
Definition of an Inflammatory Biomarker Signature in Plasma-Derived Extracellular Vesicles of Glioblastoma Patients

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Clinical Samples
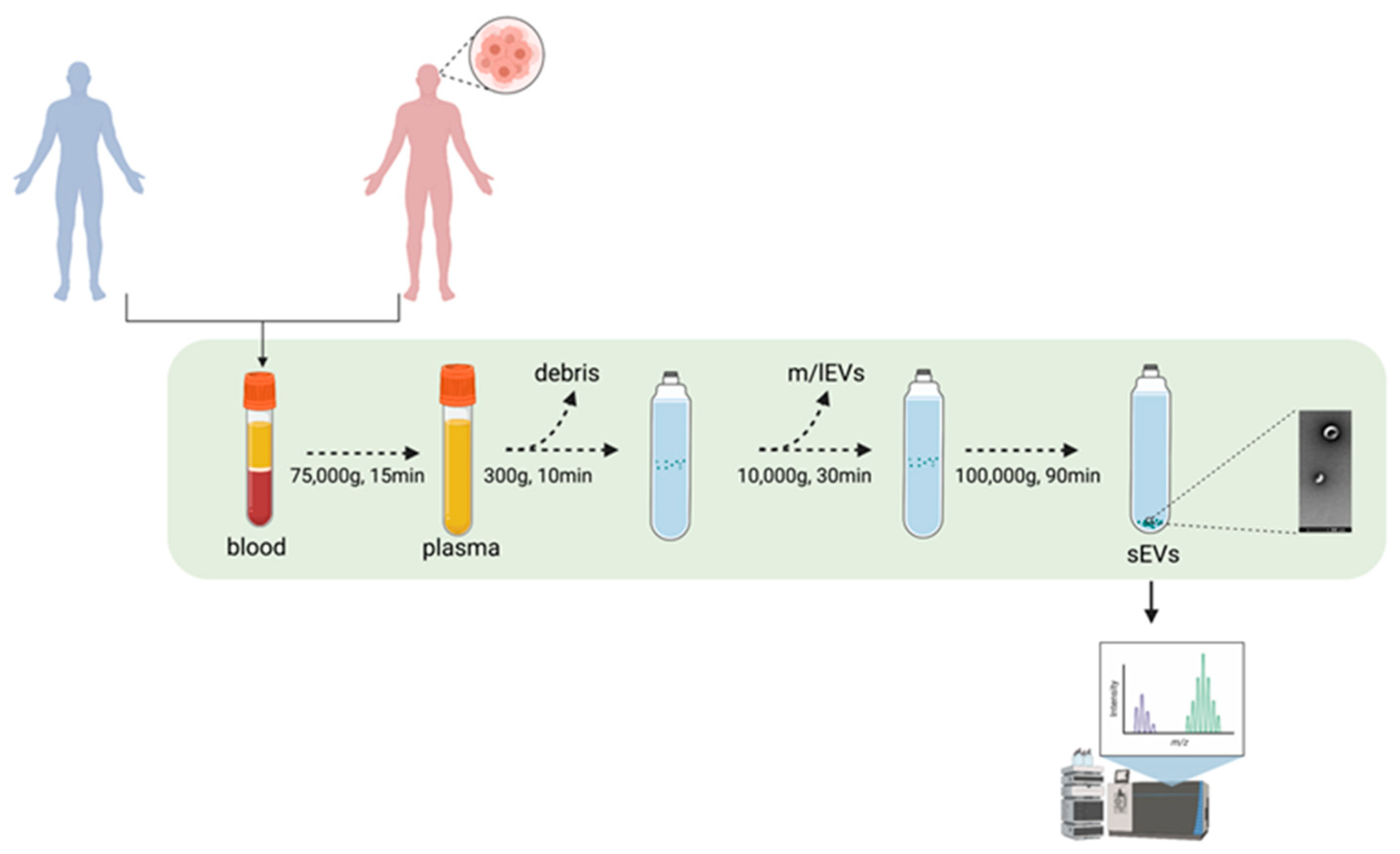
2.2. Differential Ultracentrifugation for Concentration of Extracellular Vesicles
2.3. Nanoparticles Tracking Analysis (NTA)
2.4. Coomassie Blue Staining
2.5. Western Blotting
2.6. Transmission Electron Microscopy
2.7. sEVs Analysis by ExoView
2.8. Mass Spectrometry
2.9. Proteomic Data Analysis
2.10. The Cancer Genome Atlas and Clinical Proteomic Analysis Consortium Data
2.11. Statistical Analysis
3. Results
3.1. Patients’ Clinical Characteristics
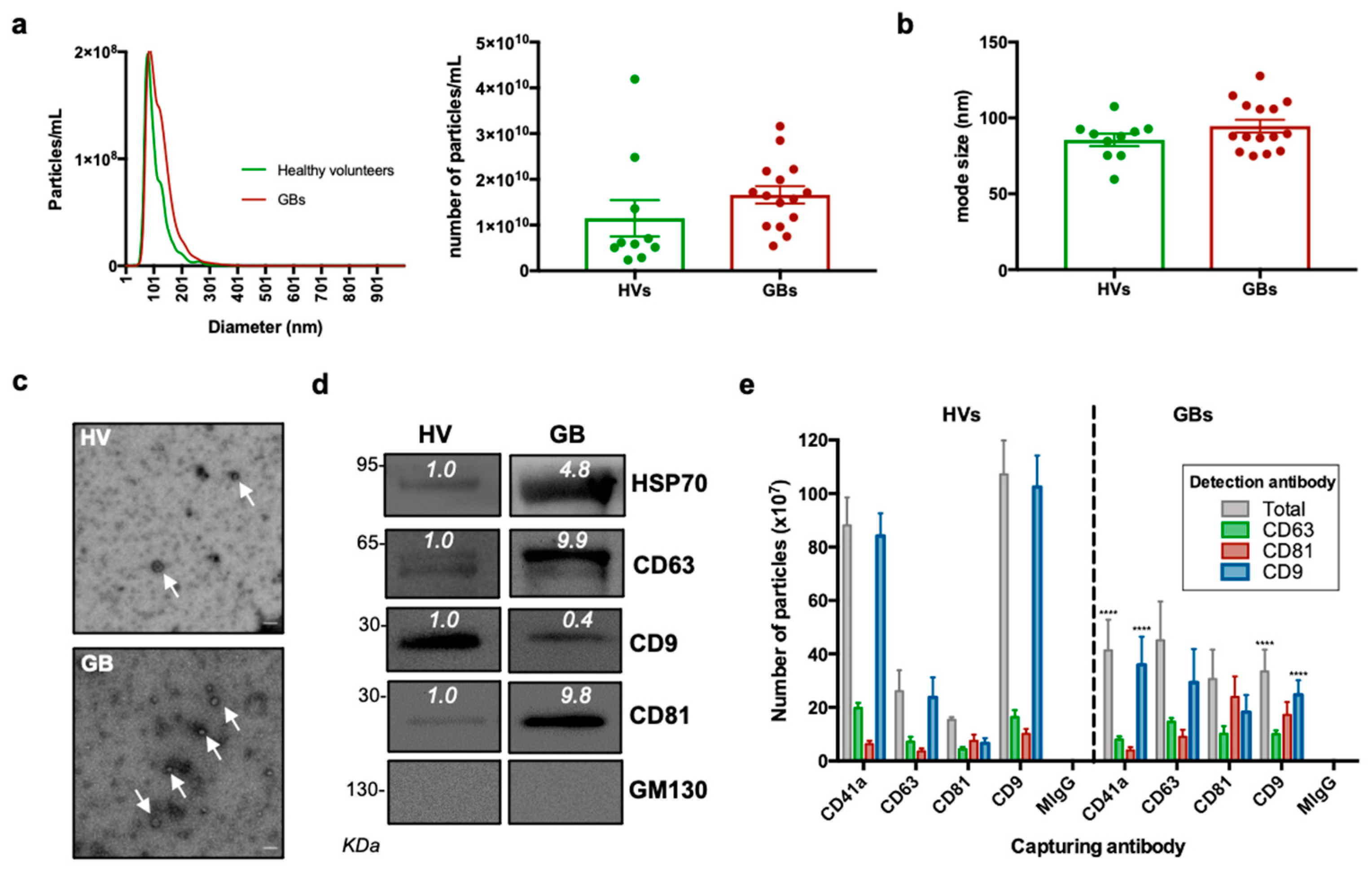
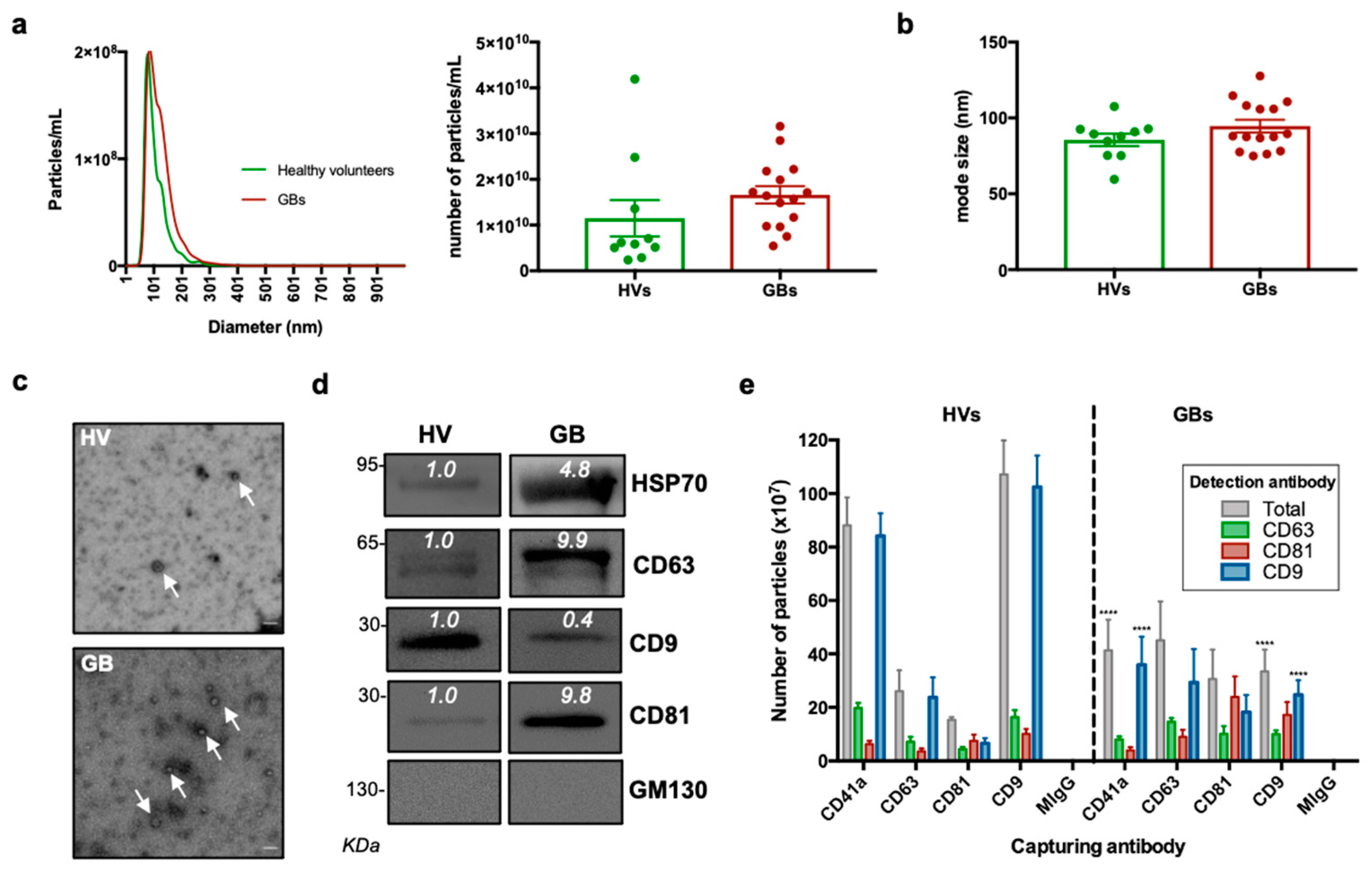
3.2. Characterisation of sEVs Isolated from Peripheral Blood
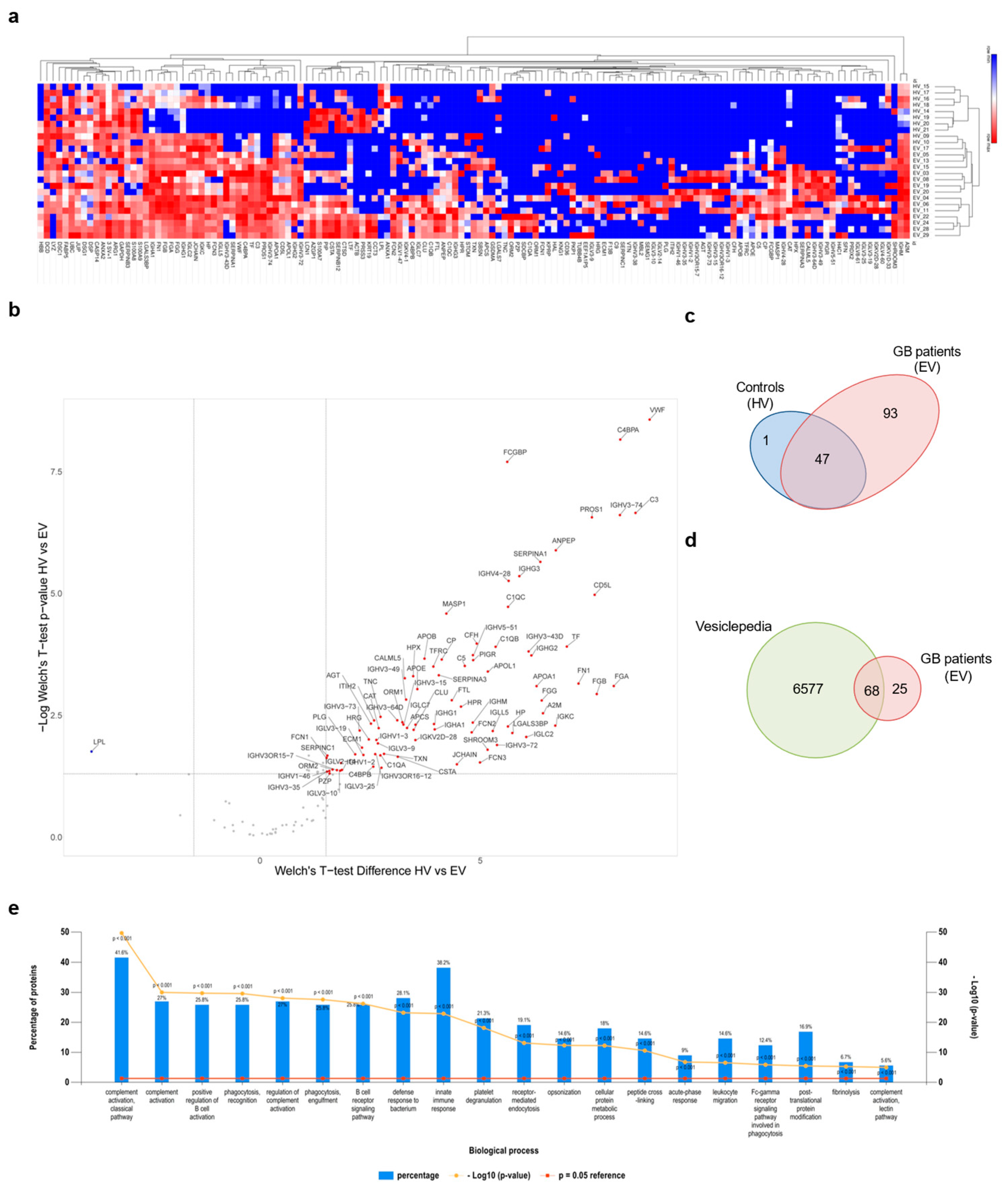
3.3. Proteomic Analysis of GB sEV Cargo Provided a GB-Related Signature
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Cruickshanks, N.; Zhang, Y.; Yuan, F.; Pahuski, M.; Gibert, M.; Abounader, R. Role and Therapeutic Targeting of the HGF/MET Pathway in Glioblastoma. Cancers 2017, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20 (Suppl. 5), S2–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohka, F.; Natsume, A.; Wakabayashi, T. Current trends in targeted therapies for glioblastoma multiforme. Neurol. Res. Int. 2012, 2012, 878425. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005, 109, 93–108. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef]
