The Occurrence of Flavonoids and Related Compounds in Cedrus brevifolia A. Henry ex Elwes & A. Henry Needles. Inhibitory Potencies on Lipoxygenase, Linoleic Acid Lipid Peroxidation and Antioxidant Activity



Abstract
:
1. Introduction
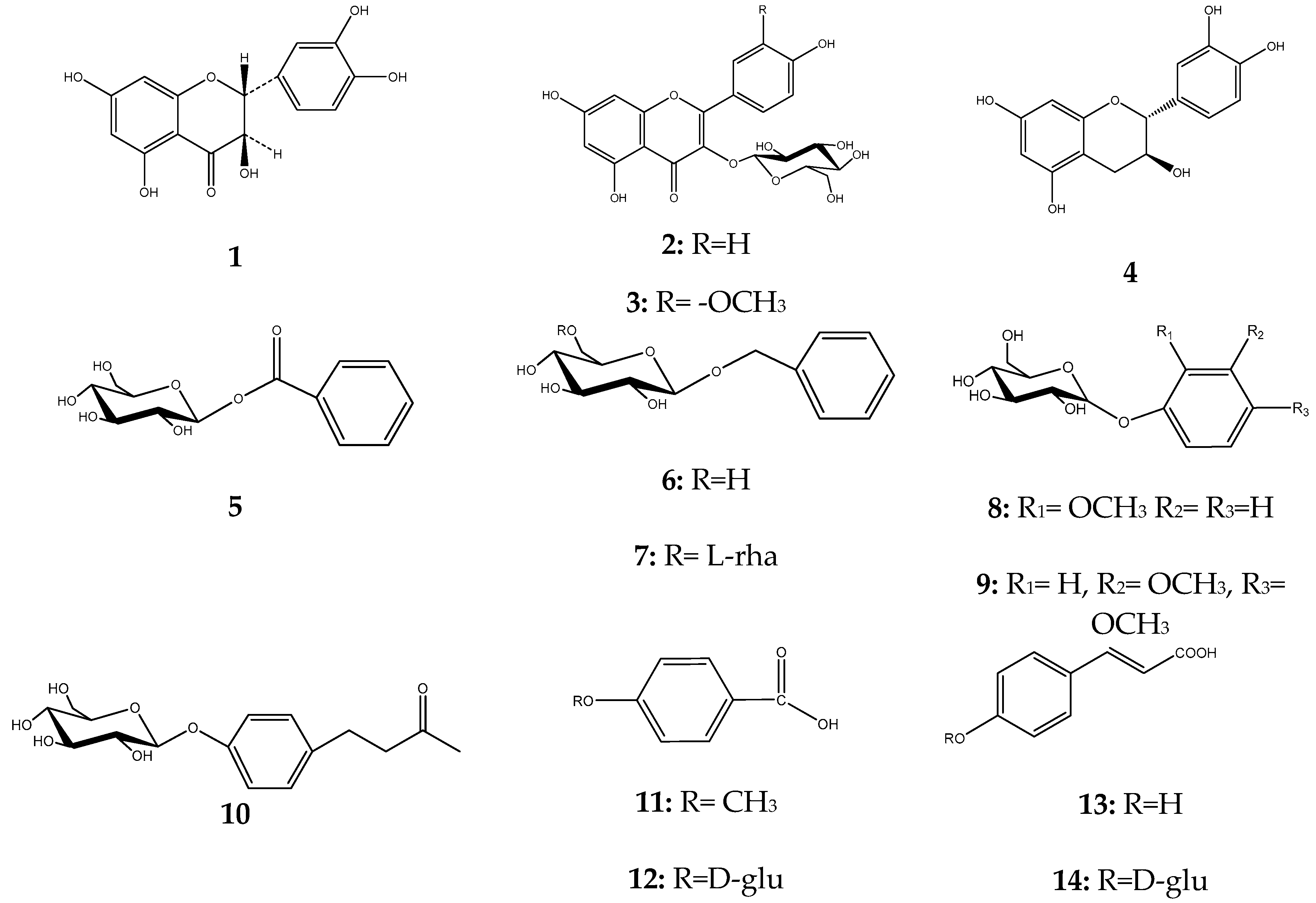
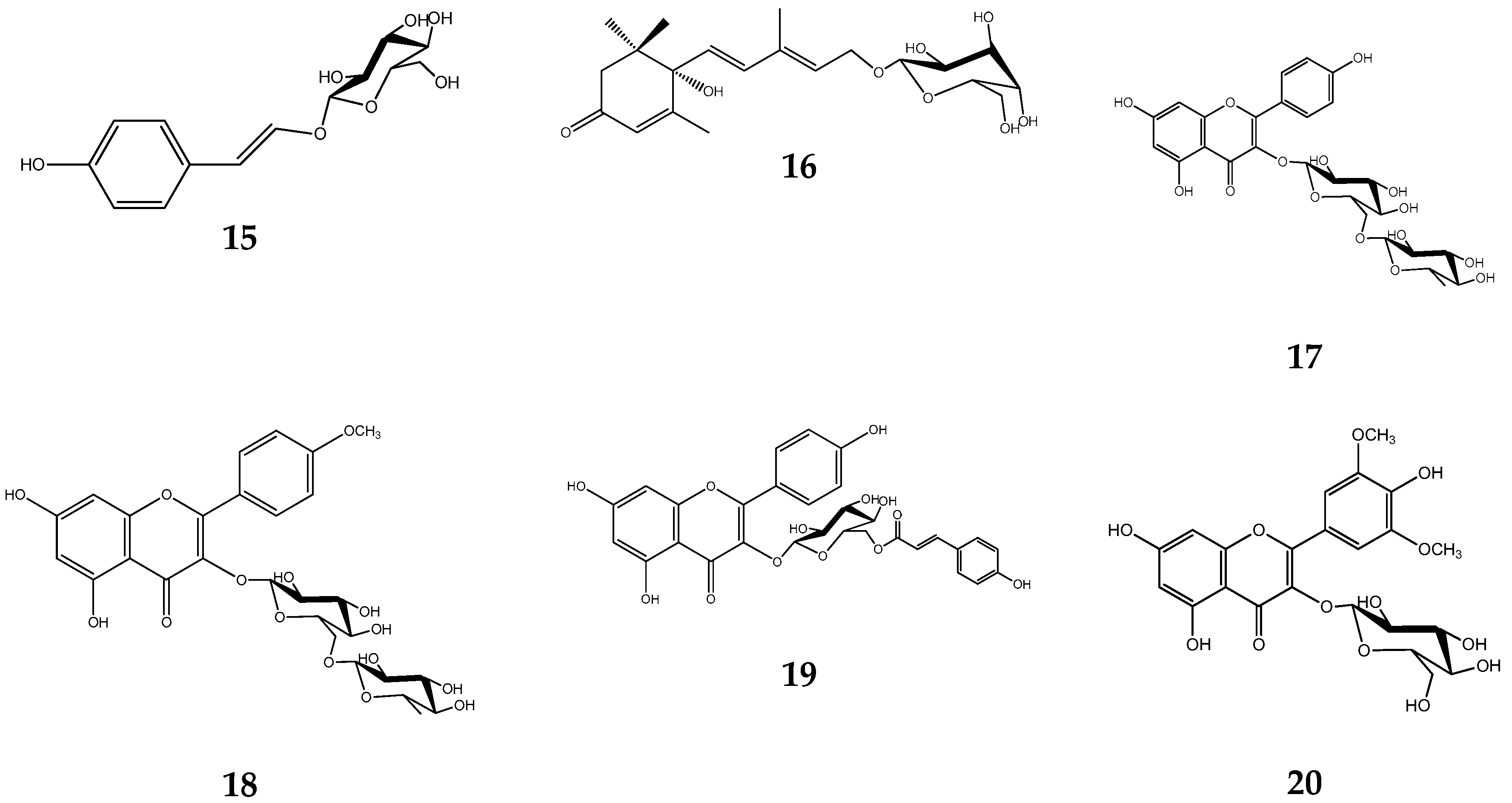
2. Results
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Equipment and Reagents
4.3. Equipment and Reagents for In Vitro Experiments
4.4. Extraction and Chromatography
4.5. DPPH Radical Scavenging Activity
4.6. AAPH Induced Linoleic Acid Lipid Peroxidation Assay
4.7. Soybean LOX Inhibition
4.8. Statistics
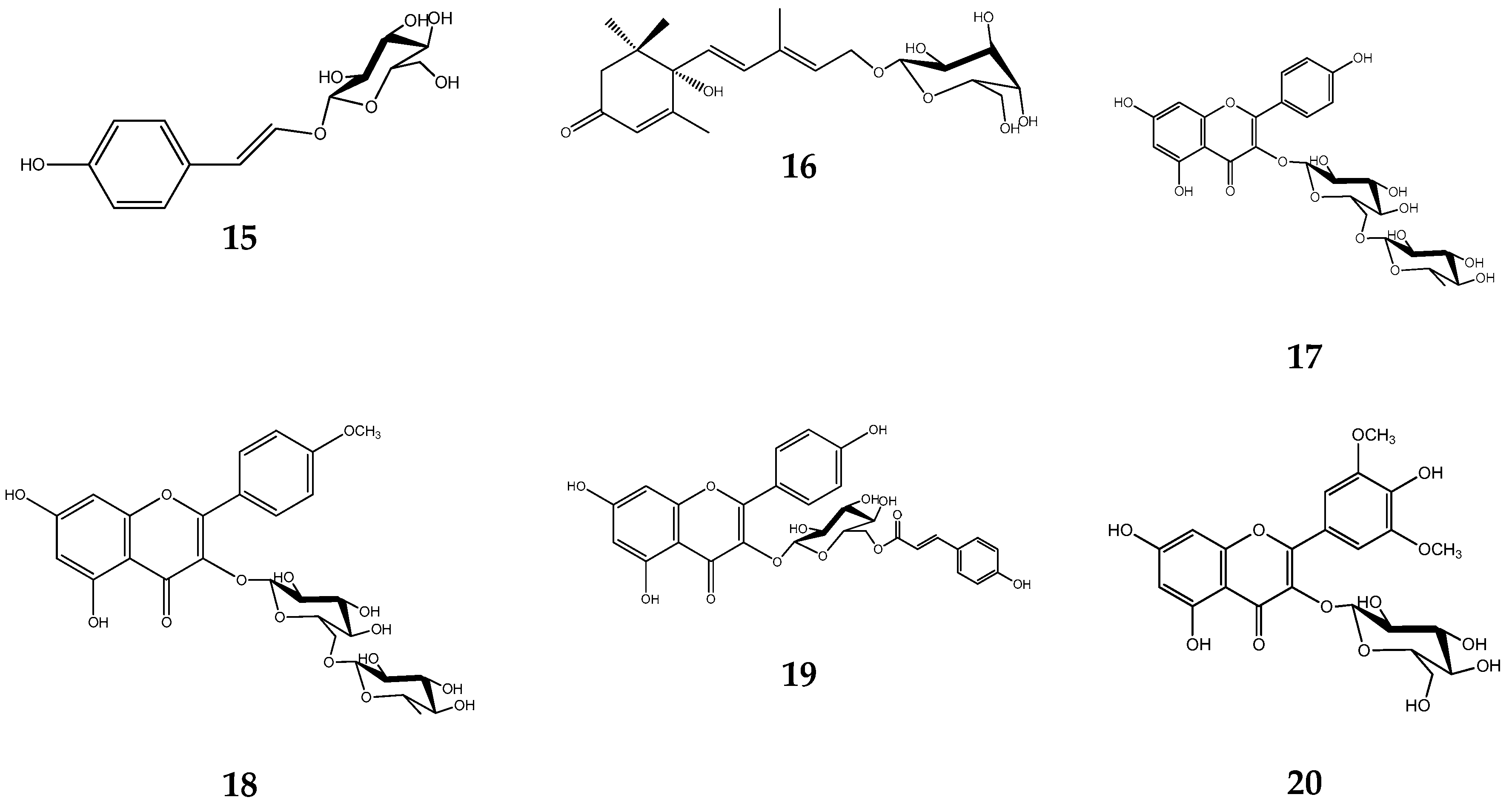
4.9. NMR Data of Compounds 1–20
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Dagher-Kharrat, M.B.; Mariette, S.; Lefèvre, F.; Fady, B.; March, G.G.; Plomion, C.; Savoure, A. Geographical diversity and genetic relationship among Cedrus species estimated by AFLP. Tree Genet. Genomes 2006, 3, 275–285. [Google Scholar] [CrossRef]
- Theophrasti, E. Opera; Wimmer, F., Ed.; Didot: Paris, France, 1866. (In Greek) [Google Scholar]
- Kurt, Y.; Kacar, M.S.; Isik, K. Traditional Tar Production from Cedrus libani A. Rich on the Taurus Mountains in Southern Turkey. Econ. Bot. 2008, 62, 615–620. [Google Scholar] [CrossRef]
