Secondary Structure of Chloroplast mRNAs In Vivo and In Vitro


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Validation of the Method
2.2. Analysis of the Secondary Structure of Start Codons
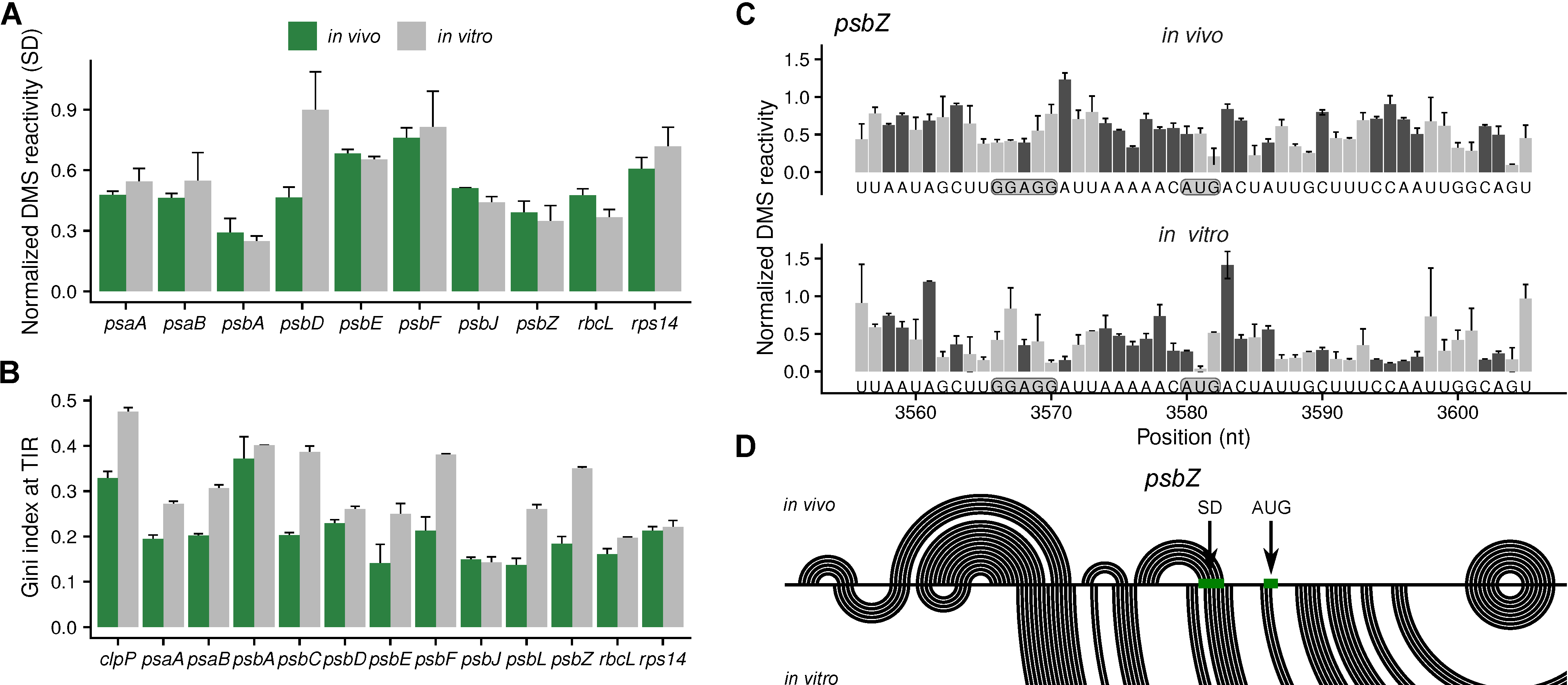
2.3. Analysis of the Secondary Structures of Shine-Dalgarno Sequences and Translation Initiation Regions
2.4. Analysis of the Secondary Structures of Coding Regions
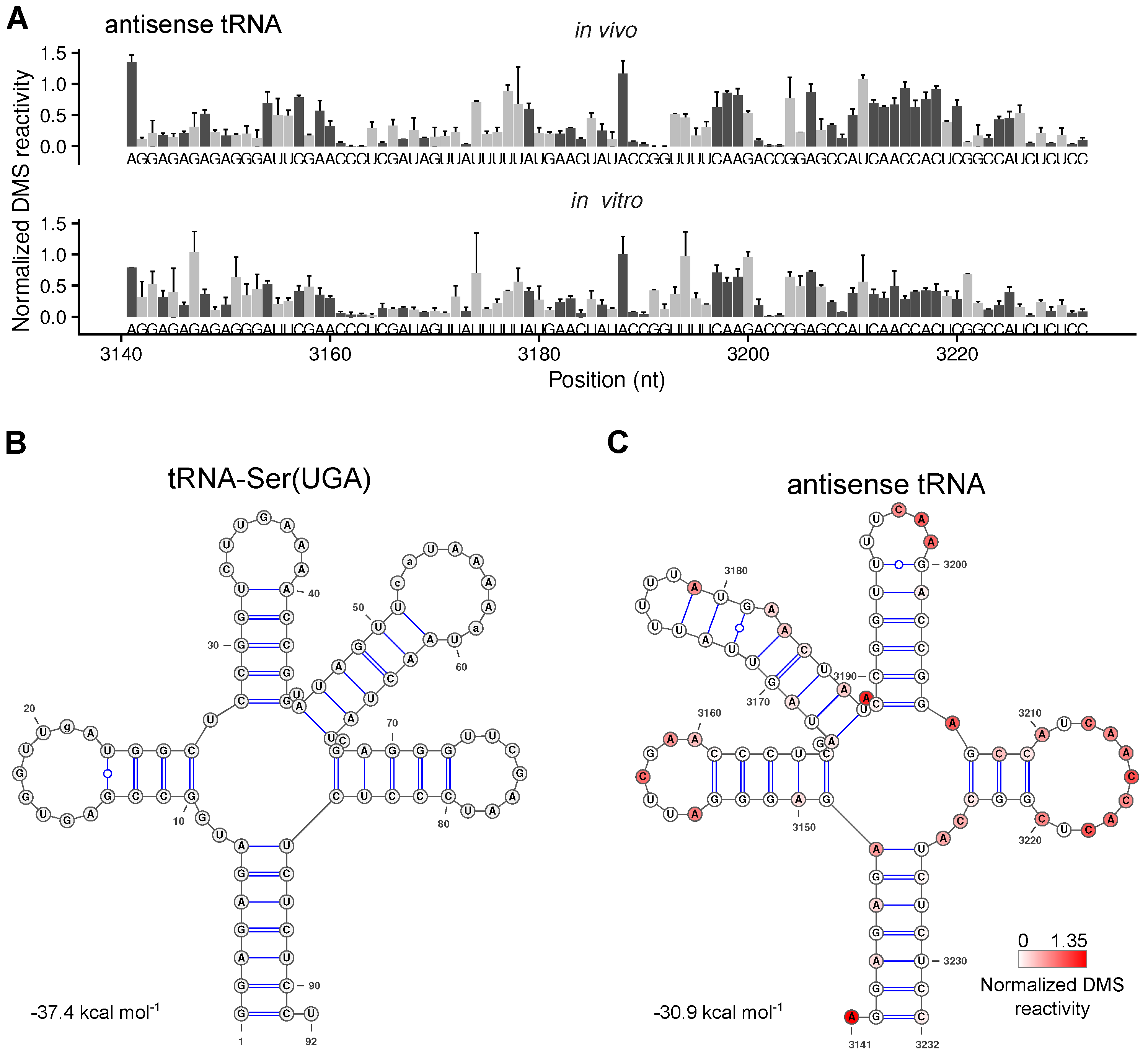
2.5. The Secondary Structure of An Antisense tRNA Sequence in the psbD/C/Z Operon
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. RNA Structure Probing with DMS
4.3. cDNA Synthesis
4.4. PCR Amplification
4.5. Data Analysis
4.6. 70S Structure Analysis
4.7. RNA Structure Prediction
4.8. Prediction of Shine-Dalgarno Sequences
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ehrenberg, M. Scientific background on the Nobel Prize in Chemistry 2009 structure and function of the ribosome. R. Swedish Acad. Sci. 2009. Available online: https://www.nobelprize.org/prizes/chemistry/2009/press-release/ https://www.nobelprize.org/uploads/2018/06/advanced-chemistryprize2009-1.pdf (accessed on 7 January 2020).
