Generation of High Yielding and Fragrant Rice (Oryza sativa L.) Lines by CRISPR/Cas9 Targeted Mutagenesis of Three Homoeologs of Cytochrome P450 Gene Family and OsBADH2 and Transcriptome and Proteome Profiling of Revealed Changes Triggered by Mutations




Abstract
1. Introduction
2. Results
2.1. Plasmid Construction and Analysis of Mutant Plants
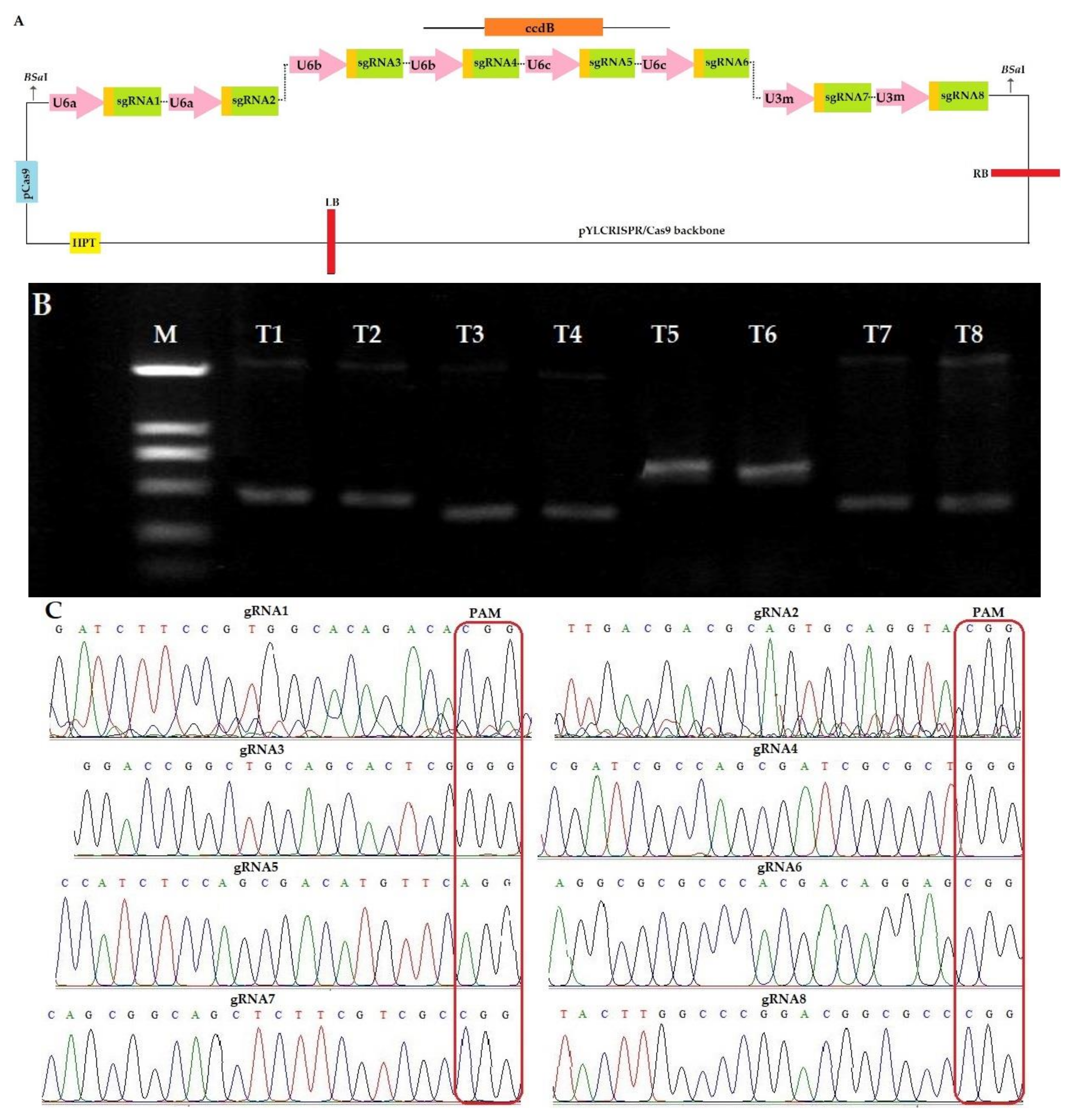
2.1.1. Construction of CRISPR-Cas9 Expression Vector
2.1.2. Identification of Mutant Plants and Sequencing Analysis of Mutation Sites
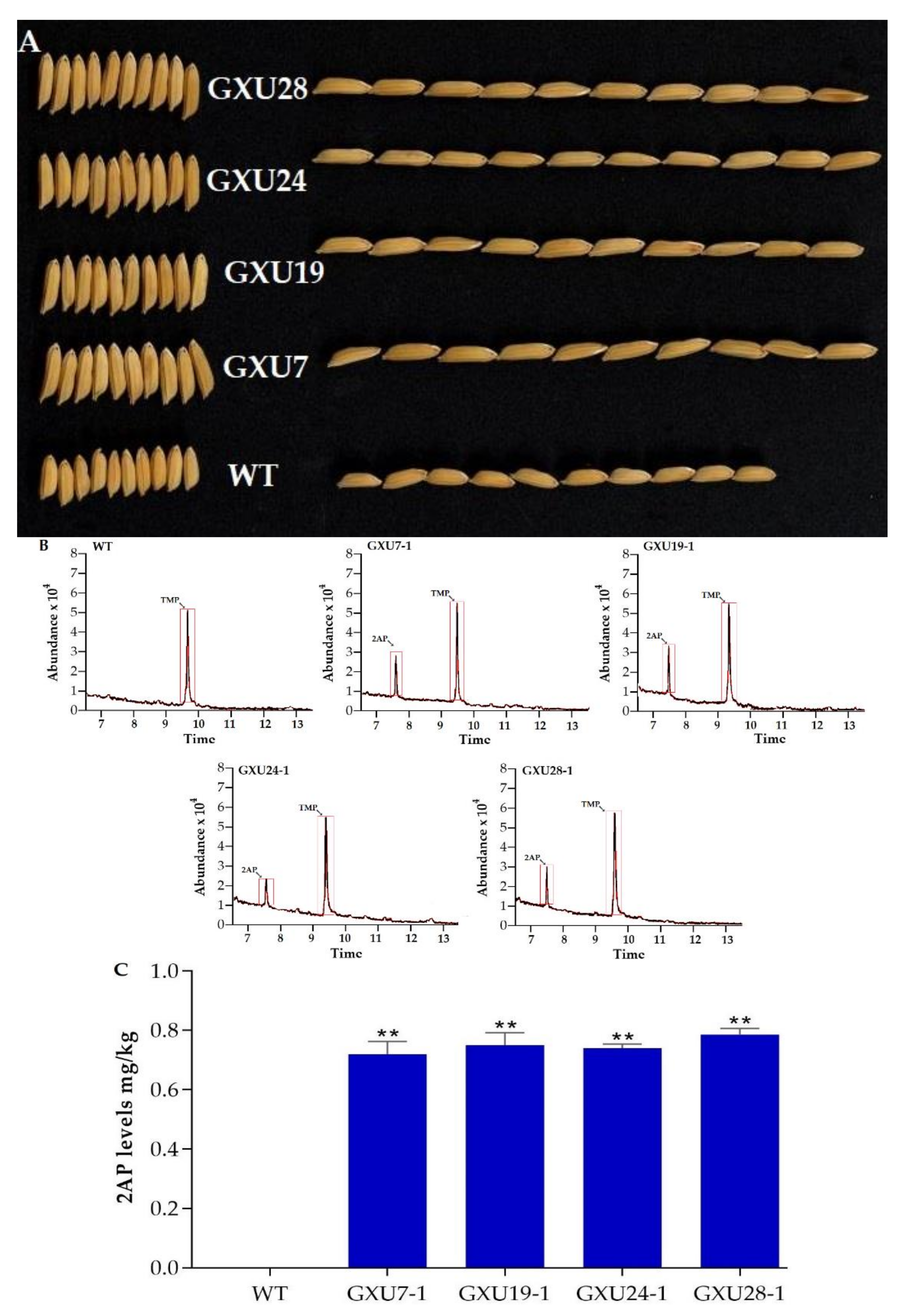
2.1.3. Agronomic Performance Assessment of Wild-Type and Mutant Lines
2.1.4. Determination of 2-AP Content in Wild-Type and Homozygous Mutant Lines
2.1.5. Segregation Ratio of Mutant Plants
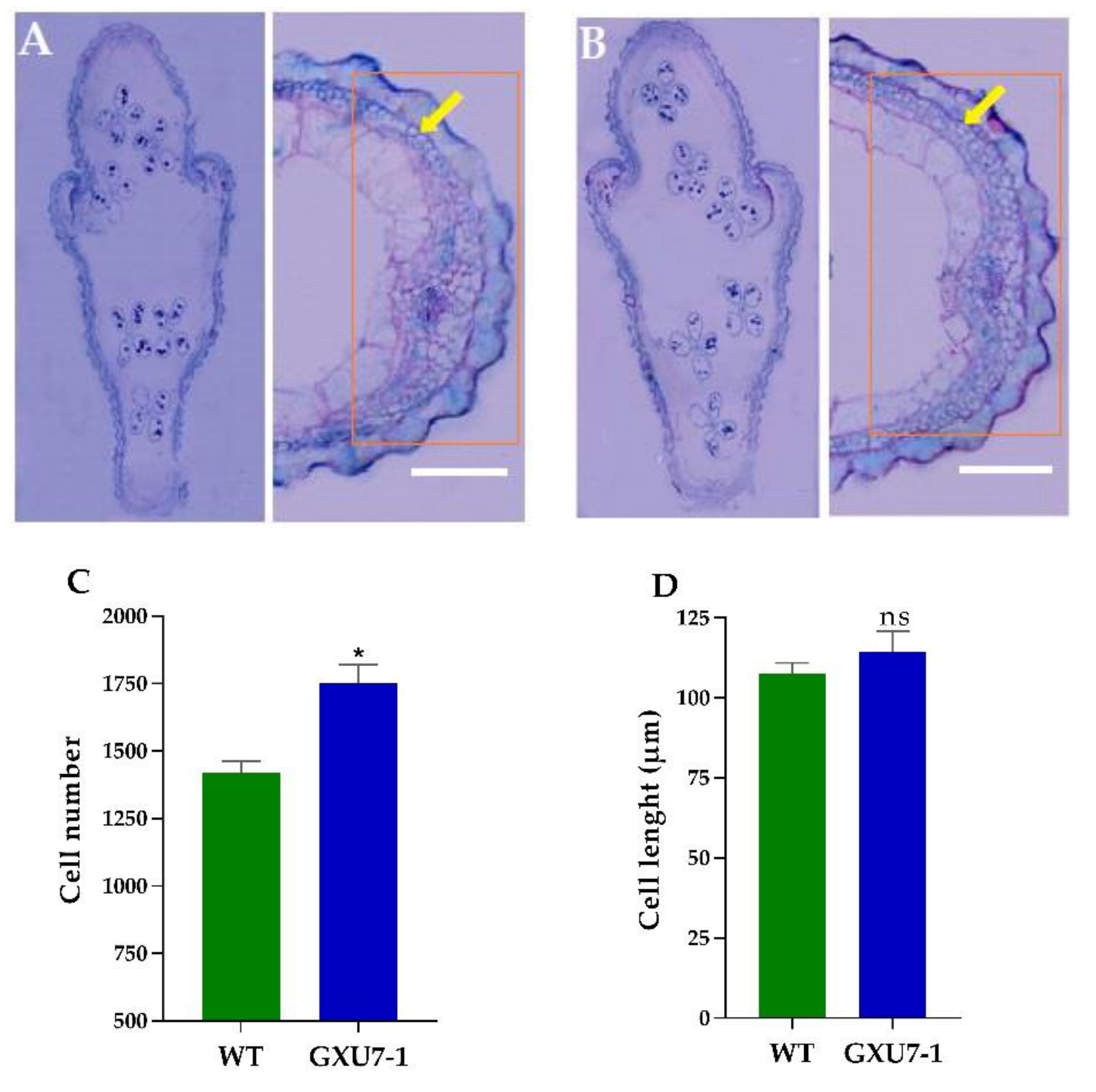
2.1.6. Anatomical Observation of WT and Mutant Line (GXU7-1) Grain
2.2. Transcriptome Analysis
2.2.1. Outcome of RNA-Sequencing Analysis
2.2.2. Functional Classification and Pathway Enrichment of DEGs.
2.2.3. Hub-Gene Network Analysis
2.3. Proteomic Analysis
2.3.1. Basic Information and Analysis of Identified Peptides and Proteins
2.3.2. Gene Ontology (GO) and Pathway Enrichment Analysis of DEPs
2.3.3. Hub-Protein Analysis
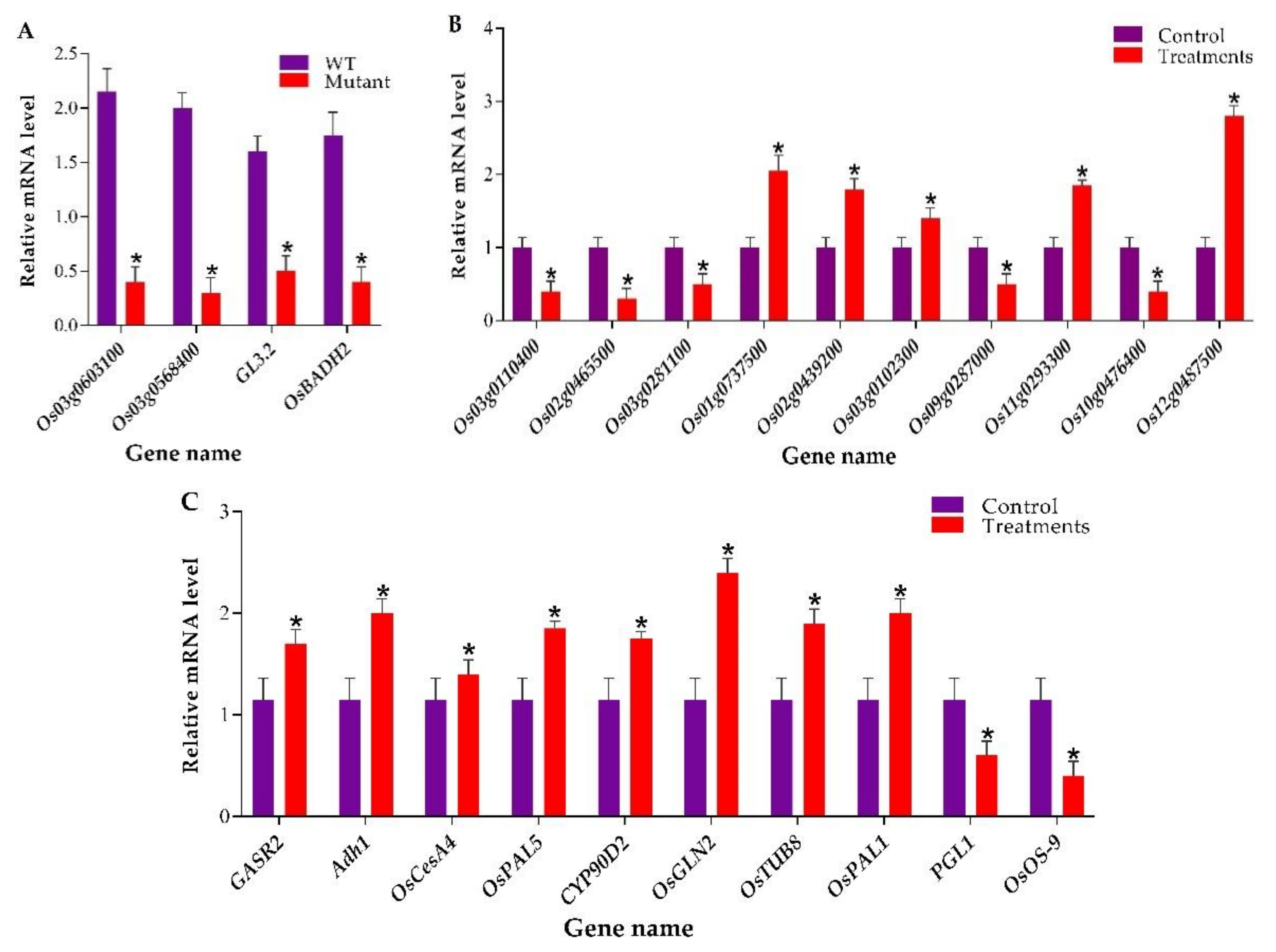
2.4. RT-qPCR-based Analysis of Target Genes and Validation of Transcriptome and Proteomic Data
3. Discussion
3.1. CRISPR Mutants Exhibited Inheritable Mutations, Increased Grain Yield, and Fragrant Phenotype
3.2. Cross-Regulation of Genes and Proteins Related to Cell Cycle, Grain Development and Cytochrome P450
4. Material and Methods
4.1. Experimental Material and Growth Conditions
4.2. SgRNAs Selection and Off-targets Prediction
4.3. Preparation of Templates and Generation of sgRNA Expression Cassettes
4.4. Assembly of Multiplex sgRNA Expression Cassette into CRISPR/Cas9Pubi-H Binary Vector
4.5. Transformation of Escherichia coli and Confirmation of Ligated Products
4.6. Rice Transformation and On/Off-target Mutation Analysis
4.7. Phenotyping and Biochemical Analysis
4.8. Library Preparation for RNA-Sequencing
4.9. Analysis of RNA-Sequencing Data
4.10. Protein Extraction, Digestion, and Labelling
4.11. HPLC Fractionation and LC-MS/MS Analysis
4.12. Proteomic Data Analysis
4.13. Determination of Transcript Level of Target Genes and Omics Data Validation
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Taguchi-Shiobara, F.; Kawagoe, Y.; Kato, H.; Onodera, H.; Tagiri, A.; Hara, N.; Miyao, A.; Hirochika, H.; Kitano, H.; Yano, M. A loss-of-function mutation of rice DENSE PANICLE 1 causes semi-dwarfness and slightly increased number of spikelets. Breed. Sci. 2011, 61, 17–25. [Google Scholar] [CrossRef]
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Takano-Kai, N.; Jiang, H.; Kubo, T.; Sweeney, M.; Matsumoto, T.; Kanamori, H.; Padhukasahasram, B.; Bustamante, C.; Yoshimura, A.; Doi, K. Evolutionary history of GS3, a gene conferring grain length in rice. Genetics 2009, 182, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Song, X.-J.; Huang, W.; Shi, M.; Zhu, M.-Z.; Lin, H.-X. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat. Genet. 2007, 39, 623–630. [Google Scholar] [CrossRef]
- Shomura, A.; Izawa, T.; Ebana, K.; Ebitani, T.; Kanegae, H.; Konishi, S.; Yano, M. Deletion in a gene associated with grain size increased yields during rice domestication. Nat. Genet. 2008, 40, 1023–1028. [Google Scholar] [CrossRef]
- Liu, J.; Chen, J.; Zheng, X.; Wu, F.; Lin, Q.; Heng, Y.; Tian, P.; Cheng, Z.; Yu, X.; Zhou, K. GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nat. Plants 2017, 3, 17043. [Google Scholar] [CrossRef]