- Pollio, V. Vitruvius: The Ten Books on Architecture; Harvard University Press: New York, NY, USA, 1914; pp. 154–196. [Google Scholar]
- Creţu, E.; Trifan, A.; Aprotosoaie, A.C.; Miron, A. 15-lipoxygenase inhibition, superoxide and hydroxyl radicals scavenging activities of Cedrus brevifolia bark extracts. Rev. Med. Chir. Soc. Med. Nat. Iasi. 2013, 117, 250–256. [Google Scholar]
- Hafizoglu, H.; Barne, H. Studies on the Chemistry of Cedrus libani A. Rich.-III. Oleoresin Composition of Cones and Bark from Cedrus libani. Holzforsch. Int. J. Biol. Chem. Phys. Technol. Wood 1987, 41, 141–145. [Google Scholar] [CrossRef]
- Zeng, W.C.; He, Q.; Sun, Q.; Zhong, K.; Gao, H. Antibacterial activity of water-soluble extract from pine needles of Cedrus deodara. Int. J. Food Microbiol. 2012, 153, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Usmana, A.; Vera Thossb, V.; Nur-e-Alamc, M. Isolation of Taxifolin from Trichilia emetica Whole Seeds. Am. Sci. Res. J. Eng. Technol. Sci. 2016, 21, 77–82. [Google Scholar]
- Saito, S.; Silva, G.; Santos, R.; Gosmann, G.; Pungartnik, C.; Brendel, M. Astragalin from Cassiaalata induces DNA Adducts in Vitro and Repairable DNA damage in the yeast Saccharomyces cerevisiae. Int. J. Mol. Sci. 2012, 13, 2846–2862. [Google Scholar] [CrossRef] [PubMed]
- El-Hawiet, A.M.; Toaima, S.M.; Asaad, A.M.; Radwan, M.M.; El-Sebakhy, N.A. Chemical constituents from Astragalus annularis Forssk. and A. trimestris L., Fabaceae. Rev. Bras. Pharmacogn. 2009, 20, 860–865. [Google Scholar] [CrossRef]