- Simon, T.; Gagliano, T.; Giamas, G. Direct Effects of Anti-Angiogenic Therapies on Tumor Cells: VEGF Signaling. Trends Mol. Med. 2017, 23, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, Z.; Mashimo, T.; Shen, B.; Nyagilo, J.; Wang, H.; Wang, Y.; Liu, Z.; Mulgaonkar, A.; Hu, X.L.; et al. Gliomas Interact with Non-glioma Brain Cells via Extracellular Vesicles. Cell Rep. 2020, 30, 2489–2500.e5. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.; Jackson, E.; Giamas, G. Breaking through the glioblastoma micro-environment via extracellular vesicles. Oncogene 2020, 39, 4477–4490. [Google Scholar] [CrossRef] [PubMed]
- Wendler, F.; Favicchio, R.; Simon, T.; Alifrangis, C.; Stebbing, J.; Giamas, G. Extracellular vesicles swarm the cancer microenvironment: From tumor-stroma communication to drug intervention. Oncogene 2017, 36, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.; Kumaran, A.; Veselu, D.F.; Giamas, G. Three Method-Combination Protocol for Improving Purity of Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 3071. [Google Scholar] [CrossRef]
- Yang, S.S.; Ma, S.; Dou, H.; Liu, F.; Zhang, S.Y.; Jiang, C.; Xiao, M.; Huang, Y.X. Breast cancer-derived exosomes regulate cell invasion and metastasis in breast cancer via miR-146a to activate cancer associated fibroblasts in tumor microenvironment. Exp. Cell Res. 2020, 391, 111983. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Pinioti, S.; Schellenberger, P.; Rajeeve, V.; Wendler, F.; Cutillas, P.R.; King, A.; Stebbing, J.; Giamas, G. Shedding of bevacizumab in tumour cells-derived extracellular vesicles as a new therapeutic escape mechanism in glioblastoma. Mol. Cancer 2018, 17, 132. [Google Scholar] [CrossRef]
- Palinski, W.; Monti, M.; Camerlingo, R.; Iacobucci, I.; Bocella, S.; Pinto, F.; Iannuzzi, C.; Mansueto, G.; Pignatiello, S.; Fazioli, F.; et al. Lysosome purinergic receptor P2X4 regulates neoangiogenesis induced by microvesicles from sarcoma patients. Cell Death Dis. 2021, 12, 797. [Google Scholar] [CrossRef]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles Released by Glioblastoma Cells Stimulate Normal Astrocytes to Acquire a Tumor-Supportive Phenotype Via p53 and MYC Signaling Pathways. Mol. Neurobiol. 2019, 56, 4566–4581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Xu, J.; Chen, Z.; Wang, H.; Xue, H.; Yang, C.; Guo, Q.; Qi, Y.; Guo, X.; Qian, M.; et al. Transfer of MicroRNA via Macrophage-Derived Extracellular Vesicles Promotes Proneural-to-Mesenchymal Transition in Glioma Stem Cells. Cancer Immunol. Res. 2020, 8, 966–981. [Google Scholar] [CrossRef]
- Lane, R.; Simon, T.; Vintu, M.; Solkin, B.; Koch, B.; Stewart, N.; Benstead-Hume, G.; Pearl, F.M.G.; Critchley, G.; Stebbing, J.; et al. Cell-derived extracellular vesicles can be used as a biomarker reservoir for glioblastoma