- Li, Y.; Fan, C.; Xing, Y.; Jiang, Y.; Luo, L.; Sun, L.; Shao, D.; Xu, C.; Li, X.; Xiao, J. Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat. Genet. 2011, 43, 1266–1269. [Google Scholar] [CrossRef]
- Wang, S.; Wu, K.; Yuan, Q.; Liu, X.; Liu, Z.; Lin, X.; Zeng, R.; Zhu, H.; Dong, G.; Qian, Q. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 2012, 44, 950–954. [Google Scholar] [CrossRef]
- Wang, Y.; Xiong, G.; Hu, J.; Jiang, L.; Yu, H.; Xu, J.; Fang, Y.; Zeng, L.; Xu, E.; Xu, J. Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat. Genet. 2015, 47, 944–948. [Google Scholar] [CrossRef]
- Xue, W.; Xing, Y.; Weng, X.; Zhao, Y.; Tang, W.; Wang, L.; Zhou, H.; Yu, S.; Xu, C.; Li, X. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat. Genet. 2008, 40, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Wang, J.; Zhu, X.; Hao, W.; Wang, L.; Li, Q.; Zhang, L.; He, W.; Lu, B.; Lin, H. Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat. Genet. 2008, 40, 1370–1374. [Google Scholar] [CrossRef]
- Tan, L.; Li, X.; Liu, F.; Sun, X.; Li, C.; Zhu, Z.; Fu, Y.; Cai, H.; Wang, X.; Xie, D. Control of a key transition from prostrate to erect growth in rice domestication. Nat. Genet. 2008, 40, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Ben-Haj-Salah, H.; Tardieu, F. Temperature affects expansion rate of maize leaves without change in spatial distribution of cell length (analysis of the coordination between cell division and cell expansion). Plant Physiol. 1995, 109, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, G.; Han, Y.; Usman, B.; Liu, F.; Qin, B.; Li, R. Knockout of OsPRP1, a gene encoding proline-rich protein, confers enhanced cold sensitivity in rice (Oryza sativa L.) at the seedling stage. 3 Biotech 2019, 9, 254. [Google Scholar] [CrossRef]
- Han, Y.; Luo, D.; Usman, B.; Nawaz, G.; Zhao, N.; Liu, F.; Li, R. Development of high yielding glutinous cytoplasmic male sterile rice (Oryza sativa L.) lines through CRISPR/Cas9 based mutagenesis of Wx and TGW6 and proteomic analysis of anther. Agronomy 2018, 8, 290. [Google Scholar] [CrossRef]
- Han, Y.; Teng, K.; Nawaz, G.; Feng, X.; Usman, B.; Wang, X.; Luo, L.; Zhao, N.; Liu, Y.; Li, R. Generation of semi-dwarf rice (Oryza sativa L.) lines by CRISPR/Cas9-directed mutagenesis of OsGA20ox2 and proteomic analysis of unveiled changes caused by mutations. 3 Biotech 2019, 9, 387. [Google Scholar] [CrossRef]
- Liao, S.; Qin, X.; Luo, L.; Han, Y.; Wang, X.; Usman, B.; Nawaz, G.; Zhao, N.; Liu, Y.; Li, R. CRISPR/Cas9-Induced Mutagenesis of Semi-Rolled Leaf1, 2 Confers Curled Leaf Phenotype and Drought Tolerance by Influencing Protein Expression Patterns and ROS Scavenging in Rice (Oryza sativa L.). Agronomy 2019, 9, 728. [Google Scholar] [CrossRef]
- Jezussek, M.; Juliano, B.O.; Schieberle, P. Comparison of key aroma compounds in cooked brown rice varieties based on aroma extract dilution analyses. J. Agric. Food Chem. 2002, 50, 1101–1105. [Google Scholar] [CrossRef]
- Kovach, M.J.; Calingacion, M.N.; Fitzgerald, M.A.; McCouch, S.R. The origin and evolution of fragrance in rice (Oryza sativa L.). Proc. Natl. Acad. Sci. USA 2009, 106, 14444–14449. [Google Scholar] [CrossRef]
- Shao, G.; Tang, A.; Tang, S.; Luo, J.; Jiao, G.; Wu, J.; Hu, P. A new deletion mutation of fragrant gene and the development of three molecular markers for fragrance in rice. Plant Breed. 2011, 130, 172–176. [Google Scholar] [CrossRef]
- Shan, Q.; Zhang, Y.; Chen, K.; Zhang, K.; Gao, C. Creation of fragrant rice by targeted knockout of the OsBADH2 gene using TALEN technology. Plant Biotechnol. J. 2015, 13, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R.; Ming, R.; Alam, M.; Schuler, M.A. Comparison of cytochrome P450 genes from six plant genomes. Trop. Plant Biol. 2008, 1, 216–235. [Google Scholar] [CrossRef]