- Lee, S.I.; Yang, J.H.; Kim, D.K. Antioxidant Flavonoids from the Twigs of Stewartia koreana. Biomol. Ther. 2010, 18, 191–196. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Chang, F.-R.; Lu, M.-C.; Hsieh, P.-W.; Wu, M.-J.; Du, Y.-C.; Wu, Y.-C. New Benzoyl Glucosides and Cytotoxic Pterosin Sesquiterpenes from Pteris ensiformis Burm. Molecules 2008, 13, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Schwab, W.; Schreier, P. Aryl β-d-glucosides from Carica papaya fruit. Phytochemistry 1987, 27, 1813–1816. [Google Scholar] [CrossRef]
- Hamerski, L.; Bomm, M.D.; Silva, D.H.S.; Claudia, M.; Young, M.; Furlan, M.; Eberlin, M.N.; Gamboa, I.C.; Cavalheiro, A.J.; Bolzani, S. Phenylpropanoid glucosides from leaves of Coussarea Hydrangeifolia (Rubiaceae). Phytochemistry 2005, 66, 1927–1932. [Google Scholar] [CrossRef] [PubMed]
- Fujimatu, E.; Ishikawa, T.; Kitajima, J. Aromatic compound glucosides, alkyl glucoside and glucide from the fruit of Anisei. Phytochemistry 2003, 63, 609–616. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, J.; Xiao, L.; Zeng, G.Z.; Sun, Z.H.; Tan, N.H. A New Phenolic Glycoside from Chamaecyparis obtuse var. Breviramea f. crippsii. Molecules 2013, 18, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Pabst, A.; Barron, D.; Adda, J.; Schreier, P. Phenylbutan-2-one β-d-glucosides from raspberry fruit. Phytochemistry 1990, 29, 3853–3858. [Google Scholar] [CrossRef]
