
Transcriptional Dynamics Underlying Somatic Embryogenesis in Coffea canephora

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
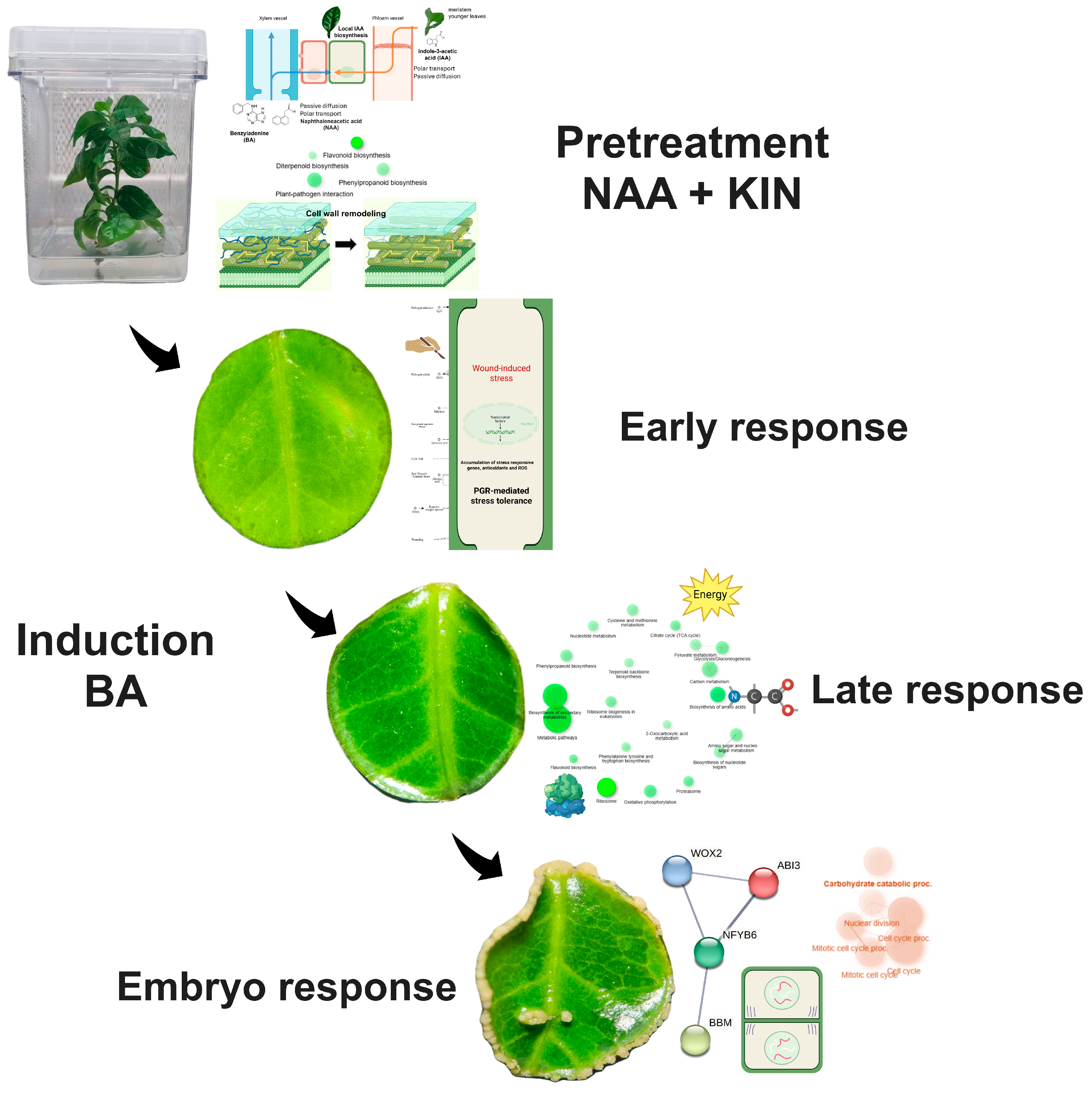
2.1. SE of C. canephora as a Study Model
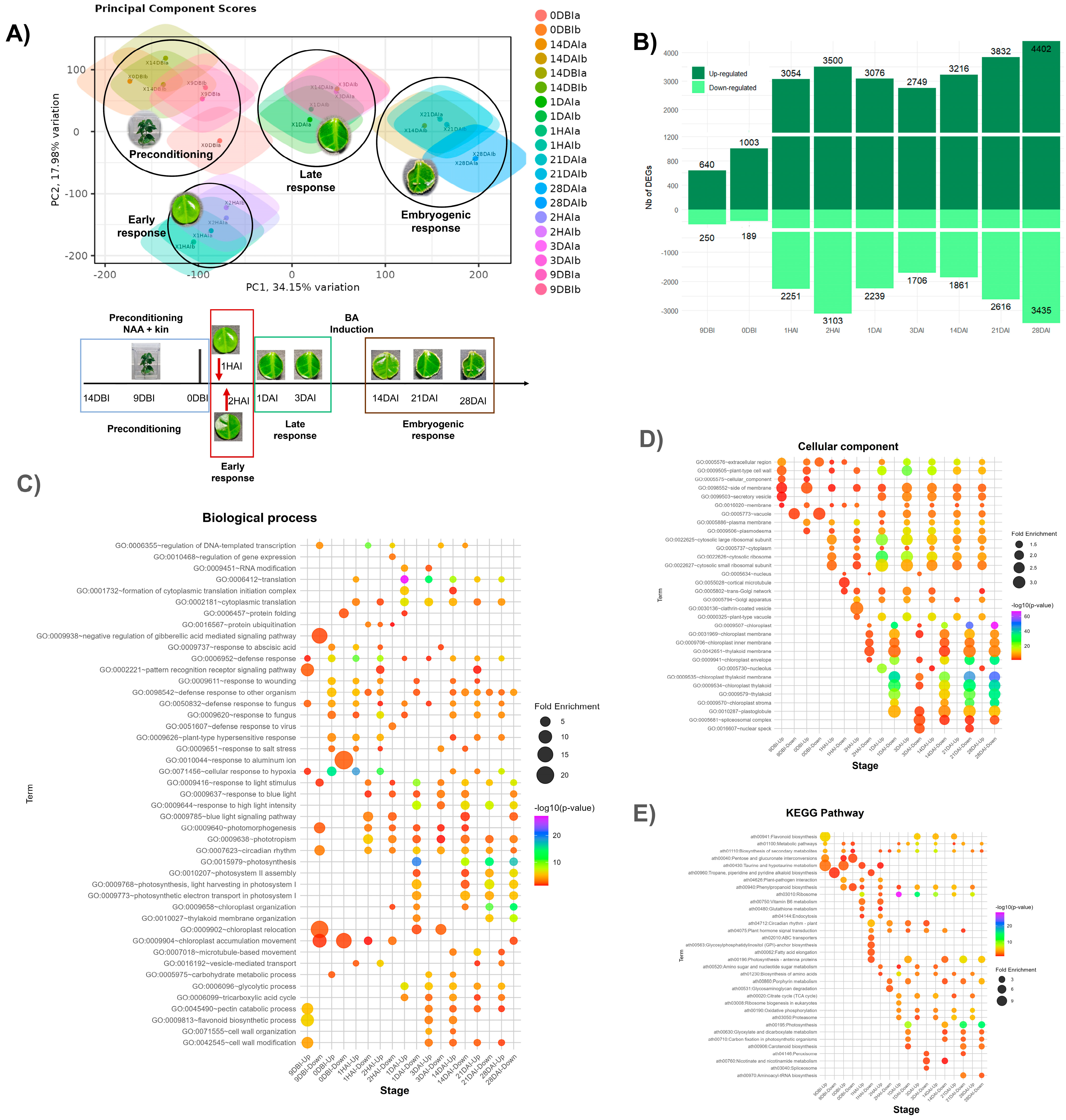
2.2. Global Transcriptomic Analysis of C. canephora SE
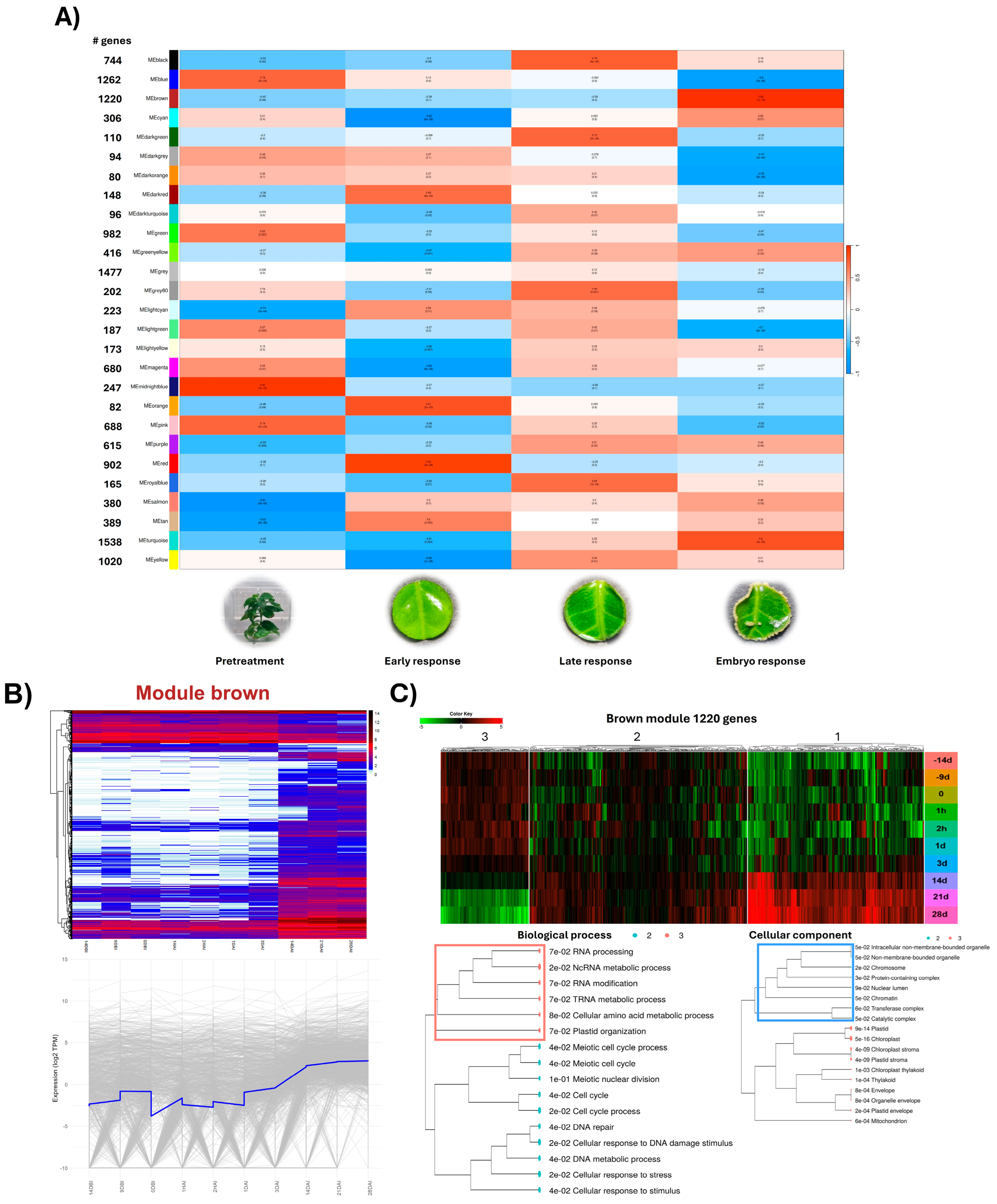
2.3. Identification of Stage-Specific Coexpression Networks Associated with SE of C. canephora
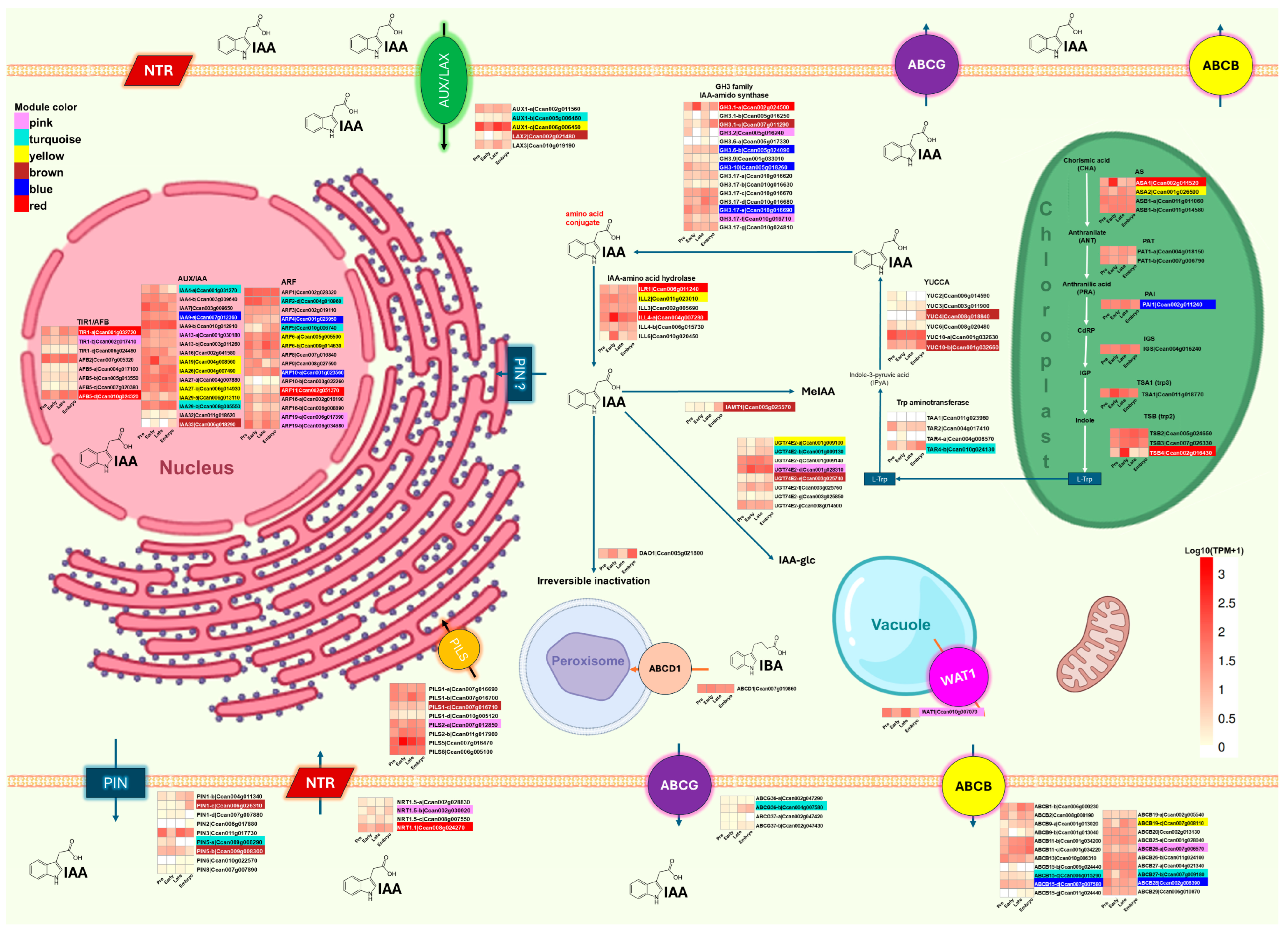
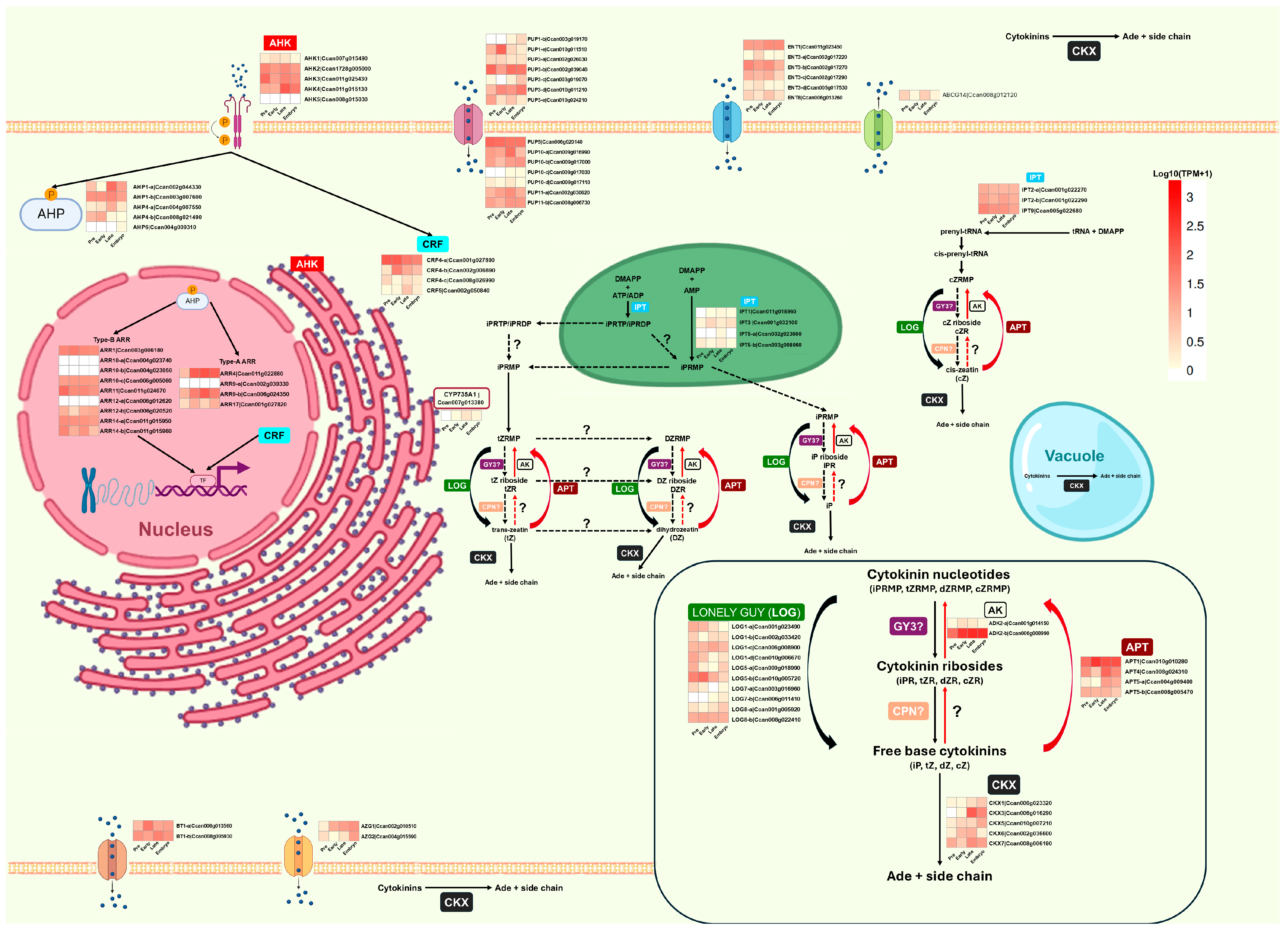
2.4. Expression of Genes Involved in Auxin and Cytokinin Metabolism During SE of C. canephora
2.4.1. Expression of Auxin Biosynthesis and Signaling Genes
2.4.2. Expression of Cytokinin Biosynthesis and Signaling Genes
3. Materials and Methods
3.1. Biological Material and Growth Conditions
3.2. RNA Extraction and Sequencing
3.3. Bioinformatic Analysis
3.4. Identification of Auxin and Cytokinin Metabolism Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quiroz-Figueroa, F.R.; Rojas-Herrera, R.; Galaz-Ávalos, R.M.; Loyola-Vargas, V.M. Embryo production through somatic embryogenesis can be used to study cell differentiation in plants. Plant Cell Tiss. Org. Cult. 2006, 86, 285–301. [Google Scholar] [CrossRef]
