scholar] [CrossRef]
- Oh, I.H.; Hur, J.K.; Ryoo, J.H.; Jung, J.Y.; Park, S.K.; Yang, H.J.; Choi, J.M.; Jung, K.W.; Won, Y.J.; Oh, C.M. Very high high-density lipoprotein cholesterol is associated with increased all-cause mortality in South Koreans. Atherosclerosis 2019, 283, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Han, K.; Park, S.H.; Kim, M.K.; Yoon, K.H.; Lee, S.H. High-Density Lipoprotein Cholesterol and the Risk of Myocardial Infarction, Stroke, and Cause-Specific Mortality: A Nationwide Cohort Study in Korea. J. Lipid Atheroscler. 2021, 10, 74–87. [Google Scholar] [CrossRef]
- Singh, K.; Rohatgi, A. Examining the paradox of high high-density lipoprotein and elevated cardiovascular risk. J. Thorac. Dis. 2018, 10, 109–112. [Google Scholar] [CrossRef]
- Zhang, Y.; Da Silva, J.R.; Reilly, M.; Billheimer, J.T.; Rothblat, G.H.; Rader, D.J. Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J. Clin. Investig. 2005, 115, 2870–2874. [Google Scholar] [CrossRef]
- Braun, A.; Trigatti, B.L.; Post, M.J.; Sato, K.; Simons, M.; Edelberg, J.M.; Rosenberg, R.D.; Schrenzel, M.; Krieger, M. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ. Res. 2002, 90, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Tang, W.H.; Mosior, M.K.; Huang, Y.; Wu, Y.; Matter, W.; Gao, V.; Schmitt, D.; Didonato, J.A.; Fisher, E.A.; et al. Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1696–1705. [Google Scholar] [CrossRef]
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Mucksavage, M.L.; Luben, R.; Billheimer, J.; et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: A prospective case-control study. Lancet Diabetes Endocrinol. 2015, 3, 507–513. [Google Scholar] [CrossRef]
- Khera, A.V.; Demler, O.V.; Adelman, S.J.; Collins, H.L.; Glynn, R.J.; Ridker, P.M.; Rader, D.J.; Mora, S. Cholesterol Efflux Capacity, High-Density Lipoprotein Particle Number, and Incident Cardiovascular Events: An Analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2017, 135, 2494–2504. [Google Scholar] [CrossRef]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, W.; Zheng, P.; Brubaker, G.; Smith, J.D. Identification of the cAMP-responsive enhancer of the murine ABCA1 gene: Requirement for CREB1 and STAT3/4 elements. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 527–533. [Google Scholar] [CrossRef]
- de la Llera-Moya, M.; Drazul-Schrader, D.; Asztalos, B.F.; Cuchel, M.; Rader, D.J.; Rothblat, G.H. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 796–801. [Google Scholar] [CrossRef]
- Monette, J.S.; Hutchins, P.M.; Ronsein, G.E.; Wimberger, J.; Irwin, A.D.; Tang, C.; Sara, J.D.; Shao, B.; Vaisar, T.; Lerman, A.; et al. Patients with Coronary Endothelial Dysfunction Have Impaired Cholesterol Efflux Capacity and Reduced HDL Particle Concentration. Circ. Res. 2016, 119, 83–90. [Google Scholar] [CrossRef]
- Hunjadi, M.; Lamina, C.; Kahler, P.; Bernscherer, T.; Viikari, J.; Lehtimaki, T.; Kahonen, M.; Hurme, M.; Juonala, M.; Taittonen, L.; et al. HDL cholesterol efflux capacity is inversely associated with subclinical cardiovascular risk markers in young adults: The cardiovascular risk in Young Finns study. Sci. Rep. 2020, 10, 19223. [Google Scholar] [CrossRef]
- Shea, S.; Stein, J.H.; Jorgensen, N.W.; McClelland, R.L.; Tascau, L.; Shrager, S.; Heinecke, J.W.; Yvan-Charvet, L.; Tall, A.R. Cholesterol Mass Efflux Capacity, Incident Cardiovascular Disease, and Progression of Carotid Plaque. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 89–96. [Google Scholar] [CrossRef]