- Schuler, M.A.; Werck-Reichhart, D. Functional genomics of P450s. Ann. Rev. Plant Biol. 2003, 54, 629–667. [Google Scholar] [CrossRef] [PubMed]
- Adamski, N.M.; Anastasiou, E.; Eriksson, S.; O’Neill, C.M.; Lenhard, M. Local maternal control of seed size by KLUH/CYP78A5-dependent growth signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 20115–20120. [Google Scholar] [CrossRef] [PubMed]
- Stransfeld, L.; Eriksson, S.; Adamski, N.M.; Breuninger, H.; Lenhard, M. KLUH/CYP78A5 promotes organ growth without affecting the size of the early primordium. Plant Signal. Behav. 2010, 5, 982–984. [Google Scholar] [CrossRef][Green Version]
- Fang, W.; Wang, Z.; Cui, R.; Li, J.; Li, Y. Maternal control of seed size by EOD3/CYP78A6 in Arabidopsis thaliana. Plant J. 2012, 70, 929–939. [Google Scholar] [CrossRef]
- Sotelo-Silveira, M.; Cucinotta, M.; Chauvin, A.-L.; Montes, R.A.C.; Colombo, L.; Marsch-Martínez, N.; De Folter, S. Cytochrome P450 CYP78A9 is involved in Arabidopsis reproductive development. Plant Physiol. 2013, 162, 779–799. [Google Scholar] [CrossRef]
- Miyoshi, K.; Ahn, B.-O.; Kawakatsu, T.; Ito, Y.; Itoh, J.-I.; Nagato, Y.; Kurata, N. PLASTOCHRON1, a timekeeper of leaf initiation in rice, encodes cytochrome P450. Proc. Natl. Acad. Sci. USA 2004, 101, 875–880. [Google Scholar] [CrossRef]
- Xu, F.; Fang, J.; Ou, S.; Gao, S.; Zhang, F.; Du, L.; Xiao, Y.; Wang, H.; Sun, X.; Chu, J. Variations in CYP78A13 coding region influence grain size and yield in rice. Plant Cell Environ. 2015, 38, 800–811. [Google Scholar] [CrossRef]
- Hirochika, H.; Guiderdoni, E.; An, G.; Hsing, Y.-i.; Eun, M.Y.; Han, C.-d.; Upadhyaya, N.; Ramachandran, S.; Zhang, Q.; Pereira, A. Rice mutant resources for gene discovery. Plant Mol. Biol. 2004, 54, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Ichikawa, H. Gene overexpression resources in cereals for functional genomics and discovery of useful genes. Front. Plant Sci. 2016, 7, 1359. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Haque, E.; Taniguchi, H.; Hassan, M.; Bhowmik, P.; Karim, M.R.; Śmiech, M.; Zhao, K.; Rahman, M.; Islam, T. Application of CRISPR/Cas9 genome editing technology for the improvement of crops cultivated in tropical climates: Recent progress, prospects, and challenges. Front. Plant Sci. 2018, 9, 617. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, D.; Ramasamy, K.; Sellamuthu, G.; Jayabalan, S.; Venkataraman, G. CRISPR for crop improvement: An update review. Front. Plant Sci. 2018, 9, 985. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Joshi, R.K.; Zhao, K. Genome editing in rice: Recent advances, challenges, and future implications. Front. Plant Sci. 2018, 9, 1361. [Google Scholar] [CrossRef]
- Li, M.; Li, X.; Zhou, Z.; Wu, P.; Fang, M.; Pan, X.; Lin, Q.; Luo, W.; Wu, G.; Li, H. Reassessment of the four yield-related genes Gn1a, DEP1, GS3, and IPA1 in rice using a CRISPR/Cas9 system. Front. Plant Sci. 2016, 7, 377. [Google Scholar] [CrossRef]
- Zhou, J.; Xin, X.; He, Y.; Chen, H.; Li, Q.; Tang, X.; Zhong, Z.; Deng, K.; Zheng, X.; Akher, S.A. Multiplex QTL editing of grain-related genes improves yield in elite rice varieties. Plant Cell Rep. 2019, 38, 475–485. [Google Scholar] [CrossRef]
- Xu, R.; Yang, Y.; Qin, R.; Li, H.; Qiu, C.; Li, L.; Wei, P.; Yang, J. Rapid improvement of grain weight via highly efficient CRISPR/Cas9-mediated multiplex genome editing in rice. J. Genet. Genomic. 2016, 43, 529–532. [Google Scholar] [CrossRef]