- Dolce, N.R.; Medina, R.D.; Terada, G.; González-Arnao, M.T.; Flachsland, E.A. In Vitro propagation and germplasm conservation of wild orchids from South America. In Orchid Biology: Recent Trends & Challenges; Khasim, S.M., Hegde, S.N., González-Arnao, M.T., Thammasiri, K., Eds.; Springer: Singapore, 2020; pp. 37–94. [Google Scholar] [CrossRef]
- Lema-Rumińska, J.; Kulus, D. Micropropagation of Cacti—A Review. Haseltonia 2014, 2014, 46–63. [Google Scholar] [CrossRef]
- Litz, R.E.; Moon, P.A.; Benson, E.M.; Stewart, J.; Chávez, V.M. A biotechnology strategy for medium-and long-term conservation of cycads. Bot. Rev. 2004, 70, 39–46. [Google Scholar] [CrossRef]
- Khan, T.; Reddy, V.; Leelavathi, S. High-frequency regeneration via somatic embryogenesis of an elite recalcitrant cotton genotype (Gossypium hirsutum L.) and efficient Agrobacterium-mediated transformation. Plant Cell Tiss. Org. Cult. 2010, 101, 323–330. [Google Scholar] [CrossRef]
- Lowe, K.; La Rota, M.; Hoerster, G.; Hastings, C.; Wang, N.; Chamberlin, M.; Wu, E.; Jones, T.; Gordon-Kamm, W. Rapid genotype “independent” Zea mays L. (maize) transformation via direct somatic embryogenesis. In Vitro Cell Dev. Biol. Plant 2018, 54, 240–252. [Google Scholar] [CrossRef]