- Bieri, P.; Leibundgut, M.; Saurer, M.; Boehringer, D.; Ban, N. The complete structure of the chloroplast 70S ribosome in complex with translation factor pY. EMBO J. 2017, 36, 475–486. [Google Scholar] [CrossRef]
- Perez Boerema, A.; Aibara, S.; Paul, B.; Tobiasson, V.; Kimanius, D.; Forsberg, B.O.; Wallden, K.; Lindahl, E.; Amunts, A. Structure of the chloroplast ribosome with chl-RRF and hibernation-promoting factor. Nat. Plants 2018, 4, 212–217. [Google Scholar] [CrossRef]
- Giegé, R.; Jühling, F.; Pütz, J.; Stadler, P.; Sauter, C.; Florentz, C. Structure of transfer RNAs: Similarity and variability. Wiley Interdiscip. Rev. RNA 2012, 3, 37–61. [Google Scholar] [CrossRef]
- Pinker, F.; Bonnard, G.; Gobert, A.; Gutmann, B.; Hammani, K.; Sauter, C.; Gegenheimer, P.A.; Giegé, P. PPR proteins shed a new light on RNase P biology. RNA Biol. 2013, 10, 1457–1468. [Google Scholar] [CrossRef]
- Ziehe, D.; Dünschede, B.; Schünemann, D. From bacteria to chloroplasts: Evolution of the chloroplast SRP system. Biol. Chem. 2017, 398, 653–661. [Google Scholar] [CrossRef]
- Scharff, L.B.; Childs, L.; Walther, D.; Bock, R. Local absence of secondary structure permits translation of mRNAs that lack ribosome-binding sites. PLoS Genet. 2011, 7, e1002155. [Google Scholar] [CrossRef]
- Zhang, J.; Ruf, S.; Hasse, C.; Childs, L.; Scharff, L.B.; Bock, R. Identification of cis-elements conferring high levels of gene expression in non-green plastids. Plant J. 2012, 72, 115–128. [Google Scholar] [CrossRef]
- Scharff, L.B.; Ehrnthaler, M.; Janowski, M.; Childs, L.H.; Hasse, C.; Gremmels, J.; Ruf, S.; Zoschke, R.; Bock, R. Shine-dalgarno sequences play an essential role in the translation of plastid mRNAs in tobacco. Plant Cell 2017, 29, 3085–3101. [Google Scholar] [CrossRef]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-sequence determinants of gene expression in Escherichia coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef]
- Mustoe, A.M.; Busan, S.; Rice, G.M.; Hajdin, C.E.; Peterson, B.K.; Ruda, V.M.; Kubica, N.; Nutiu, R.; Baryza, J.L.; Weeks, K.M. Pervasive regulatory functions of mrna structure revealed by high-resolution SHAPE probing. Cell 2018, 173, 181–195.e18. [Google Scholar] [CrossRef]
- Stampacchia, O.; Girard-Bascou, J.; Zanasco, J.L.; Zerges, W.; Bennoun, P.; Rochaix, J.-D. A nuclear-encoded function essential for translation of the chloroplast psaB mRNA in chlamydomonas. Plant Cell 1997, 9, 773–782. [Google Scholar]
- Klinkert, B.; Elles, I.; Nickelsen, J. Translation of chloroplast psbD mRNA in Chlamydomonas is controlled by a secondary RNA structure blocking the AUG start codon. Nucleic Acids Res. 2006, 34, 386–394. [Google Scholar] [CrossRef]
- Prikryl, J.; Rojas, M.; Schuster, G.; Barkan, A. Mechanism of RNA stabilization and translational activation by a pentatricopeptide repeat protein. Proc. Natl. Acad. Sci. USA 2011, 108, 415–420. [Google Scholar] [CrossRef]
- Hammani, K.; Cook, W.B.; Barkan, A. RNA binding and RNA remodeling activities of the half-a-tetratricopeptide (HAT) protein HCF107 underlie its effects on gene expression. Proc. Natl. Acad. Sci. USA 2012, 109, 5651–5656. [Google Scholar] [CrossRef]
- Verhounig, A.; Karcher, D.; Bock, R. Inducible gene expression from the plastid genome by a synthetic riboswitch. Proc. Natl. Acad. Sci. USA 2010, 107, 6204–6209. [Google Scholar] [CrossRef]