- Lan, P.; Li, W.; Schmidt, W. Complementary proteome and transcriptome profiling in phosphate-deficient Arabidopsis roots reveals multiple levels of gene regulation. Mol. Cell. Protem. 2012, 11, 1156–1166. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, X.; Xu, B.; Li, Y.; Ma, Y.; Wang, G. Phenotype and transcriptome analysis reveals chloroplast development and pigment biosynthesis together influenced the leaf color formation in mutants of Anthurium andraeanum ‘Sonate’. Front. Plant Sci. 2015, 6, 139. [Google Scholar] [CrossRef] [PubMed]
- Hellinger, R.; Koehbach, J.; Soltis, D.E.; Carpenter, E.J.; Wong, G.K.-S.; Gruber, C.W. Peptidomics of circular cysteine-rich plant peptides: Analysis of the diversity of cyclotides from viola tricolor by transcriptome and proteome mining. J. Proteome Res. 2015, 14, 4851–4862. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.H.M.; Cho, K.; Choi, J.-S.; Bae, K.-H.; Komatsu, S.; Uozumi, N.; Woo, S.H. The wheat chloroplastic proteome. J. Proteom. 2013, 93, 326–342. [Google Scholar] [CrossRef]
- Cho, K.; Agrawal, G.K.; Shibato, J.; Jung, Y.-H.; Kim, Y.-K.; Nahm, B.H.; Jwa, N.-S.; Tamogami, S.; Han, O.; Kohda, K. Survey of differentially expressed proteins and genes in jasmonic acid treated rice seedling shoot and root at the proteomics and transcriptomics levels. J. Proteome Res. 2007, 6, 3581–3603. [Google Scholar] [CrossRef]
- Cho, K.; Shibato, J.; Agrawal, G.K.; Jung, Y.-H.; Kubo, A.; Jwa, N.-S.; Tamogami, S.; Satoh, K.; Kikuchi, S.; Higashi, T. Integrated transcriptomics, proteomics, and metabolomics analyses to survey ozone responses in the leaves of rice seedling. J. Proteome Res. 2008, 7, 2980–2998. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Tao, Y.; Zhao, K.; Shao, J.; Li, X.; Liu, G.; Liu, S.; Zhu, L. Proteomic and transcriptomic analysis of rice mature seed-derived callus differentiation. Proteomics 2007, 7, 755–768. [Google Scholar] [CrossRef]
- Soyk, S.; Lemmon, Z.H.; Oved, M.; Fisher, J.; Liberatore, K.L.; Park, S.J.; Goren, A.; Jiang, K.; Ramos, A.; van der Knaap, E. Bypassing negative epistasis on yield in tomato imposed by a domestication gene. Cell 2017, 169, 1142–1155. [Google Scholar] [CrossRef]
- Jacob, P.; Avni, A.; Bendahmane, A. Translational research: Exploring and creating genetic diversity. Trends Plant Sci. 2018, 23, 42–52. [Google Scholar] [CrossRef]
- Shan, Q.; Wang, Y.; Li, J.; Gao, C. Genome editing in rice and wheat using the CRISPR/Cas system. Nat. Protoc. 2014, 9, 2395–2410. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Shan, Q.; Wang, Y.; Chen, K.; Liang, Z.; Li, J.; Zhang, Y.; Zhang, K.; Liu, J.; Voytas, D.F.; Zheng, X. Rapid and efficient gene modification in rice and Brachypodium using TALENs. Mol. Plant 2013, 6, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R.; Schuler, M.A.; Paquette, S.M.; Werck-Reichhart, D.; Bak, S. Comparative genomics of rice and Arabidopsis. Analysis of 727 cytochrome P450 genes and pseudogenes from a monocot and a dicot. Plant Physiol. 2004, 135, 756–772. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, B.; Weeks, D.P.; Spalding, M.H.; Yang, B. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res. 2014, 42, 10903–10914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, J.; Wei, P.; Zhang, B.; Gou, F.; Feng, Z.; Mao, Y.; Yang, L.; Zhang, H.; Xu, N. The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol. J. 2014, 12, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, T. CRISPR/Cas9 in rice can induce new mutations in later generations, leading to chimerism and unpredicted segregation of the targeted mutation. Mol. Breed. 2016, 36, 165. [Google Scholar] [CrossRef]