- Pathi, K.M.; Tula, S.; Tuteja, N. High frequency regeneration via direct somatic embryogenesis and efficient Agrobacterium- mediated genetic transformation of tobacco. Plant Signal Behav. 2013, 8, e24354. [Google Scholar] [CrossRef]
- Loyola-Vargas, V.M.; Ochoa-Alejo, N. Somatic embryogenesis. An overview. In Somatic Embryogenesis. Fundamental Aspects and Applications; Loyola-Vargas, V.M., Ochoa-Alejo, N., Eds.; Springer: Cham, Switzerland, 2016; pp. 1–10. [Google Scholar] [CrossRef]
- Garcês, H.M.P.; Champagne, C.E.; Townsley, B.T.; Park, S.; Malho, R.; Pedroso, M.C.; Harada, J.J.; Sinha, N.R. Evolution of asexual reproduction in leaves of the genus Kalanchoë. Proc. Natl. Acad. Sci. USA 2007, 104, 15578–15583. [Google Scholar] [CrossRef]
- Garcês, H.M.P.; Sinha, N. The ‘mother of thousands’ (Kalanchoë daigremontiana): A plant model for asexual reproduction and CAM studies. Cold Spring Harb. Protoc. 2009, 10, pdb-emo133. [Google Scholar] [CrossRef]
- Loyola-Vargas, V.M. The history of somatic embryogenesis. In Somatic Embryogenesis. Fundamental Aspects and Applications; Loyola-Vargas, V.M., Ochoa-Alejo, N., Eds.; Springer: Cham, Switzerland, 2016; pp. 11–22. [Google Scholar] [CrossRef]