- Gawroński, P.; Jensen, P.E.; Karpiński, S.; Leister, D.; Scharff, L.B. Pausing of chloroplast ribosomes is induced by multiple features and is linked to the assembly of photosynthetic complexes. Plant Physiol. 2018, 176, 2557–2569. [Google Scholar] [CrossRef]
- Strobel, E.J.; Yu, A.M.; Lucks, J.B. High-throughput determination of RNA structures. Nat. Rev. Genet. 2018, 19, 615–634. [Google Scholar] [CrossRef]
- Mitchell, D.; Assmann, S.M.; Bevilacqua, P.C. Probing RNA structure in vivo. Curr. Opin. Struct. Biol. 2019, 59, 151–158. [Google Scholar] [CrossRef]
- Wells, S.E.; Hughes, J.M.; Igel, A.H.; Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Methods Enzymol. 2000, 318, 479–493. [Google Scholar]
- Ding, Y.; Tang, Y.; Kwok, C.K.; Zhang, Y.; Bevilacqua, P.C.; Assmann, S.M. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014, 505, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.; Ritchey, L.E.; Park, H.; Babitzke, P.; Assmann, S.M.; Bevilacqua, P.C. Glyoxals as in vivo RNA structural probes of guanine base-pairing. RNA 2018, 24, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.Y.; Sexton, A.N.; Culligan, W.J.; Simon, M.D. Carbodiimide reagents for the chemical probing of RNA structure in cells. RNA 2019, 25, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Merino, E.J.; Wilkinson, K.A.; Coughlan, J.L.; Weeks, K.M. RNA structure analysis at single nucleotide resolution by Selective 2′-Hydroxyl Acylation and Primer Extension (SHAPE). J. Am. Chem. Soc. 2005, 127, 4223–4231. [Google Scholar] [CrossRef]
- McGinnis, J.L.; Dunkle, J.A.; Cate, J.H.D.; Weeks, K.M. The mechanisms of RNA SHAPE chemistry. J. Am. Chem. Soc. 2012, 134, 6617–6624. [Google Scholar] [CrossRef]
- Spitale, R.C.; Crisalli, P.; Flynn, R.A.; Torre, E.A.; Kool, E.T.; Chang, H.Y. RNA SHAPE analysis in living cells. Nat. Chem. Biol. 2013, 9, 18–20. [Google Scholar] [CrossRef]
- Siegfried, N.A.; Busan, S.; Rice, G.M.; Nelson, J.A.E.; Weeks, K.M. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods 2014, 11, 959–965. [Google Scholar] [CrossRef]
- Zubradt, M.; Gupta, P.; Persad, S.; Lambowitz, A.M.; Weissman, J.S.; Rouskin, S. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 2017, 14, 75–82. [Google Scholar] [CrossRef]
- Smola, M.J.; Weeks, K.M. In-cell RNA structure probing with SHAPE-MaP. Nat. Protoc. 2018, 13, 1181–1195. [Google Scholar] [CrossRef]
- Mustoe, A.M.; Lama, N.N.; Irving, P.S.; Olson, S.W.; Weeks, K.M. RNA base-pairing complexity in living cells visualized by correlated chemical probing. Proc. Natl. Acad. Sci. USA 2019, 116, 24574–24582. [Google Scholar] [CrossRef]
- Su, P.-H.; Lai, Y.-H. A reliable and non-destructive method for monitoring the stromal pH in isolated chloroplasts using a fluorescent pH probe. Front. Plant Sci. 2017, 8, 2079. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Shi, J.; Bhushan, S. Unique localization of the plastid-specific ribosomal proteins in the chloroplast ribosome small subunit provides mechanistic insights into the chloroplastic translation. Nucleic Acids Res. 2017, 45, 8581–8595. [Google Scholar] [CrossRef]
- Ruwe, H.; Wang, G.; Gusewski, S.; Schmitz-Linneweber, C. Systematic analysis of plant mitochondrial and chloroplast small RNAs suggests organelle-specific mRNA stabilization mechanisms. Nucleic Acids Res. 2016, 44, 7406–7417. [Google Scholar] [CrossRef]
- Schuster, M.; Gao, Y.; Schöttler, M.A.; Bock, R.; Zoschke, R. Limited responsiveness of chloroplast gene expression during acclimation to high light in tobacco. Plant Physiol. 2020, 182, 424–435. [Google Scholar] [CrossRef]