- Zheng, L.; Nukuna, B.; Brennan, M.L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, D.; Fu, X.; Thomson, L.; Fox, P.L.; et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Investig. 2004, 114, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, Z.; Riwanto, M.; Gao, S.; Levison, B.S.; Gu, X.; Fu, X.; Wagner, M.A.; Besler, C.; Gerstenecker, G.; et al. Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J. Clin. Investig. 2013, 123, 3815–3828. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.Q.; Brubaker, G.; Wu, Z.; Zheng, L.; Willard, B.; Kinter, M.; Hazen, S.L.; Smith, J.D. Apolipoprotein A-I tryptophan substitution leads to resistance to myeloperoxidase-mediated loss of function. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; DiDonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef]
- Undurti, A.; Huang, Y.; Lupica, J.A.; Smith, J.D.; DiDonato, J.A.; Hazen, S.L. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J. Biol. Chem. 2009, 284, 30825–30835. [Google Scholar] [CrossRef] [PubMed]
- Van Lenten, B.J.; Hama, S.Y.; de Beer, F.C.; Stafforini, D.M.; McIntyre, T.M.; Prescott, S.M.; La Du, B.N.; Fogelman, A.M.; Navab, M. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J. Clin. Investig. 1995, 96, 2758–2767. [Google Scholar] [CrossRef]
- Seetharam, D.; Mineo, C.; Gormley, A.K.; Gibson, L.L.; Vongpatanasin, W.; Chambliss, K.L.; Hahner, L.D.; Cummings, M.L.; Kitchens, R.L.; Marcel, Y.L.; et al. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ. Res. 2006, 98, 63–72. [Google Scholar] [CrossRef]
- Mineo, C.; Deguchi, H.; Griffin, J.H.; Shaul, P.W. Endothelial and antithrombotic actions of HDL. Circ. Res. 2006, 98, 1352–1364. [Google Scholar] [CrossRef]
- Badrnya, S.; Assinger, A.; Volf, I. Native high density lipoproteins (HDL) interfere with platelet activation induced by oxidized low density lipoproteins (OxLDL). Int. J. Mol. Sci. 2013, 14, 10107–10121. [Google Scholar] [CrossRef]
- Hewing, B.; Parathath, S.; Barrett, T.; Chung, W.K.; Astudillo, Y.M.; Hamada, T.; Ramkhelawon, B.; Tallant, T.C.; Yusufishaq, M.S.; Didonato, J.A.; et al. Effects of native and myeloperoxidase-modified apolipoprotein a-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Raper, A.C.; Conlon, D.M.; Pryma, D.A.; Freifelder, R.H.; Poria, R.; Cromley, D.; Li, X.; Dunbar, R.L.; French, B.; et al. A novel approach to measuring macrophage-specific reverse cholesterol transport in vivo in humans. J. Lipid Res. 2017, 58, 752–762. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Chapman, M.J.; Fazio, S.; Hussain, M.M.; Kontush, A.; Krauss, R.M.; Otvos, J.D.; Remaley, A.T.; Schaefer, E.J. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin. Chem. 2011, 57, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Asztalos, B.F.; Sloop, C.H.; Wong, L.; Roheim, P.S. Two-dimensional electrophoresis of plasma lipoproteins: Recognition of new apo A-I-containing subpopulations. Biochim. Biophys. Acta 1993, 1169, 291–300. [Google Scholar] [CrossRef]
- Asztalos, B.F.; de la Llera-Moya, M.; Dallal, G.E.; Horvath, K.V.; Schaefer, E.J.; Rothblat, G.H. Differential effects of HDL subpopulations on cellular ABCA1- and SR-BI-mediated cholesterol efflux. J. Lipid Res. 2005, 46, 2246–2253. [Google Scholar] [CrossRef]