- Asghar, S.; Ghori, N.; Hyat, F.; Li, Y.; Chen, C. Use of auxin and cytokinin for somatic embryogenesis in plant: A story from competence towards completion. Plant Growth Regul. 2023, 99, 413–428. [Google Scholar] [CrossRef]
- Skoog, F.; Miller, C.O. Chemical regulation of growth and organ formation in plant tissues cultured in vitro. Symp. Soc. Exp. Biol. 1957, 11, 118–131. [Google Scholar]
- Wójcik, A.M.; Wójcikowska, B.; Gaj, M.D. Current perspectives on the auxin-mediated genetic network that controls the induction of somatic embryogenesis in plants. Int. J. Mol. Sci. 2020, 21, 1333. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowska, B.; Wójcik, A.M.; Gaj, M.D. Epigenetic regulation of auxin-induced somatic embryogenesis in plants. Int. J. Mol. Sci. 2020, 21, 2307. [Google Scholar] [CrossRef]
- Bai, B.; Su, Y.H.; Yuan, J.; Zhang, X.S. Induction of somatic embryos in Arabidopsis requires local YUCCA expression mediated by the down-regulation of ethylene biosynthesis. Mol. Plant 2013, 6, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wrobel-Marek, J.; Heidmann, I.; Horstman, A.; Chen, B.; Reis, R.; Angenent, G.C.; Boutilier, K. Auxin biosynthesis maintains embryo identity and growth during BABY BOOM-induced somatic embryogenesis. Plant Physiol. 2022, 188, 1095–1110. [Google Scholar] [CrossRef]