tumor subtyping. Commun. Biol. 2019, 2, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Zheng, Z.; Guan, J.; Qi, D.; Zhou, S.; Shen, X.; Wang, F.; Wenkert, D.; Kirmani, B.; Solouki, T.; et al. Identification of a panel of genes as a prognostic biomarker for glioblastoma. EBioMedicine 2018, 37, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Cuperlovic-Culf, M.; Khieu, N.H.; Surendra, A.; Hewitt, M.; Charlebois, C.; Sandhu, J.K. Analysis and Simulation of Glioblastoma Cell Lines-Derived Extracellular Vesicles Metabolome. Metabolites 2020, 10, 88. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Shao, H.; Chung, J.; Lee, K.; Balaj, L.; Min, C.; Carter, B.S.; Hochberg, F.H.; Breakefield, X.O.; Lee, H.; Weissleder, R. Chip-based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat. Commun. 2015, 6, 6999. [Google Scholar] [CrossRef] [Green Version]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringner, M.; Morgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.F.; Liao, F.; Wu, H.; Dai, J. Glioma stem cells-derived exosomal miR-26a promotes angiogenesis of microvessel endothelial cells in glioma. J. Exp. Clin. Cancer Res. 2019, 38, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Haraszti, R.A.; Didiot, M.C.; Sapp, E.; Leszyk, J.; Shaffer, S.A.; Rockwell, H.E.; Gao, F.; Narain, N.R.; DiFiglia, M.; Kiebish, M.A.; et al. High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J. Extracell. Vesicles 2016, 5, 32570. [Google Scholar] [CrossRef]