- Sexton, T.B.; Christopher, D.A.; Mullet, J.E. Light-induced switch in barley psbD-psbC promoter utilization: A novel mechanism regulating chloroplast gene expression. EMBO J. 1990, 9, 4485–4494. [Google Scholar] [CrossRef]
- Nagashima, A.; Hanaoka, M.; Shikanai, T.; Fujiwara, M.; Kanamaru, K.; Takahashi, H.; Tanaka, K. The multiple-stress responsive plastid sigma factor, SIG5, directs activation of the psbD Blue Light-Responsive Promoter (BLRP) in Arabidopsis thaliana. Plant Cell Physiol. 2004, 45, 357–368. [Google Scholar] [CrossRef]
- Gamble, P.E.; Sexton, T.B.; Mullet, J.E. Light-dependent changes in psbD and psbC transcripts of barley chloroplasts: Accumulation of two transcripts maintains psbD and psbC translation capability in mature chloroplasts. EMBO J. 1988, 7, 1289–1297. [Google Scholar] [CrossRef]
- Yao, W.B.; Meng, B.Y.; Tanaka, M.; Sugiura, M. An additional promoter within the protein-coding region of the psbD-psbC gene cluster in tobacco chloroplast DNA. Nucleic Acids Res. 1989, 17, 9583–9591. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. bioRxiv 2019, 614032. [Google Scholar] [CrossRef]
- Zhelyazkova, P.; Sharma, C.M.; Förstner, K.U.; Liere, K.; Vogel, J.; Börner, T. The primary transcriptome of barley chloroplasts: Numerous noncoding RNAs and the dominating role of the plastid-encoded RNA polymerase. Plant Cell 2012, 24, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Zhelyazkova, P.; Hammani, K.; Rojas, M.; Voelker, R.; Vargas-Suárez, M.; Börner, T.; Barkan, A. Protein-mediated protection as the predominant mechanism for defining processed mRNA termini in land plant chloroplasts. Nucleic Acids Res. 2012, 40, 3092–3105. [Google Scholar] [CrossRef]
- Kwok, C.K.; Ding, Y.; Tang, Y.; Assmann, S.M.; Bevilacqua, P.C. Determination of in vivo RNA structure in low-abundance transcripts. Nat. Commun. 2013, 4, 2971. [Google Scholar] [CrossRef] [PubMed]
- Talkish, J.; May, G.; Lin, Y.; Woolford, J.L.; McManus, C.J. Mod-seq: High-throughput sequencing for chemical probing of RNA structure. RNA 2014, 20, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, K.; Zhang, D.; Wan, Y.; Wen, Y.; Lu, Q.; Wang, L. High-throughput m6A-seq reveals RNA m6A methylation patterns in the chloroplast and mitochondria transcriptomes of Arabidopsis thaliana. PLoS ONE 2017, 12, e0185612. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.L.; David, R.; Searle, I.R. Conservation of tRNA and rRNA 5-methylcytosine in the kingdom Plantae. BMC Plant Biol. 2015, 15, 199. [Google Scholar] [CrossRef]
- Song, J.; Yi, C. Chemical modifications to RNA: A new layer of gene expression regulation. ACS Chem. Biol. 2017, 12, 316–325. [Google Scholar] [CrossRef]
- Sun, L.; Xu, Y.; Bai, S.; Bai, X.; Zhu, H.; Dong, H.; Wang, W.; Zhu, X.; Hao, F.; Song, C.-P. Transcriptome-wide analysis of pseudouridylation of mRNA and non-coding RNAs in Arabidopsis. J. Exp. Bot. 2019, 70, 5089–5600. [Google Scholar] [CrossRef]
- Small, I.D.; Schallenberg-Rüdinger, M.; Takenaka, M.; Mireau, H.; Ostersetzer-Biran, O. Plant organellar RNA editing: What 30 years of research has revealed. Plant J. 2019, 1216. [Google Scholar] [CrossRef]
- Barkan, A. Expression of plastid genes: Organelle-specific elaborations on a prokaryotic scaffold. Plant Physiol. 2011, 155, 1520–1532. [Google Scholar] [CrossRef]