- Nagasawa, N.; Hibara, K.I.; Heppard, E.P.; Vander Velden, K.A.; Luck, S.; Beatty, M.; Nagato, Y.; Sakai, H. GIANT EMBRYO encodes CYP78A13, required for proper size balance between embryo and endosperm in rice. Plant J. 2013, 75, 592–605. [Google Scholar] [CrossRef]
- Yang, W.; Gao, M.; Yin, X.; Liu, J.; Xu, Y.; Zeng, L.; Li, Q.; Zhang, S.; Wang, J.; Zhang, X. Control of rice embryo development, shoot apical meristem maintenance, and grain yield by a novel cytochrome p450. Mol. Plant 2013, 6, 1945–1960. [Google Scholar] [CrossRef]
- Anastasiou, E.; Kenz, S.; Gerstung, M.; MacLean, D.; Timmer, J.; Fleck, C.; Lenhard, M. Control of plant organ size by KLUH/CYP78A5-dependent intercellular signaling. Develop. Cell 2007, 13, 843–856. [Google Scholar] [CrossRef]
- Wang, J.-W.; Schwab, R.; Czech, B.; Mica, E.; Weigel, D. Dual effects of miR156-targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size in Arabidopsis thaliana. Plant Cell 2008, 20, 1231–1243. [Google Scholar] [CrossRef]
- Tamiru, M.; Undan, J.R.; Takagi, H.; Abe, A.; Yoshida, K.; Undan, J.Q.; Natsume, S.; Uemura, A.; Saitoh, H.; Matsumura, H. A cytochrome P450, OsDSS1, is involved in growth and drought stress responses in rice (Oryza sativa L.). Plant Mol. Biol. 2015, 88, 85–99. [Google Scholar] [CrossRef]
- Zondlo, S.C.; Irish, V.F. CYP78A5 encodes a cytochrome P450 that marks the shoot apical meristem boundary in Arabidopsis. Plant J. 1999, 19, 259–268. [Google Scholar] [CrossRef]
- Ito, T.; Meyerowitz, E.M. Overexpression of a gene encoding a cytochrome P450, CYP78A9, induces large and seedless fruit in Arabidopsis. Plant Cell 2000, 12, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Morinaka, Y.; Ohnishi, T.; Sunohara, H.; Fujioka, S.; Ueguchi-Tanaka, M.; Mizutani, M.; Sakata, K.; Takatsuto, S.; Yoshida, S. Erect leaves caused by brassinosteroid deficiency increase biomass production and grain yield in rice. Nat. Biotechnol. 2006, 24, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Umeda, M.; Umeda-Hara, C.; Yamaguchi, M.; Hashimoto, J.; Uchimiya, H. Differential expression of genes for cyclin-dependent protein kinases in rice plants. Plant Physiol. 1999, 119, 31–40. [Google Scholar] [CrossRef]
- Endo, M.; Nakayama, S.; Umeda-Hara, C.; Ohtsuki, N.; Saika, H.; Umeda, M.; Toki, S. CDKB2 is involved in mitosis and DNA damage response in rice. Plant J. 2012, 69, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Suzuki, Y.; Yoshizawa, R.; Kanno, K.; Makino, A. Effect of individual suppression of RBCS multigene family on Rubisco contents in rice leaves. Plant Cell Environ. 2012, 35, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Thirumurugan, T.; Ito, Y.; Kubo, T.; Serizawa, A.; Kurata, N. Identification, characterization and interaction of HAP family genes in rice. Mol. Genet. Genom. 2008, 279, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Xu, J.; Guo, H.; Jiang, L.; Chen, S.; Yu, C.; Zhou, Z.; Hu, P.; Zhai, H.; Wan, J. DTH8 suppresses flowering in rice, influencing plant height and yield potential simultaneously. Plant Physiol. 2010, 153, 1747–1758. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Ding, Y.; Tan, L.; Fu, Y.; Liu, F.; Zhu, Z.; Sun, X.; Sun, X.; Gu, P.; Cai, H. LHD1, an allele of DTH8/Ghd8, controls late heading date in common wild rice (Oryza rufipogon) F. J. Integr. Plant Biol. 2012, 54, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Song, W.Y.; Lee, H.S.; Jin, S.R.; Ko, D.; Martinoia, E.; Lee, Y.; An, G.; Ahn, S.N. Rice PCR1 influences grain weight and Zn accumulation in grains. Plant Cell Environ. 2015, 38, 2327–2339. [Google Scholar] [CrossRef]