- Osti, D.; Del Bene, M.; Rappa, G.; Santos, M.; Matafora, V.; Richichi, C.; Faletti, S.; Beznoussenko, G.V.; Mironov, A.; Bachi, A.; et al. Clinical Significance of Extracellular Vesicles in Plasma from Glioblastoma Patients. Clin. Cancer Res. 2019, 25, 266–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, C.M.; Toms, S.A. The Role of Circulating MicroRNA in Glioblastoma Liquid Biopsy. World Neurosurg. 2020, 138, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Morishita, M.; Takahashi, Y.; Nishikawa, M.; Takakura, Y. Pharmacokinetics of Exosomes-An Important Factor for Elucidating the Biological Roles of Exosomes and for the Development of Exosome-Based Therapeutics. J. Pharm. Sci. 2017, 106, 2265–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ditsiou, A.; Cilibrasi, C.; Simigdala, N.; Papakyriakou, A.; Milton-Harris, L.; Vella, V.; Nettleship, J.E.; Lo, J.H.; Soni, S.; Smbatyan, G.; et al. The structure-function relationship of oncogenic LMTK3. Sci. Adv. 2020, 6, eabc3099. [Google Scholar] [CrossRef]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Funrich. Available online: http://www.funrich.org/ (accessed on 22 November 2021).
- Gliovis. Available online: http://gliovis.bioinfo.cnio.es (accessed on 22 November 2021).
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Morpheus. Available online: https://software.broadinstitute.org/morpheus (accessed on 22 November 2021).