- Wójcikowska, B.; Jaskóla, K.; Gasiorek, P.; Meus, M.; Nowak, K.; Gaj, M.D. LEAFY COTYLEDON2 (LEC2) promotes embryogenic induction in somatic tissues of Arabidopsis, via YUCCA-mediated auxin biosynthesis. Planta 2013, 238, 425–440. [Google Scholar] [CrossRef]
- Grzyb, M.; Kalandyk, A.; Mikula, A. Effect of TIBA, fluridone and salicylic acid on somatic embryogenesis and endogenous hormone and sugar contents in the tree fern Cyathea delgadii Sternb. Acta Physiol. Plant. 2018, 40, 1. [Google Scholar] [CrossRef]
- Uc-Chuc, M.Á.; Pérez-Hernández, C.A.; Galaz-Ávalos, R.M.; Brito-Argáez, L.; Aguilar-Hernández, V.; Loyola-Vargas, V.M. YUCCA-mediated biosynthesis of the auxin IAA is required during the somatic embryogenic induction process in Coffea canephora. Int. J. Mol. Sci. 2020, 21, 4751. [Google Scholar] [CrossRef]
- Horstman, A.; Bemer, M.; Boutilier, K. A transcriptional view on somatic embryogenesis. Regeneration 2017, 4, 201–216. [Google Scholar] [CrossRef]
- Loyola-Vargas, V.M.; Avilez-Montalvo, J.R.; Avilez-Montalvo, R.N.; Márquez-López, R.E.; Galaz-Ávalos, R.M.; Mellado-Mojica, E. Somatic embryogenesis in Coffea spp. In Somatic Embryogenesis. Fundamental Aspects and Applications; Loyola-Vargas, V.M., Ochoa-Alejo, N., Eds.; Springer: Handel, Switzerland, 2016; pp. 241–266. [Google Scholar] [CrossRef]