- Ruwe, H.; Kupsch, C.; Teubner, M.; Schmitz-Linneweber, C. The RNA-recognition motif in chloroplasts. J. Plant Physiol. 2011, 168, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Hammani, K.; Bonnard, G.; Bouchoucha, A.; Gobert, A.; Pinker, F.; Salinas, T.; Giegé, P. Helical repeats modular proteins are major players for organelle gene expression. Biochimie 2014, 100, 141–150. [Google Scholar] [CrossRef]
- Manavski, N.; Schmid, L.-M.; Meurer, J. RNA-stabilization factors in chloroplasts of vascular plants. Essays Biochem. 2018, 62, 51–64. [Google Scholar] [PubMed]
- Jiang, J.; Chai, X.; Manavski, N.; Williams-Carrier, R.; He, B.; Brachmann, A.; Ji, D.; Ouyang, M.; Liu, Y.; Barkan, A.; et al. An RNA chaperone-like protein plays critical roles in chloroplast mRNA stability and translation in Arabidopsis and Maize. Plant Cell 2019, 31, 1308–1327. [Google Scholar] [CrossRef] [PubMed]
- McDermott, J.J.; Civic, B.; Barkan, A. Effects of RNA structure and salt concentration on the affinity and kinetics of interactions between pentatricopeptide repeat proteins and their RNA ligands. PLoS ONE 2018, 13, e0209713. [Google Scholar] [CrossRef]
- Takyar, S.; Hickerson, R.P.; Noller, H.F. mRNA helicase activity of the ribosome. Cell 2005, 120, 49–58. [Google Scholar] [CrossRef]
- Horlitz, M.; Klaff, P. Gene-specific trans-regulatory functions of magnesium for chloroplast mRNA stability in higher plants. J. Biol. Chem. 2000, 275, 35638–35645. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, M.; Wang, T.; Zhang, Y.; Zhang, X. Genome-wide probing RNA structure with the modified DMS-MaPseq in Arabidopsis. Methods 2019, 155, 30–40. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Morgan, M.; Pagès, H.; Obenchain, V.; Hayden, N. Rsamtools: Binary alignment (BAM), FASTA, variant call (BCF), and tabix file import. R package version 2.2.3. 2020. Available online: http://bioconductor.org/packages/Rsamtools (accessed on 4 March 2020).
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Turner, D.H.; Watson, R.M. RNA secondary structure prediction. Curr. Protoc. Nucleic Acid Chem. 2016, 67, 11.2.1–11.2.19. [Google Scholar] [CrossRef] [PubMed]
- Lai, D.; Proctor, J.R.; Zhu, J.Y.A.; Meyer, I.M. R-CHIE: A web server and R package for visualizing RNA secondary structures. Nucleic Acids Res. 2012, 40, e95. [Google Scholar] [CrossRef] [PubMed]
- Darty, K.; Denise, A.; Ponty, Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 2009, 25, 1974–1975. [Google Scholar] [CrossRef] [PubMed]
- Starmer, J.; Stomp, A.; Vouk, M.; Bitzer, D. Predicting shine-dalgarno sequence locations exposes genome annotation errors. PLoS Comput. Biol. 2006, 2, e57. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gawroński, P.; Pałac, A.; Scharff, L.B. Secondary Structure of Chloroplast mRNAs In Vivo and In Vitro. Plants 2020, 9, 323. https://doi.org/10.3390/plants9030323
Gawroński P, Pałac A, Scharff LB. Secondary Structure of Chloroplast mRNAs In Vivo and In Vitro. Plants. 2020; 9(3):323. https://doi.org/10.3390/plants9030323
Chicago/Turabian StyleGawroński, Piotr, Aleksandra Pałac, and Lars B. Scharff. 2020. "Secondary Structure of Chloroplast mRNAs In Vivo and In Vitro" Plants 9, no. 3: 323. https://doi.org/10.3390/plants9030323
APA StyleGawroński, P., Pałac, A., & Scharff, L. B. (2020). Secondary Structure of Chloroplast mRNAs In Vivo and In Vitro. Plants, 9(3), 323. https://doi.org/10.3390/plants9030323