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 2020, 10, 20560. [Google Scholar] [CrossRef]
- Chen, F.; Chandrashekar, D.S.; Varambally, S.; Creighton, C.J. Pan-cancer molecular subtypes revealed by mass-spectrometry-based proteomic characterization of more than 500 human cancers. Nat. Commun. 2019, 10, 5679. [Google Scholar] [CrossRef] [Green Version]
- Loo, H.K.; Mathen, P.; Lee, J.; Camphausen, K. Circulating biomarkers for high-grade glioma. Biomark Med. 2019, 13, 161–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardelli, A.; Pantel, K. Liquid Biopsies, What We Do Not Know (Yet). Cancer Cell 2017, 31, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallawaaratchy, D.M.; Hallal, S.; Russell, B.; Ly, L.; Ebrahimkhani, S.; Wei, H.; Christopherson, R.I.; Buckland, M.E.; Kaufman, K.L. Comprehensive proteome profiling of glioblastoma-derived extracellular vesicles identifies markers for more aggressive disease. J. Neurooncol. 2017, 131, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Hallal, S.; Russell, B.P.; Wei, H.; Lee, M.Y.T.; Toon, C.W.; Sy, J.; Shivalingam, B.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles from Neurosurgical Aspirates Identifies Chaperonin Containing TCP1 Subunit 6A as a Potential Glioblastoma Biomarker with Prognostic Significance. Proteomics 2019, 19, e1800157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathan, M.; Fonseka, P.; Chitti, S.V.; Kang, T.; Sanwlani, R.; Van Deun, J.; Hendrix, A.; Mathivanan, S. Vesiclepedia 2019: A compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019, 47, D516–D519. [Google Scholar] [CrossRef] [Green Version]
- Dobra, G.; Bukva, M.; Szabo, Z.; Bruszel, B.; Harmati, M.; Gyukity-Sebestyen, E.; Jenei, A.; Szucs, M.; Horvath, P.; Biro, T.; et al. Small Extracellular Vesicles Isolated from Serum May Serve as Signal-Enhancers for the Monitoring of CNS Tumors. Int. J. Mol. Sci. 2020, 21, 5359. [Google Scholar] [CrossRef] [PubMed]
- Hallal, S.; Azimi, A.; Wei, H.; Ho, N.; Lee, M.Y.T.; Sim, H.W.; Sy, J.; Shivalingam, B.; Buckland, M.E.; Alexander-Kaufman, K.L. A Comprehensive Proteomic SWATH-MS Workflow for Profiling Blood Extracellular Vesicles: A New Avenue for Glioma Tumour Surveillance. Int. J. Mol. Sci. 2020, 21, 4754. [Google Scholar] [CrossRef] [PubMed]
- Marfia, G.; Navone, S.E.; Fanizzi, C.; Tabano, S.; Pesenti, C.; Abdel Hadi, L.; Franzini, A.; Caroli, M.; Miozzo, M.; Riboni, L.; et al. Prognostic value of preoperative von Willebrand factor plasma levels in patients with Glioblastoma. Cancer Med. 2016, 5, 1783–1790. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.; Coquerel, B.; Petit, A.; Kassim, Y.; Demange, E.; Le Cerf, D.; Perrot, V.; Vannier, J.P. Direct effect of bevacizumab on glioblastoma cell lines in vitro. Neuromolecular Med. 2014, 16, 752–771. [Google Scholar] [CrossRef]
- Leon, S.P.; Folkerth, R.D.; Black, P.M. Microvessel density is a prognostic indicator for patients with astroglial brain tumors. Cancer 1996, 77, 362–372. [Google Scholar] [CrossRef]
- Yang, X.; Sun, H.J.; Li, Z.R.; Zhang, H.; Yang, W.J.; Ni, B.; Wu, Y.Z. Gastric cancer-associated enhancement of von Willebrand factor is regulated by vascular endothelial growth factor and related to disease severity. BMC Cancer 2015, 15, 80. [Google Scholar] [CrossRef] [Green Version]
- Greco, F.; Anastasi, F.; Pardini, L.F.; Dilillo, M.; Vannini, E.; Baroncelli, L.; Caleo, M.; McDonnell, L.A. Longitudinal Bottom-Up Proteomics of Serum, Serum Extracellular Vesicles, and Cerebrospinal Fluid Reveals Candidate Biomarkers for Early Detection of Glioblastoma in a Murine Model. Molecules 2021, 26, 5992. [Google Scholar] [CrossRef] [PubMed]
- Revel, M.; Daugan, M.V.; Sautes-Fridman, C.; Fridman, W.H.; Roumenina, L.T. Complement System: Promoter or Suppressor of Cancer Progression? Antibodies 2020, 9, 57. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautes-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef] [Green Version]