- Asztalos, B.F.; Batista, M.; Horvath, K.V.; Cox, C.E.; Dallal, G.E.; Morse, J.S.; Brown, G.B.; Schaefer, E.J. Change in alpha1 HDL concentration predicts progression in coronary artery stenosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 847–852. [Google Scholar] [CrossRef]
- Asztalos, B.F.; Collins, D.; Cupples, L.A.; Demissie, S.; Horvath, K.V.; Bloomfield, H.E.; Robins, S.J.; Schaefer, E.J. Value of high-density lipoprotein (HDL) subpopulations in predicting recurrent cardiovascular events in the Veterans Affairs HDL Intervention Trial. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2185–2191. [Google Scholar] [CrossRef]
- Lounila, J.; Ala-Korpela, M.; Jokisaari, J.; Savolainen, M.J.; Kesaniemi, Y.A. Effects of orientational order and particle size on the NMR line positions of lipoproteins. Phys. Rev. Lett. 1994, 72, 4049–4052. [Google Scholar] [CrossRef]
- El Harchaoui, K.; Arsenault, B.J.; Franssen, R.; Despres, J.P.; Hovingh, G.K.; Stroes, E.S.; Otvos, J.D.; Wareham, N.J.; Kastelein, J.J.; Khaw, K.T.; et al. High-density lipoprotein particle size and concentration and coronary risk. Ann. Intern. Med. 2009, 150, 84–93. [Google Scholar] [CrossRef]
- Mora, S.; Otvos, J.D.; Rifai, N.; Rosenson, R.S.; Buring, J.E.; Ridker, P.M. Lipoprotein particle profiles by nuclear magnetic resonance compared with standard lipids and apolipoproteins in predicting incident cardiovascular disease in women. Circulation 2009, 119, 931–939. [Google Scholar] [CrossRef]
- Singh, K.; Chandra, A.; Sperry, T.; Joshi, P.H.; Khera, A.; Virani, S.S.; Ballantyne, C.M.; Otvos, J.D.; Dullaart, R.P.F.; Gruppen, E.G.; et al. Associations Between High-Density Lipoprotein Particles and Ischemic Events by Vascular Domain, Sex, and Ethnicity: A Pooled Cohort Analysis. Circulation 2020, 142, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Shah, A.S.; Sexmith, H.; Gordon, S.M. The HDL Proteome Watch: Compilation of studies leads to new insights on HDL function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1867, 159072. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Collier, T.S.; Dai, D.L.Y.; Chen, V.; Hollander, Z.; Ng, R.T.; McManus, B.M.; Balshaw, R.; Apostolidou, S.; Penn, M.S.; et al. Development and Validation of Apolipoprotein AI-Associated Lipoprotein Proteome Panel for the Prediction of Cholesterol Efflux Capacity and Coronary Artery Disease. Clin. Chem. 2019, 65, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Cavigiolio, G.; Geier, E.G.; Shao, B.; Heinecke, J.W.; Oda, M.N. Exchange of apolipoprotein A-I between lipid-associated and lipid-free states: A potential target for oxidative generation of dysfunctional high density lipoproteins. J. Biol. Chem. 2010, 285, 18847–18857. [Google Scholar] [CrossRef]
- Borja, M.S.; Zhao, L.; Hammerson, B.; Tang, C.; Yang, R.; Carson, N.; Fernando, G.; Liu, X.; Budamagunta, M.S.; Genest, J.; et al. HDL-apoA-I exchange: Rapid detection and association with atherosclerosis. PLoS ONE 2013, 8, e71541. [Google Scholar] [CrossRef]
- Borja, M.S.; Ng, K.F.; Irwin, A.; Hong, J.; Wu, X.; Isquith, D.; Zhao, X.Q.; Prazen, B.; Gildengorin, V.; Oda, M.N.; et al. HDL-apolipoprotein A-I exchange is independently associated with cholesterol efflux capacity. J. Lipid Res. 2015, 56, 2002–2009. [Google Scholar] [CrossRef]
- Heier, M.; Ofstad, A.P.; Borja, M.S.; Brunborg, C.; Endresen, K.; Gullestad, L.; Birkeland, K.I.; Johansen, O.E.; Oda, M.N. High-density lipoprotein function is associated with atherosclerotic burden and cardiovascular outcomes in type 2 diabetes. Atherosclerosis 2019, 282, 183–187. [Google Scholar] [CrossRef]