- Marquesa, D.D.; Machado, M.I.L.; de Carvalhob, M.G.; da C. Meleirab, L.A.; Braz-Filhoc, R. Isoflavonoids and Triterpenoids Isolated from Pterodon Polygalaeflorus. J. Braz. Chem. Soc. 1998, 9, 295–301. [Google Scholar] [CrossRef]
- Fiorentino, A.; Abrosca, B.; Pacifico, S.; Mastellone, C.; Piscopo, V.; Caputo, R.; Monaco, P. Isolation and Structure Elucidation of Antioxidant Polyphenols from Quince (Cydonia vulgaris) Peels. J. Agric. Food Chem. 2008, 56, 2660–2667. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Rahman, M.; Jabbar, A.; Rashid, M. Phytochemical and biological investigations of Albizzia lebbeck Benth. Boletín Latinoamericano y del Caribe de Plantas Medicinales y Aromáticas 2008, 7, 273–278. [Google Scholar]
- Johnssona, P.; Peerlkampa, N.; Kamal-Eldina, A.; Andersson, R.; Anderssona, R.; Lundgren, L.; Aman, P. Polymeric fractions containing phenol glucosides in flaxseed. Food Chem. 2002, 76, 207–212. [Google Scholar] [CrossRef]
- Abdel-Kade, M.S. Two New Nor-phenylpropanoid Glucosides and Hemipholin from the Flowers of Ononis vaginalis. J. Braz. Chem. Soc. 1997, 8, 637–639. [Google Scholar] [CrossRef]