- Bouwens van der Vlis, T.A.M.; Kros, J.M.; Mustafa, D.A.M.; van Wijck, R.T.A.; Ackermans, L.; van Hagen, P.M.; van der Spek, P.J. The complement system in glioblastoma multiforme. Acta Neuropathol. Commun. 2018, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Ogata, H.; Morikawa, M.; Iijima, S.; Harada, N.; Yoshida, T.; Brown, W.R.; Inoue, N.; Hamada, Y.; Ishii, H.; et al. Distribution and partial characterisation of IgG Fc binding protein in various mucin producing cells and body fluids. Gut 2002, 51, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Wang, L.; Yang, J.; Hu, P.; Zhang, C.; Tong, S.; Liu, Z.; Tian, D. Identification of tumor antigens and immune subtypes in lower grade gliomas for mRNA vaccine development. J. Transl. Med. 2021, 19, 352. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Zhao, Z.; Hu, H.; Zhu, Y.; Zhang, W.; Tang, Q.; Huang, R.; Gao, F.; Zou, C.; Wang, G.; et al. IgG Fc Binding Protein (FCGBP) is Down-Regulated in Metastatic Lesions and Predicts Survival in Metastatic Colorectal Cancer Patients. Onco Targets Ther. 2021, 14, 967–977. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Liu, H.; Zhang, Q.; Song, H.; Tang, J.; Fu, J.; Wang, X. FcGBP was upregulated by HPV infection and correlated to longer survival time of HNSCC patients. Oncotarget 2017, 8, 86503–86514. [Google Scholar] [CrossRef]
- Zhuang, Q.; Shen, A.; Liu, L.; Wu, M.; Shen, Z.; Liu, H.; Cheng, Y.; Lin, X.; Wu, X.; Lin, W.; et al. Prognostic and immunological roles of Fc fragment of IgG binding protein in colorectal cancer. Oncol. Lett. 2021, 22, 526. [Google Scholar] [CrossRef]
- Li, X.; Meng, Y. Analyses of metastasis-associated genes in IDH wild-type glioma. BMC Cancer 2020, 20, 1114. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.M.; Keating, A.K. TAM receptor tyrosine kinases: Expression, disease and oncogenesis in the central nervous system. Brain Res. 2014, 1542, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Onken, J.; Vajkoczy, P.; Torka, R.; Hempt, C.; Patsouris, V.; Heppner, F.L.; Radke, J. Phospho-AXL is widely expressed in glioblastoma and associated with significant shorter overall survival. Oncotarget 2017, 8, 50403–50414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashiyama, M.; Doi, O.; Kodama, K.; Yokouchi, H.; Tateishi, R. An evaluation of the prognostic significance of alpha-1-antitrypsin expression in adenocarcinomas of the lung: An immunohistochemical analysis. Br. J. Cancer 1992, 65, 300–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara, E.; Ito, H.; Taniyama, K.; Yokozaki, H.; Hata, J. Alpha 1-antitrypsin, alpha 1-antichymotrypsin, and alpha 2-macroglobulin in human gastric carcinomas: A retrospective immunohistochemical study. Hum. Pathol. 1984, 15, 957–964. [Google Scholar] [CrossRef]
- Karashima, S.; Kataoka, H.; Itoh, H.; Maruyama, R.; Koono, M. Prognostic significance of alpha-1-antitrypsin in early stage of colorectal carcinomas. Int. J. Cancer 1990, 45, 244–250. [Google Scholar] [CrossRef]
- Kwon, C.H.; Park, H.J.; Lee, J.R.; Kim, H.K.; Jeon, T.Y.; Jo, H.J.; Kim, D.H.; Kim, G.H.; Park, D.Y. Serpin peptidase inhibitor clade A member 1 is a biomarker of poor prognosis in gastric cancer. Br. J. Cancer 2014, 111, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.H.; Park, H.J.; Choi, J.H.; Lee, J.R.; Kim, H.K.; Jo, H.J.; Kim, H.S.; Oh, N.; Song, G.A.; Park, D.Y. Snail and serpinA1 promote tumor progression and predict prognosis in colorectal cancer. Oncotarget 2015, 6, 20312–20326. [Google Scholar] [CrossRef] [Green Version]
- Ookawa, S.; Wanibuchi, M.; Kataoka-Sasaki, Y.; Sasaki, M.; Oka, S.; Ohtaki, S.; Noshiro, S.; Komatsu, K.; Akiyama, Y.; Mikami, T.; et al. Digital Polymerase Chain Reaction Quantification of SERPINA1 Predicts Prognosis in High-Grade Glioma. World Neurosurg. 2018, 111, e783–e789. [Google Scholar] [CrossRef]
- Tso, C.L.; Freije, W.A.; Day, A.; Chen, Z.; Merriman, B.; Perlina, A.; Lee, Y.; Dia, E.Q.; Yoshimoto, K.; Mischel, P.S.; et al. Distinct transcription profiles of primary and secondary glioblastoma subgroups. Cancer Res. 2006, 66, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef] [Green Version]
- Maiser, A.; Dillinger, S.; Langst, G.; Schermelleh, L.; Leonhardt, H.; Nemeth, A. Super-resolution in situ analysis of active ribosomal DNA chromatin organization in the nucleolus. Sci. Rep. 2020, 10, 7462. [Google Scholar] [CrossRef]