- Wang, S.; Gulshan, K.; Brubaker, G.; Hazen, S.L.; Smith, J.D. ABCA1 mediates unfolding of apolipoprotein AI N terminus on the cell surface before lipidation and release of nascent high-density lipoprotein. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1197–1205. [Google Scholar] [CrossRef]
- Lorkowski, S.W.; Brubaker, G.; Li, L.; Li, X.S.; Hazen, S.L.; Smith, J.D. A Novel Cell-Free Fluorescent Assay for HDL Function: Low Apolipoprotein A1 Exchange Rate Associated with Increased Incident Cardiovascular Events. J. Appl. Lab. Med. 2020, 5, 544–557. [Google Scholar] [CrossRef]
- Lorkowski, S.W.; Brubaker, G.; Rotroff, D.M.; Kashyap, S.R.; Bhatt, D.L.; Nissen, S.E.; Schauer, P.R.; Aminian, A.; Smith, J.D. Bariatric Surgery Improves HDL Function Examined by ApoA1 Exchange Rate and Cholesterol Efflux Capacity in Patients with Obesity and Type 2 Diabetes. Biomolecules 2020, 10, 551. [Google Scholar] [CrossRef]
- Harada, A.; Toh, R.; Murakami, K.; Kiriyama, M.; Yoshikawa, K.; Miwa, K.; Kubo, T.; Irino, Y.; Mori, K.; Tanaka, N.; et al. Cholesterol Uptake Capacity: A New Measure of HDL Functionality for Coronary Risk Assessment. J. Appl. Lab. Med. 2017, 2, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, Y.; Lai, S.J.; Yamazaki, A.; Nakamura, A.; Ohkawa, R.; Yano, K.; Kameda, T.; Okubo, S.; Shimano, S.; Hagihara, M.; et al. Validation and application of a novel cholesterol efflux assay using immobilized liposomes as a substitute for cultured cells. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Barnett, J.; Fong, L.G. High-density lipoprotein inhibits the oxidative modification of low-density lipoprotein. Biochim. Biophys. Acta 1990, 1044, 275–283. [Google Scholar] [CrossRef]
- Mackness, M.I.; Arrol, S.; Abbott, C.; Durrington, P.N. Protection of low-density lipoprotein against oxidative modification by high-density lipoprotein associated paraoxonase. Atherosclerosis 1993, 104, 129–135. [Google Scholar] [CrossRef]
- Navab, M.; Hama, S.Y.; Hough, G.P.; Subbanagounder, G.; Reddy, S.T.; Fogelman, A.M. A cell-free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J. Lipid Res. 2001, 42, 1308–1317. [Google Scholar] [CrossRef]
- Wang, N.; Lan, D.; Chen, W.; Matsuura, F.; Tall, A.R. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 9774–9779. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Pagler, T.; Gautier, E.L.; Avagyan, S.; Siry, R.L.; Han, S.; Welch, C.L.; Wang, N.; Randolph, G.J.; Snoeck, H.W.; et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 2010, 328, 1689–1693. [Google Scholar] [CrossRef]
- Westerterp, M.; Murphy, A.J.; Wang, M.; Pagler, T.A.; Vengrenyuk, Y.; Kappus, M.S.; Gorman, D.J.; Nagareddy, P.R.; Zhu, X.; Abramowicz, S.; et al. Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ. Res. 2013, 112, 1456–1465. [Google Scholar] [CrossRef]
- Westerterp, M.; Tsuchiya, K.; Tattersall, I.W.; Fotakis, P.; Bochem, A.E.; Molusky, M.M.; Ntonga, V.; Abramowicz, S.; Parks, J.S.; Welch, C.L.; et al. Deficiency of ATP-Binding Cassette Transporters A1 and G1 in Endothelial Cells Accelerates Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1328–1337. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; la Bastide-van Gemert, S.; Wang, N.; et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef]