- Heang, D.; Sassa, H. Antagonistic actions of HLH/bHLH proteins are involved in grain length and weight in rice. PLoS ONE 2012, 7, e31325. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Ni, S.; Wang, J.; Zhang, B.; Xu, R.; Wang, Y.; Chen, H.; Zhu, X.; Li, Y. Regulation of OsGRF4 by OsmiR396 controls grain size and yield in rice. Nat. Plants 2015, 2, 15203–15207. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.; Ohta, D. Diversification of P450 genes during land plant evolution. Ann. Rev. Plant Biol. 2010, 61, 291–315. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, S.; Ashikari, M.; Fujioka, S.; Takatsuto, S.; Yoshida, S.; Yano, M.; Yoshimura, A.; Kitano, H.; Matsuoka, M.; Fujisawa, Y. A novel cytochrome P450 is implicated in brassinosteroid biosynthesis via the characterization of a rice dwarf mutant, dwarf11, with reduced seed length. Plant Cell 2005, 17, 776–790. [Google Scholar] [CrossRef]
- Wei, X.; Jiao, G.; Lin, H.; Sheng, Z.; Shao, G.; Xie, L.; Tang, S.; Xu, Q.; Hu, P. GRAIN INCOMPLETE FILLING 2 regulates grain filling and starch synthesis during rice caryopsis development. J. Integr. Plant Biol. 2017, 59, 134–153. [Google Scholar] [CrossRef]
- Nakagawa, H.; Tanaka, A.; Tanabata, T.; Ohtake, M.; Fujioka, S.; Nakamura, H.; Ichikawa, H.; Mori, M. Short grain1 decreases organ elongation and brassinosteroid response in rice. Plant Physiol. 2012, 158, 1208–1219. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, K.; Ai, J.; Deng, X.; Hong, Y.; Wang, X. Patatin-related phospholipase A, pPLAIIIα, modulates the longitudinal growth of vegetative tissues and seeds in rice. J. Exp. Bot. 2015, 66, 6945–6955. [Google Scholar] [CrossRef]
- Heang, D.; Sassa, H. An atypical bHLH protein encoded by POSITIVE REGULATOR OF GRAIN LENGTH 2 is involved in controlling grain length and weight of rice through interaction with a typical bHLH protein APG. Breed. Sci. 2012, 62, 133–141. [Google Scholar] [CrossRef]
- Feng, Z.; Wu, C.; Wang, C.; Roh, J.; Zhang, L.; Chen, J.; Zhang, S.; Zhang, H.; Yang, C.; Hu, J. SLG controls grain size and leaf angle by modulating brassinosteroid homeostasis in rice. J. Exp. Bot. 2016, 67, 4241–4253. [Google Scholar] [CrossRef]
- Rossi, V.; Varotto, S. Insights into the G1/S transition in plants. Planta 2002, 215, 345–356. [Google Scholar] [CrossRef]
- Burssens, S.; de Almeida Engler, J.; Beeckman, T.; Richard, C.; Shaul, O.; Ferreira, P.; Van Montagu, M.; Inzé, D. Developmental expression of the Arabidopsis thaliana CycA2; 1 gene. Planta 2000, 211, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Stals, H.; Inzé, D. When plant cells decide to divide. Trends Plant Sci. 2001, 6, 359–364. [Google Scholar] [CrossRef]
- Potuschak, T.; Doerner, P. Cell cycle controls: Genome-wide analysis in Arabidopsis. Curr. Opin. Plant Biol. 2001, 4, 501–506. [Google Scholar] [CrossRef]
- Criqui, M.C.; Genschik, P. Mitosis in plants: How far we have come at the molecular level? Curr. Opin. Plant Biol. 2002, 5, 487–493. [Google Scholar] [CrossRef]
- Umeda, M.; Iwamoto, N.; Umeda-Hara, C.; Yamaguchi, M.; Hashimoto, J.; Uchimiya, H. Molecular characterization of mitotic cyclins in rice plants. Mol. Gen. Genet. 1999, 262, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, P. Targeting of nucleus-encoded proteins to chloroplasts in plants. New Phytol. 2008, 179, 257–285. [Google Scholar] [CrossRef]
- Chen, M.-H.; Huang, L.-F.; Li, H.-m.; Chen, Y.-R.; Yu, S.-M. Signal peptide-dependent targeting of a rice α-amylase and cargo proteins to plastids and extracellular compartments of plant cells. Plant Physiol. 2004, 135, 1367–1377. [Google Scholar] [CrossRef]