- Lutz, A.; Winterhalter, P. Isolation of Additional Carotenoid Metabolites from Quince Fruit (Cydonia oblonga Mill.). J. Agric. Food Chem. 1992, 40, 1116–1120. [Google Scholar] [CrossRef]
- Akkola, E.K.; Süntara, I.; Keleşb, H.; Sezika, E.; Gürlerc, G. Bioassay-guided isolation and characterization of wound healer compounds from Morus nigra L. (Moraceae). Rec. Nat. Prod. 2015, 9, 484–495. [Google Scholar]
- Moustafa, A.M.Y.; Khodair, A.I.; Saleh, M.A. Isolation, structural elucidation of flavonoid constituents from Leptadenia pyrotechnica and evaluation of their toxicity and antitumor activity. Pharm. Biol. 2009, 47, 539–552. [Google Scholar] [CrossRef]
- Mekhelfi, T.; Kerbab, K.; Guella, G.; Zaiter, L.; Benayache, S.; Benayache, F. Phytochemical constituents of Thymelaea microphylla Coss. et Dur. from Algeria. Pharm. Lett. 2014, 6, 152–156. [Google Scholar]
- Gutzeit, D.; Wray, V.; Winterhalter, P.; Jerz, G. Preparative Isolation and Purification of Flavonoids and Protocatechuic Acid from Sea Buckthorn Juice Concentrate (Hippophaë rhamnoides L. ssp. rhamnoides) by High-Speed Counter-Current Chromatography. Chromatographia 2007, 65, 1–7. [Google Scholar] [CrossRef]
- Koleva, I.I.; van Beek, T.A.; Linssen, J.P.H.; de Groot, A.; Evstatieva, L.N. Screening of Plant Extracts for Antioxidant Activity: A Comparative Study on Three Testing Methods. Phytochem. Anal. 2001, 13, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Liegois, C.; Lermusieau, G.; Colin, S.J. Measuring Antioxidant Efficiency of Wort, Malt, and Hops against the 2,2′-Azobis(2-amidinopropane) Dihydrochloride-Induced Oxidation of an Aqueous Dispersion of Linoleic Acid. Agric. Food Chem. 2000, 48, 1129–1134. [Google Scholar] [CrossRef]
- Mabry, T.G.; Markham, K.R.; Thomas, M.B. The Systematic Identification of Flavonoids; Springer: New York, NY, USA, 1970. [Google Scholar]
- Coll, J.C.; Bowden, B.F. The application of vacuum liquid chromatography to the separation of terpene mixtures. J. Nat. Prod. 1986, 49, 934–936. [Google Scholar] [CrossRef]
- Neu, R. Chelate von Diarylborsäuren mit aliphatischen Oxyalkylaminen als Reagenzien für den Nachweis von Oxyphenyl-benzo-γ-pyronen. Naturwissenschaften 1957, 44, 181–183. [Google Scholar] [CrossRef]
- Pontiki, E.; Hadjipavlou-Litina, D.; Litinas, K.; Nicolotti, O.; Carotti, A. Design, synthesis and pharmacological evaluation of novel acrylic acid derivatives acting as lipoxygenase and cyclooxygenase-1 inhibitors with antioxidant and anti-inflammatory activities. Eur. J. Med. Chem. 2011, 46, 191–200. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
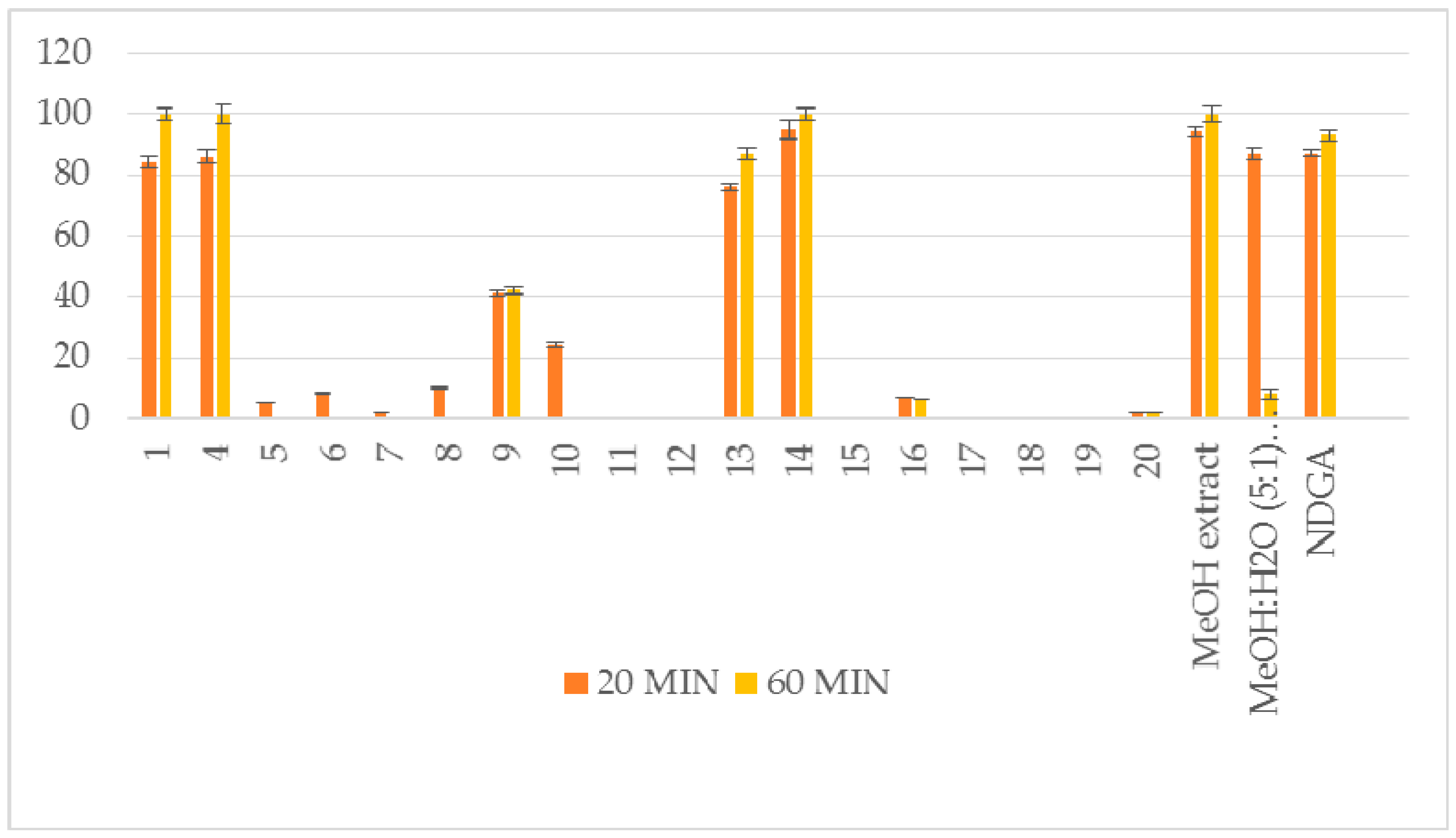
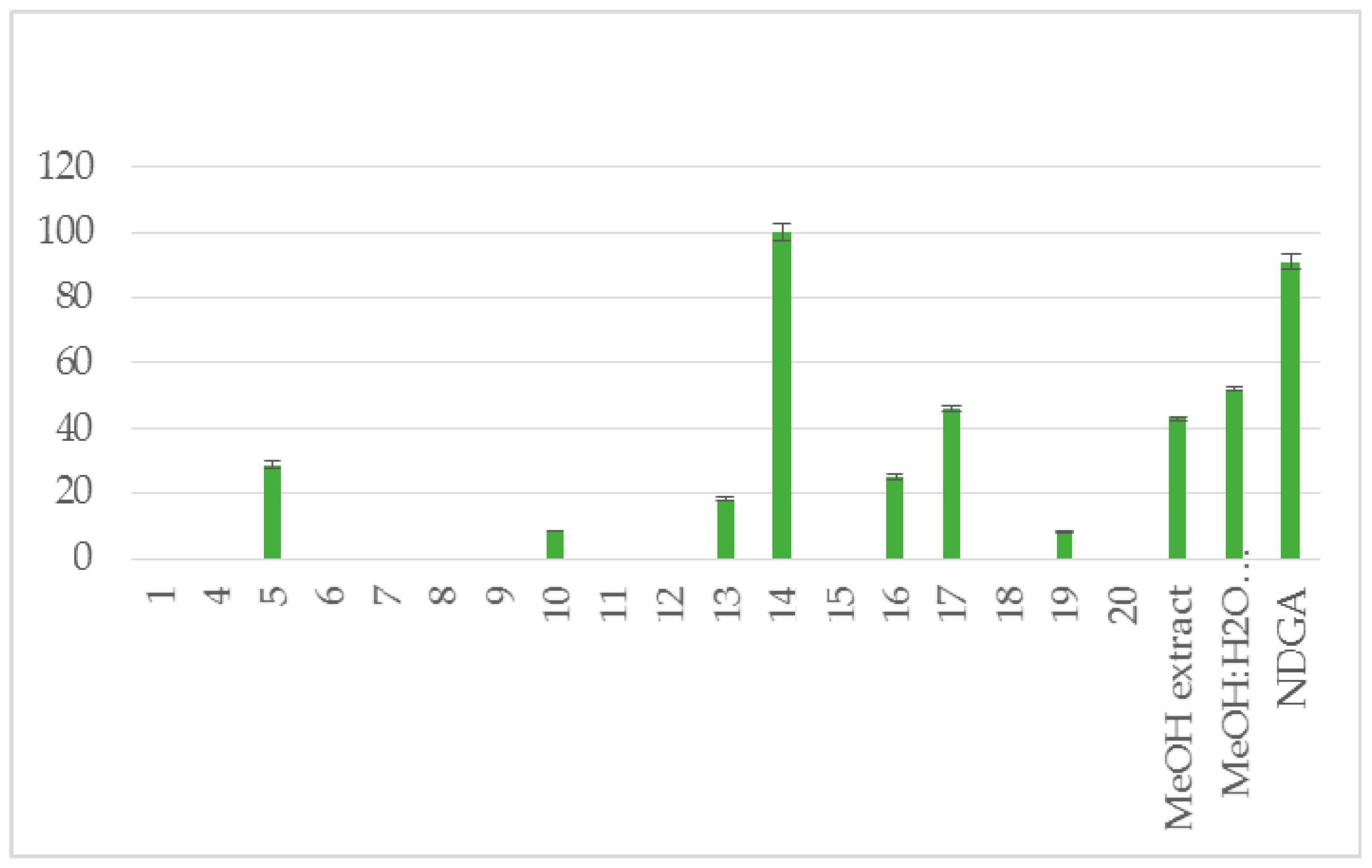
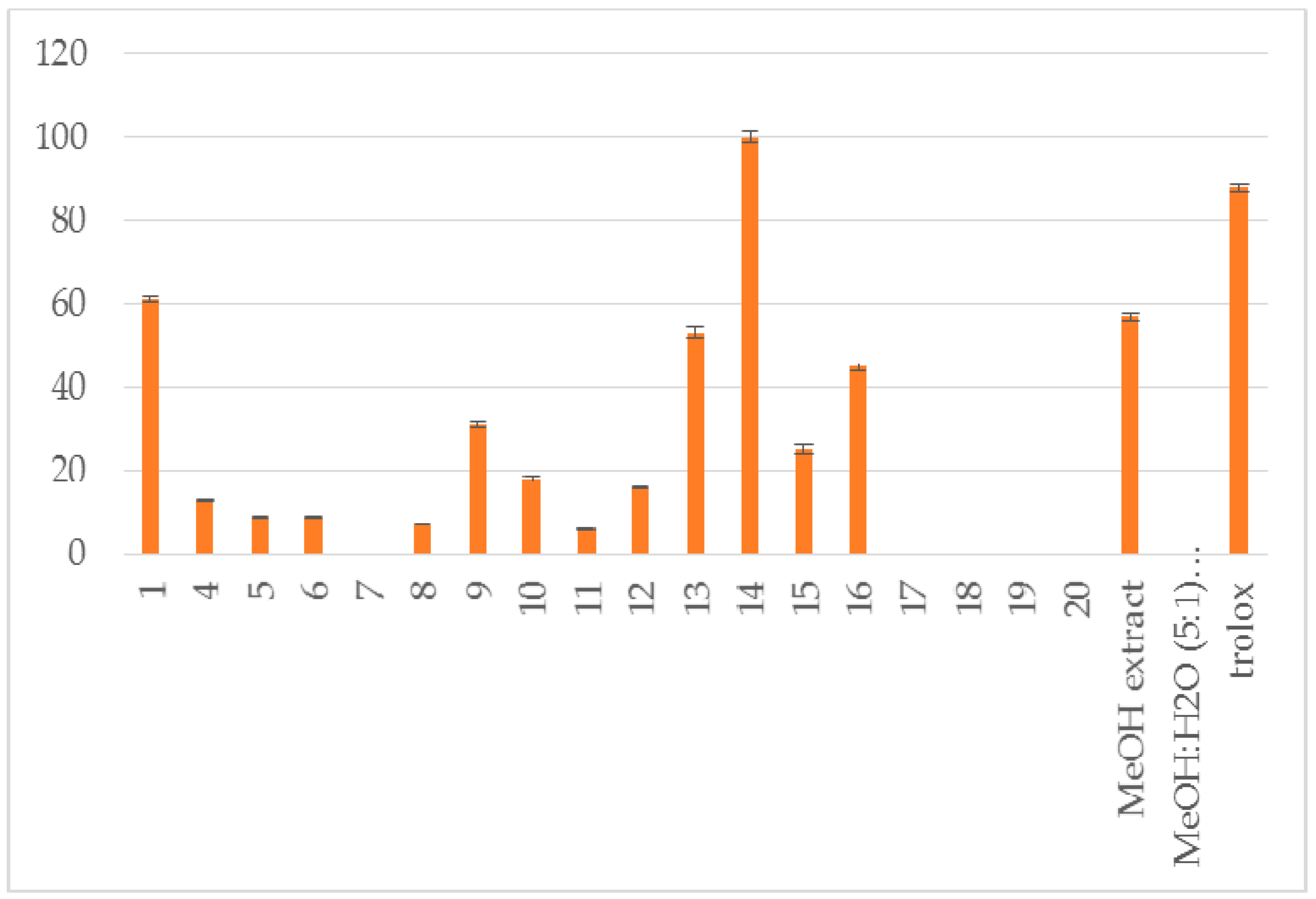
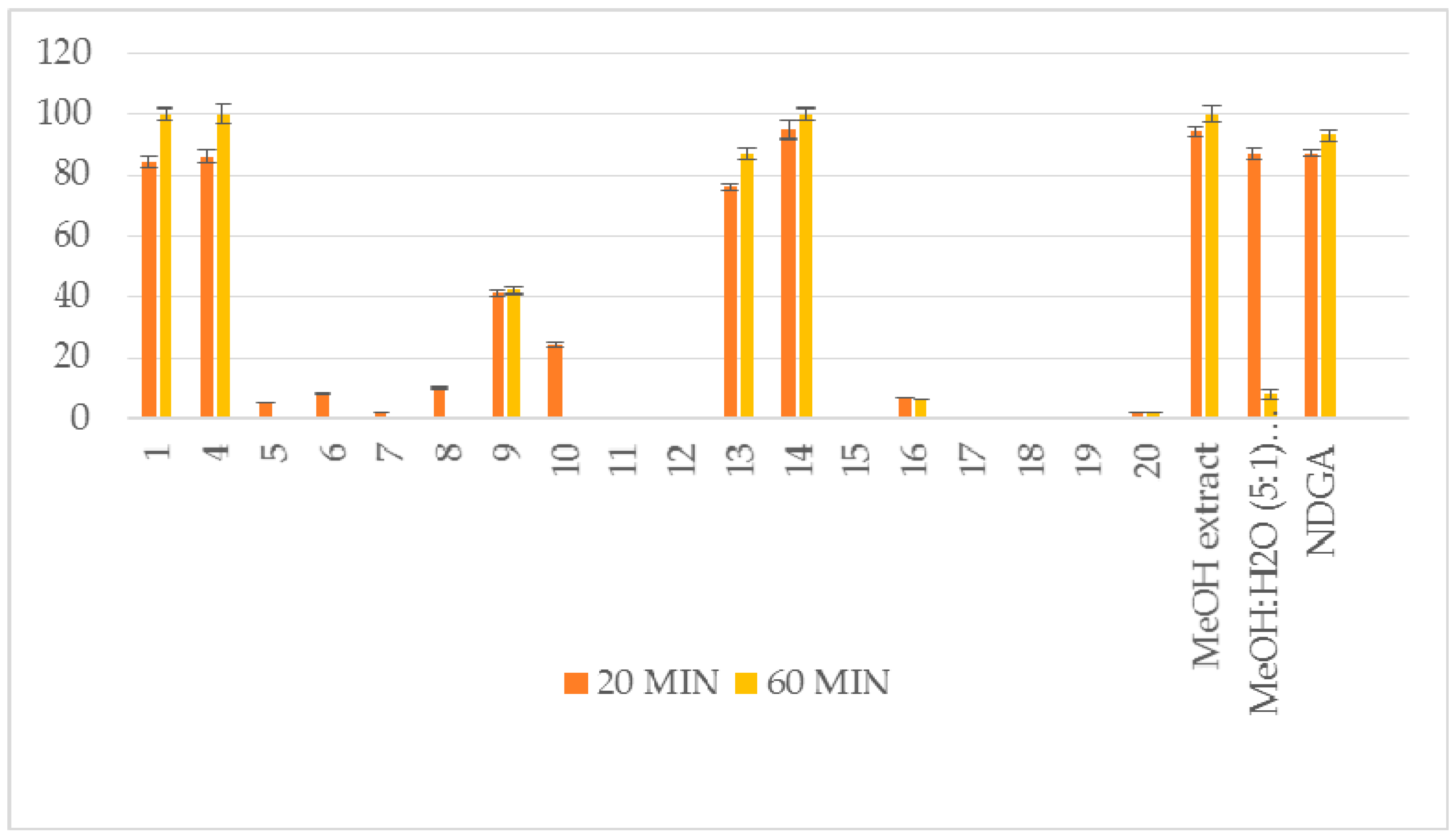