- Méndez-Hernández, H.A.; Galaz-Ávalos, R.M.; Quintana-Escobar, A.O.; Pech-Hoil, R.; Collí-Rodríguez, A.M.; Salas-Peraza, I.Q.; Loyola-Vargas, V.M. In vitro conversion of Coffea spp. somatic embryos in SETISTM bioreactor system. Plants 2023, 12, 3055. [Google Scholar] [CrossRef]
- Etienne, H.; Breton, D.; Breitler, J.C.; Bertrand, B.; Déchamp, E.; Awada, R.; Marraccini, P.; Léran, S.; Alpizar, E.; Campa, C.; et al. Coffee somatic embryogenesis: How did research, experience gained and innovations promote the commercial propagation of elite clones from the two cultivated species. Front. Plant Sci. 2018, 9, 1630. [Google Scholar] [CrossRef]
- Staritsky, G. Embryoid formation in callus tissues of coffee. Acta Bot. Neerl. 1970, 19, 509–514. [Google Scholar] [CrossRef]
- Söndahl, M.R.; Sharp, W.R. High frequency induction of somatic embryos in cultured leaf explants of Coffea arabica L. Z. Pflanzenphysiol. 1977, 81, 395–408. [Google Scholar] [CrossRef]
- Behera, P.P.; Sivasankarreddy, K.; Prasanna, V.S.S.V. Somatic embryogenesis and plant regeneration in horticultural crops. In Commercial Scale Tissue Culture for Horticulture and Plantation Crops; Gupta, S., Chaturvedi, P., Eds.; Springer: Singapore, 2022; pp. 197–217. [Google Scholar] [CrossRef]