- Bochem, A.E.; van der Valk, F.M.; Tolani, S.; Stroes, E.S.; Westerterp, M.; Tall, A.R. Increased Systemic and Plaque Inflammation in ABCA1 Mutation Carriers with Attenuation by Statins. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1663–1669. [Google Scholar] [CrossRef]
- Fotakis, P.; Kothari, V.; Thomas, D.G.; Westerterp, M.; Molusky, M.M.; Altin, E.; Abramowicz, S.; Wang, N.; He, Y.; Heinecke, J.W.; et al. Anti-Inflammatory Effects of HDL (High-Density Lipoprotein) in Macrophages Predominate Over Proinflammatory Effects in Atherosclerotic Plaques. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e253–e272. [Google Scholar] [CrossRef]


{kind=link}
| Clinical Study [Reference] | HDL Function Assay | Outcome |
|---|---|---|
| Dallas Heart Study [56] | CEC using J774 macrophages and Bodipy-cholesterol | Inverse correlation between CEC and incident cardiovascular events |
| Epic-Norfolk Study [54] | CEC using J774 macrophages and 3H-cholesterol | Inverse correlation between CEC and incident CHD |
| GeneBank [53] | CEC using RAW macrophages and 3H-cholesterol | Positive correlation between CEC and incident MACE |
| Jupiter rial [55] | CEC using J774 macrophages and 3H-cholesterol | Inverse correlation between CEC and incident CVD at statin therapy but not at baseline |
| MESA study [61] | Cholesterol mass efflux capacity using THP-1 differentiated macrophages | Inverse correlation between cholesterol mass efflux capacity and incident CVD |
| Assay | Methodology | Clinical Relevance | Practicality |
|---|---|---|---|
| NMR spectroscopy | Quantify the number and size of HDLs | Inverse association between HDL-particle concentration and CHD | Requires NMR spectroscopy |
| ApoA1-associated proteome panel | Generate a multivariate algorithm for CHD prediction through multiprotein analysis | Predictive for CHD | High-throughput, requires mass spectrometry |
| ApoA1 exchange assay | Quantify spin-labeled lipid-free apoA1 probe exchanged into HDL by electron paramagnetic resonance spectrometry | Inverse association between apoA1 exchange and prevalent CHD in small clinical studies | High-throughput, requires an electron paramagnetic resonance spectrometer |
| ApoA1 exchange rate | Quantify the rate of NBD/Alexa647-labeled lipid-free apoA1 probe exchanged into HDL | Inverse association between apoA1 exchange rate and incident MACE | High-throughput |
| Cholesterol uptake capacity | Detect the capacity of HDL to take up Bodipy-labeled cholesterol from liposomes | Inversely associated with the requirement for revascularization in a small clinical study | High-throughput |
| Cholesterol exchange assay | Quantify Bodipy-labeled cholesterol from liposomes exchanged into HDL | Lacks clinical evidence | High-throughput |
| Antioxidant assay | Detect the ability of HDL in preventing the formation of oxidized phospholipids | HDLs from CHD patients lose antioxidant function in small clinical samples | High-throughput |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorkowski, S.W.; Smith, J.D. HDL Is Not Dead Yet. Biomedicines 2022, 10, 128. https://doi.org/10.3390/biomedicines10010128
Lorkowski SW, Smith JD. HDL Is Not Dead Yet. Biomedicines. 2022; 10(1):128. https://doi.org/10.3390/biomedicines10010128
Chicago/Turabian StyleLorkowski, Shuhui Wang, and Jonathan D. Smith. 2022. "HDL Is Not Dead Yet" Biomedicines 10, no. 1: 128. https://doi.org/10.3390/biomedicines10010128
APA StyleLorkowski, S. W., & Smith, J. D. (2022). HDL Is Not Dead Yet. Biomedicines, 10(1), 128. https://doi.org/10.3390/biomedicines10010128