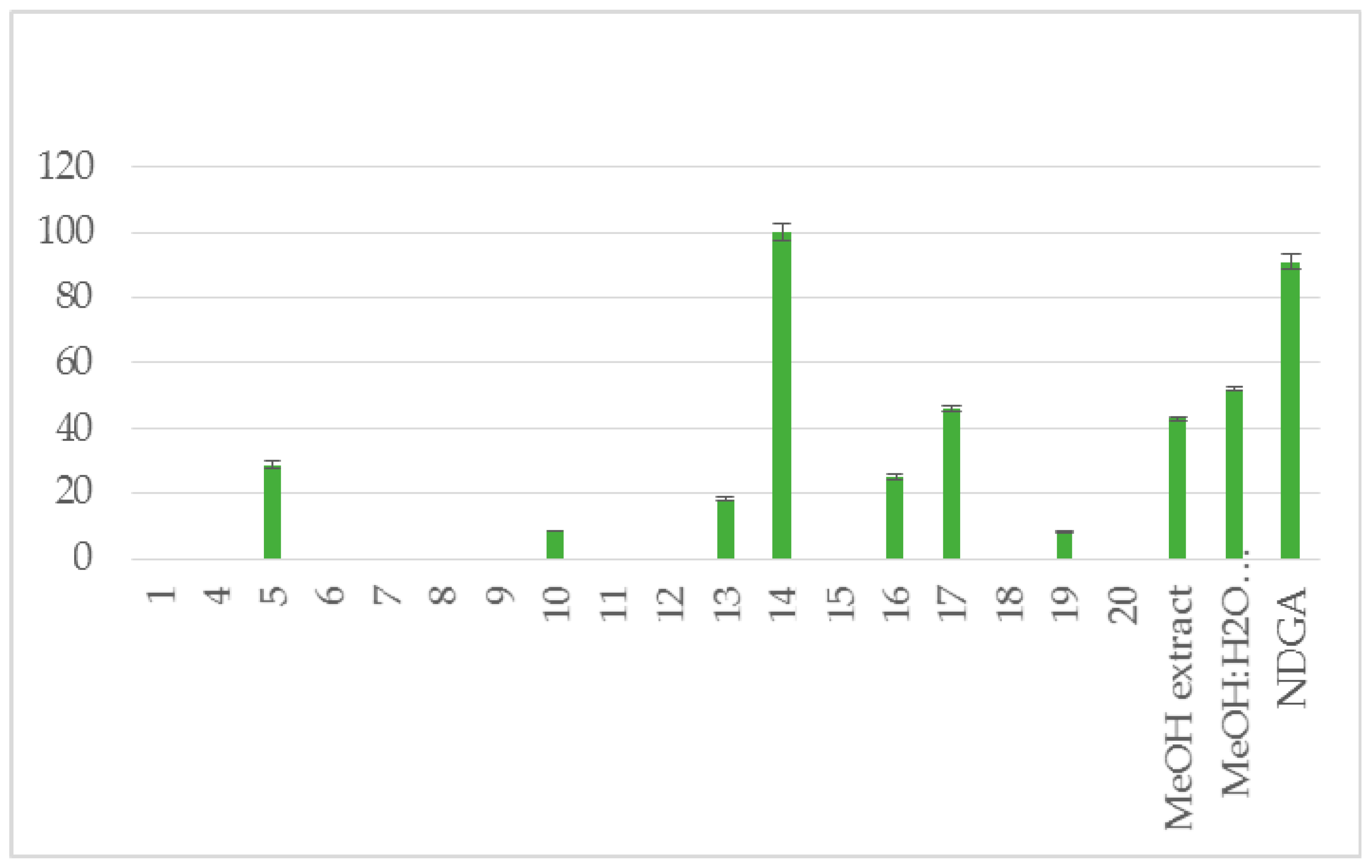
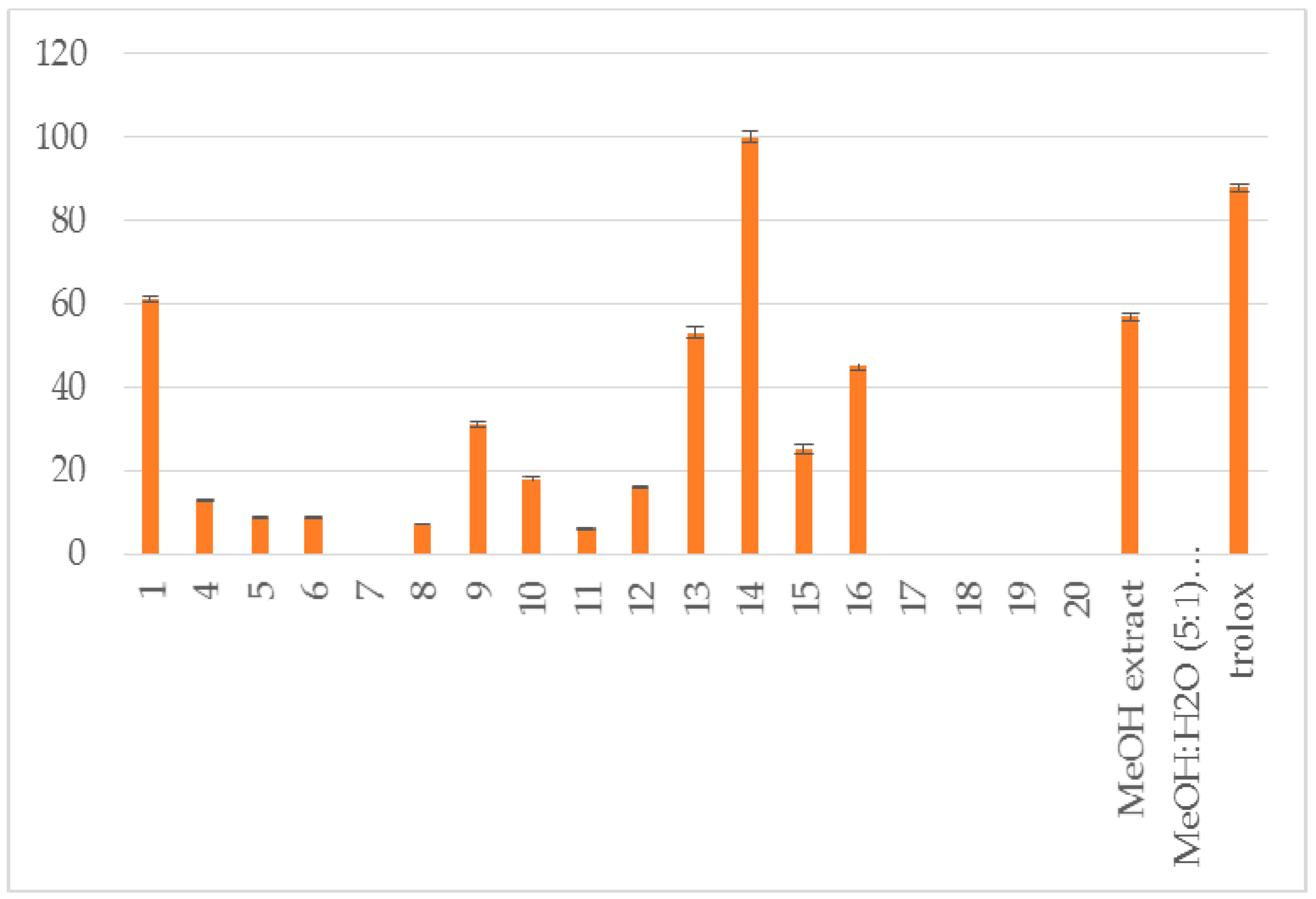
| Compound | RA # % ± SD, DPPH, (20 min) | RA # % ± SD, DPPH, (60 min) | % LOX ± SD Inhbt @ (0.1 mM) | A-LP % ± SD @ (0.1 mM) |
|---|---|---|---|---|
| 1 | 84 ± 1.8 * | 100 ± 2.1 ** | no | 61 ± 0.6 ** |
| 4 | 86 ± 2.2 ** | 100 ± 3.1 ** | no | 13 ± 0.3 * |
| 5 | 5 ± 0.1 * | no | 29 ± 1.1 ** | 9 ± 0.1 * |
| 6 | 8 ± 0.3 ** | no | no | 9 ± 0.1 * |
| 7 | 2 ± 0.0 * | no | no | no |
| 8 | 9.8 ± 0.4 * | no | no | 7 ± 0.1 * |
| 9 | 41 ± 1.0 ** | 42 ± 1.3 ** | no | 31 ± 0.7 * |
| 10 | 24 ± 0.8 ** | no | 8.5 ± 0.1 ** | 18 ± 0.6 ** |
| 11 | no | no | no | 6 ± 0.1 * |
| 12 | no | no | no | 16 ± 0.1 * |
| 13 | 76 ± 1.1 ** | 87 ± 1.9 ** | 18 ± 0.6 ** | 53 ± 1.2 ** |
| 14 | 95 ± 3.2 ** | 100 ± 2.1 ** | 100 ± 2.5 ** | 100 ± 1.4 ** |
| 15 | no | no | no | 25 ± 1.0 * |
| 16 | 7 ± 0.1 * | 6 ± 0.0 * | 25±1.2 ** | 45 ± 0.9 ** |
| 17 | nt # | nt # | 46 ± 1.0 ** | nt # |
| 18 | nt # | nt # | no | no |
| 19 | no | no | 8 ± 0.3 * | no |
| 20 | 2 ± 0.0 * | 2 ± 0.0 * | no | no |
| MeOH extract | 94 ± 1.9 ** | 100 ± 2.5 ** | 43 ± 0.4 * | 57 ± 1.0 ** |
| MeOH:H2O (5:1) extract | 87 ± 2.1 ** | 8 ± 1.8 ** | 52 ± 0.7 ** | no |
| NDGA | 87 ± 1.1** | 93 ± 1.8 ** | 91 ± 2.3 ** | |