- Spinelli, C.; Montermini, L.; Meehan, B.; Brisson, A.R.; Tan, S.; Choi, D.; Nakano, I.; Rak, J. Molecular subtypes and differentiation programmes of glioma stem cells as determinants of extracellular vesicle profiles and endothelial cell-stimulating activities. J. Extracell. Vesicles 2018, 7, 1490144. [Google Scholar] [CrossRef]
- Werner, C.; Stangl, S.; Salvermoser, L.; Schwab, M.; Shevtsov, M.; Xanthopoulos, A.; Wang, F.; Dezfouli, A.B.; Tholke, D.; Ostheimer, C.; et al. Hsp70 in Liquid Biopsies-A Tumor-Specific Biomarker for Detection and Response Monitoring in Cancer. Cancers 2021, 13, 3706. [Google Scholar] [CrossRef] [PubMed]
- Yeung, L.; Hickey, M.J.; Wright, M.D. The Many and Varied Roles of Tetraspanins in Immune Cell Recruitment and Migration. Front. Immunol. 2018, 9, 1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.; Deng, Y.; Peng, H.; Shen, J. Role of Tetraspanins in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 723341. [Google Scholar] [CrossRef]
- Zheng, W.; Chen, Q.; Liu, H.; Hu, S.; Zhou, Y.; Bai, Y.; Zhang, J.; Pan, Y.; Shao, C. CD81 Enhances Radioresistance of Glioblastoma by Promoting Nuclear Translocation of Rad51. Cancers 2021, 13, 1988. [Google Scholar] [CrossRef] [PubMed]
- Panzarini, E.; Tacconi, S.; Carata, E.; Mariano, S.; Tata, A.M.; Dini, L. Molecular Characterization of Temozolomide-Treated and Non Temozolomide-Treated Glioblastoma Cells Released Extracellular Vesicles and Their Role in the Macrophage Response. Int. J. Mol. Sci. 2020, 21, 8353. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Number | Age | Gender | Lobe Affected | Surgery | Oncological Treatment | Recurrence | Months to Death |
|---|---|---|---|---|---|---|---|
| EV003 | 57 | F | Left parietal–occipital–temporal | 40–50% debulking | high-dose RT | yes | 7 |
| EV004 | 60 | F | Right occipital | 90% debulking | radical chemoRT | no | 7 |
| EV005 | 72 | M | Right occipital | 95% debulking | radical chemoRT; adjuvant TMZ: CCNU | yes | 19 |
| EV006 | 76 | M | Left frontal | 100% resection | n/a | no | 1 |
| EV008 | 48 | F | Left temporal | 100% resection | high-dose palliative RT | yes | 8 |
| EV011 | 64 | M | Right frontal | 100% resection | radical chemoRT; adjuvant TMZ | no | alive |
| EV013 | 65 | M | Right parietal | 70% resection | radical chemoRT; adjuvant TMZ | yes | 12 |
| EV015 | 50 | F | Right temporal | 90% resection | radical chemoRT; adjuvant TMZ | yes | 11 |
| EV017 | 64 | M | Left temporal–occipital | 70% resection | high-dose palliative chemoRT | yes | 6 |
| EV019 | 71 | M | Left temporal | 80–90% resection | high-dose RT | yes | 2 |
| EV020 | 55 | M | Right temporal | 80% resection | n/a | no | 2 |
| EV022 | 66 | F | Right frontal | 90–95% resection | radical chemoRT | yes | 11 |
| EV024 | 60 | M | Right temporal–parietal | 80% resection | high-dose RT | yes | 10 |
| EV028 | 53 | F | Right frontal | 90–95% resection | high-dose palliative RT | no | 4 |
| EV029 | 71 | M | Left temporal | 100% resection | radical chemoRT | no | alive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cilibrasi, C.; Simon, T.; Vintu, M.; Tolias, C.; Samuels, M.; Mazarakis, N.K.; Eravci, M.; Stewart, N.; Critchley, G.; Giamas, G. Definition of an Inflammatory Biomarker Signature in Plasma-Derived Extracellular Vesicles of Glioblastoma Patients. Biomedicines 2022, 10, 125. https://doi.org/10.3390/biomedicines10010125
Cilibrasi C, Simon T, Vintu M, Tolias C, Samuels M, Mazarakis NK, Eravci M, Stewart N, Critchley G, Giamas G. Definition of an Inflammatory Biomarker Signature in Plasma-Derived Extracellular Vesicles of Glioblastoma Patients. Biomedicines. 2022; 10(1):125. https://doi.org/10.3390/biomedicines10010125
Chicago/Turabian StyleCilibrasi, Chiara, Thomas Simon, Marian Vintu, Christos Tolias, Mark Samuels, Nektarios K. Mazarakis, Murat Eravci, Nicolas Stewart, Giles Critchley, and Georgios Giamas. 2022. "Definition of an Inflammatory Biomarker Signature in Plasma-Derived Extracellular Vesicles of Glioblastoma Patients" Biomedicines 10, no. 1: 125. https://doi.org/10.3390/biomedicines10010125
APA StyleCilibrasi, C., Simon, T., Vintu, M., Tolias, C., Samuels, M., Mazarakis, N. K., Eravci, M., Stewart, N., Critchley, G., & Giamas, G. (2022). Definition of an Inflammatory Biomarker Signature in Plasma-Derived Extracellular Vesicles of Glioblastoma Patients. Biomedicines, 10(1), 125. https://doi.org/10.3390/biomedicines10010125