- Wang, Z.; Hou, J.; Lu, L.; Qi, Z.; Sun, J.; Gao, W.; Meng, J.; Wang, Y.; Sun, H.; Gu, H. Small ribosomal protein subunit S7 suppresses ovarian tumorigenesis through regulation of the PI3K/AKT and MAPK pathways. PLoS ONE 2013, 8, e79117. [Google Scholar] [CrossRef]
- Moin, M.; Bakshi, A.; Saha, A.; Dutta, M.; Madhav, S.M.; Kirti, P. Rice ribosomal protein large subunit genes and their spatio-temporal and stress regulation. Front. Plant Sci. 2016, 7, 1284. [Google Scholar] [CrossRef]
- Kebeish, R.; Niessen, M.; Thiruveedhi, K.; Bari, R.; Hirsch, H.-J.; Rosenkranz, R.; Stäbler, N.; Schönfeld, B.; Kreuzaler, F.; Peterhänsel, C. Chloroplastic photorespiratory bypass increases photosynthesis and biomass production in Arabidopsis thaliana. Nat. Biotechnol. 2007, 25, 593–599. [Google Scholar] [CrossRef]
- Zhu, X.-G.; Long, S.P.; Ort, D.R. What is the maximum efficiency with which photosynthesis can convert solar energy into biomass? Curr. Opin. Biotechnol. 2008, 19, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Raines, C.A. Increasing photosynthetic carbon assimilation in C3 plants to improve crop yield: Current and future strategies. Plant Physiol. 2011, 155, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Sage, R.F.; Zhu, X.-G. Exploiting the engine of C4 photosynthesis. J. Exp. Bot. 2011, 62, 2989–3000. [Google Scholar] [CrossRef] [PubMed]
- Long, S.P.; Marshall-Colon, A.; Zhu, X.-G. Meeting the global food demand of the future by engineering crop photosynthesis and yield potential. Cell 2015, 161, 56–66. [Google Scholar] [CrossRef]
- Ort, D.R.; Merchant, S.S.; Alric, J.; Barkan, A.; Blankenship, R.E.; Bock, R.; Croce, R.; Hanson, M.R.; Hibberd, J.M.; Long, S.P. Redesigning photosynthesis to sustainably meet global food and bioenergy demand. Proc. Natl. Acad. Sci. USA 2015, 112, 8529–8536. [Google Scholar] [CrossRef]
- Simkin, A.J.; McAusland, L.; Headland, L.R.; Lawson, T.; Raines, C.A. Multigene manipulation of photosynthetic carbon assimilation increases CO2 fixation and biomass yield in tobacco. J. Exp. Bot. 2015, 66, 4075–4090. [Google Scholar] [CrossRef]
- Teng, S.; Qian, Q.; Zeng, D.; Kunihiro, Y.; Fujimoto, K.; Huang, D.; Zhu, L. QTL analysis of leaf photosynthetic rate and related physiological traits in rice (Oryza sativa L.). Euphytica 2004, 135, 1–7. [Google Scholar] [CrossRef]
- Adachi, S.; Nito, N.; Kondo, M.; Yamamoto, T.; Arai-Sanoh, Y.; Ando, T.; Ookawa, T.; Yano, M.; Hirasawa, T. Identification of chromosomal regions controlling the leaf photosynthetic rate in rice by using a progeny from japonica and high-yielding indica varieties. Plant Prod. Sci. 2011, 14, 118–127. [Google Scholar] [CrossRef]
- Gu, J.; Yin, X.; Struik, P.C.; Stomph, T.J.; Wang, H. Using chromosome introgression lines to map quantitative trait loci for photosynthesis parameters in rice (Oryza sativa L.) leaves under drought and well-watered field conditions. J. Exp. Bot. 2012, 63, 455–469. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Q.; Zhu, Q.; Liu, W.; Chen, Y.; Qiu, R.; Wang, B.; Yang, Z.; Li, H.; Lin, Y. A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol. Plant 2015, 8, 1274–1284. [Google Scholar] [CrossRef]
- China Rice Data Center. 2012. Available online: http://www.ricedata.cn/ (accessed on 9 April 2018).
- Xie, X.; Ma, X.; Zhu, Q.; Zeng, D.; Li, G.; Liu, Y.-G. CRISPR-GE: A convenient software toolkit for CRISPR-based genome editing. Mol. Plant 2017, 10, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. 1988. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 9 April 2018).