| trolox | 88 ± 0.9 ** |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Douros, A.; Hadjipavlou-Litina, D.; Nikolaou, K.; Skaltsa, H. The Occurrence of Flavonoids and Related Compounds in Cedrus brevifolia A. Henry ex Elwes & A. Henry Needles. Inhibitory Potencies on Lipoxygenase, Linoleic Acid Lipid Peroxidation and Antioxidant Activity. Plants 2018, 7, 1. https://doi.org/10.3390/plants7010001
Douros A, Hadjipavlou-Litina D, Nikolaou K, Skaltsa H. The Occurrence of Flavonoids and Related Compounds in Cedrus brevifolia A. Henry ex Elwes & A. Henry Needles. Inhibitory Potencies on Lipoxygenase, Linoleic Acid Lipid Peroxidation and Antioxidant Activity. Plants. 2018; 7(1):1. https://doi.org/10.3390/plants7010001
Chicago/Turabian StyleDouros, Andreas, Dimitra Hadjipavlou-Litina, Konstantinos Nikolaou, and Helen Skaltsa. 2018. "The Occurrence of Flavonoids and Related Compounds in Cedrus brevifolia A. Henry ex Elwes & A. Henry Needles. Inhibitory Potencies on Lipoxygenase, Linoleic Acid Lipid Peroxidation and Antioxidant Activity" Plants 7, no. 1: 1. https://doi.org/10.3390/plants7010001
APA StyleDouros, A., Hadjipavlou-Litina, D., Nikolaou, K., & Skaltsa, H. (2018). The Occurrence of Flavonoids and Related Compounds in Cedrus brevifolia A. Henry ex Elwes & A. Henry Needles. Inhibitory Potencies on Lipoxygenase, Linoleic Acid Lipid Peroxidation and Antioxidant Activity. Plants, 7(1), 1. https://doi.org/10.3390/plants7010001