- Gruber, A.R.; Bernhart, S.H.; Lorenz, R. The ViennaRNA web services. In RNA Bioinformatics; Humana Press: New York, NY, USA, 2015; pp. 307–326. [Google Scholar] [CrossRef]
- Hiei, Y.; Ohta, S.; Komari, T.; Kumashiro, T. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J. 1994, 6, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xie, X.; Ma, X.; Li, J.; Chen, J.; Liu, Y.-G. DSDecode: A web-based tool for decoding of sequencing chromatograms for genotyping of targeted mutations. Mol. Plant 2015, 8, 1431–1433. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yang, Y.; Shi, W.; Ji, Q.; He, F.; Zhang, Z.; Cheng, Z.; Liu, X.; Xu, M. Badh2, encoding betaine aldehyde dehydrogenase, inhibits the biosynthesis of 2-acetyl-1-pyrroline, a major component in rice fragrance. Plant Cell 2008, 20, 1850–1861. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, 64–70. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Xu, X.Y.; Gong, Q.Q.; Xie, C.; Fan, W.; Yang, J.L.; Lin, Q.S.; Zheng, S.J. Root proteome of rice studied by iTRAQ provides integrated insight into aluminum stress tolerance mechanisms in plants. J. Proteom. 2014, 98, 189–205. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.J.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Lu, K.; Li, T.; He, J.; Chang, W.; Zhang, R.; Liu, M.; Yu, M.; Fan, Y.; Ma, J.; Sun, W.; et al. qPrimerDB: A thermodynamics-based gene-specific qPCR primer database for 147 organisms. Nucleic Acids Res. 2018, 46, D1229–D1236. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Plants | T1 | T2 | T3 | T4 | T5 | T6 | T7 | T8 |
|---|---|---|---|---|---|---|---|---|
| Total | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 |
| Homozygous | 6 | 5 | 6 | 7 | 4 | 5 | 6 | 7 |
| Mono-allelic heterozygous | 16 | 14 | 9 | 12 | 10 | 11 | 13 | 16 |
| Bi-allelic heterozygous | 2 | - | 1 | 2 | 1 | - | 1 | - |
| Chimeric | 1 | 2 | - | - | - | 2 | 1 | - |
| Wild type | 11 | 15 | 20 | 15 | 21 | 18 | 15 | 13 |
| Generation | Line | PH (cm) | GWD (mm) | GL (mm) | GWT (g) | GNPP | SSR (%) |
|---|---|---|---|---|---|---|---|
| T1 | WT | 95.3 ± 3.2 | 3.5 ± 0.5 | 9.3 ± 0.4 | 28.1 ± 0.4 | 139 ± 9 | 91.2 ± 3.6 |
| GXU7-1 | 93.7 ± 1.2 ns | 4.4 ± 0.5 * | 11.3 ± 0.5 * | 34.1 ± 0.4 * | 139 ± 10 ns | 90.5 ± 3.3 ns | |
| GXU19-1 | 95.5 ± 2.2 ns | 4.1 ± 0.3 * | 11.2 ± 0.5 * | 34.1 ± 0.3 * | 141 ± 10 ns | 92.3 ± 4.3 ns | |
| GXU24-1 | 97.8 ± 2.1 ns | 4.2 ± 0.2 * | 11.4 ± 0.3 * | 34.4 ± 0.4 * | 143 ± 7 ns | 91.6 ± 4.3 ns | |
| GXU28-1 | 96.3 ± 1.7 ns | 4.3 ± 0.4 * | 11.2 ± 0.4 * | 34.2 ± 0.3 * | 141 ± 8 ns | 91.4 ± 4.3 ns | |
| T2 | WT | 94.9 ± 2.3 | 3.4 ± 0.4 | 9.4 ± 0.5 | 29.3 ± 0.3 | 140 ± 8 | 90.5 ± 4.4 |
| GXU7-2 | 96.7 ± 1.2 ns | 4.4 ± 0.3 * | 11.3 ± 0.4 * | 34.4 ± 0.4 * | 141 ± 9 ns | 88.9 ± 3.5 ns | |
| GXU19-2 | 95.6 ± 2.2 ns | 4.3 ± 0.4 * | 11.4 ± 0.8 * | 34.0 ± 0.4 * | 140 ± 8 ns | 91.2 ± 3.2 ns | |
| GXU24-2 | 97.5 ± 1.1 ns | 4.1 ± 0.3 * | 11.3 ± 0.5 * | 34.7 ± 0.3 * | 141 ± 7 ns | 92.1 ± 4.3 ns | |
| GXU28-2 | 96.5 ± 1.7 ns | 4.0 ± 0.5 * | 11.5 ± 0.5 * | 34.8 ± 0.3 * | 139 ± 6 ns | 90.4 ± 2.3 ns | |
| T3 | WT | 97.5 ± 2.3 | 3.6 ± 0.4 | 9.2 ± 0.4 | 28.1 ± 0.3 | 143 ± 7 | 92.5 ± 4.5 |
| GXU7-3 | 94.8 ± 3.2 ns | 4.2 ± 0.3 * | 11.2 ± 0.6 * | 34.6 ± 0.4 * | 142 ± 9 ns | 91.5 ± 3.3 ns | |
| GXU19-3 | 95.6 ± 3.1 ns | 4.4 ± 0.3 * | 11.4 ± 0.8 * | 33.9 ± 0.4 * | 139 ± 11 ns | 92.3 ± 5.3 ns | |
| GXU24-3 | 96.4 ± 3.1 ns | 4.3 ± 0.5 * | 11.3 ± 0.5 * | 34.5 ± 0.4 * | 140 ± 10 ns | 90.6 ± 3.3 ns | |
| GXU28-3 | 96.3 ± 2.9 ns | 4.2 ± 0.5 * | 11.5 ± 0.3 * | 34.8 ± 0.3 * | 140 ± 9 ns | 90.4 ± 5.3 ns |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usman, B.; Nawaz, G.; Zhao, N.; Liu, Y.; Li, R. Generation of High Yielding and Fragrant Rice (Oryza sativa L.) Lines by CRISPR/Cas9 Targeted Mutagenesis of Three Homoeologs of Cytochrome P450 Gene Family and OsBADH2 and Transcriptome and Proteome Profiling of Revealed Changes Triggered by Mutations. Plants 2020, 9, 788. https://doi.org/10.3390/plants9060788
Usman B, Nawaz G, Zhao N, Liu Y, Li R. Generation of High Yielding and Fragrant Rice (Oryza sativa L.) Lines by CRISPR/Cas9 Targeted Mutagenesis of Three Homoeologs of Cytochrome P450 Gene Family and OsBADH2 and Transcriptome and Proteome Profiling of Revealed Changes Triggered by Mutations. Plants. 2020; 9(6):788. https://doi.org/10.3390/plants9060788
Chicago/Turabian StyleUsman, Babar, Gul Nawaz, Neng Zhao, Yaoguang Liu, and Rongbai Li. 2020. "Generation of High Yielding and Fragrant Rice (Oryza sativa L.) Lines by CRISPR/Cas9 Targeted Mutagenesis of Three Homoeologs of Cytochrome P450 Gene Family and OsBADH2 and Transcriptome and Proteome Profiling of Revealed Changes Triggered by Mutations" Plants 9, no. 6: 788. https://doi.org/10.3390/plants9060788
APA StyleUsman, B., Nawaz, G., Zhao, N., Liu, Y., & Li, R. (2020). Generation of High Yielding and Fragrant Rice (Oryza sativa L.) Lines by CRISPR/Cas9 Targeted Mutagenesis of Three Homoeologs of Cytochrome P450 Gene Family and OsBADH2 and Transcriptome and Proteome Profiling of Revealed Changes Triggered by Mutations. Plants, 9(6), 788. https://doi.org/10.3390/plants9060788