- Aguilar, M.E.; Wang, X.Y.; Escalona, M.; Yan, L.; Huang, L.F. Somatic embryogenesis of Arabica coffee in temporary immersion culture: Advances, limitations, and perspectives for mass propagation of selected genotypes. Front. Plant Sci. 2022, 13, 994578. [Google Scholar] [CrossRef]
- Tajedini, S.; Goessnitzer, F.; Ingelbrecht, I.L.W. Somatic embryogenesis and temporary immersion for mass propagation of chimera-free mutant Arabica coffee plantlets. In Mutation Breeding in Coffee with Special Reference to Leaf Rust: Protocols; Ingelbrecht, I.L.W., Silva, M.d.C.L.d., Jankowicz-Cieslak, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2023; pp. 51–60. [Google Scholar] [CrossRef]
- Méndez-Hernández, H.A.; Loyola-Vargas, V.M. Scale-Up of Coffea canephora somatic embryogenesis in temporary immersion system. In Plant Cell Culture Protocols; Loyola-Vargas, V.M., Ochoa-Alejo, N., Eds.; Springer: New York, NY, USA, 2024; pp. 291–301. [Google Scholar] [CrossRef]
- Astari, R.P.; Basyuni, M.; Siregar, L.A.M.; Damanik, R.I.M.; Arifiyanto, D.; Affandi, D.; Syahputra, I. Genotypic effects on accelerated propagation of oil palm breeding materials selected (Elaeis guineensis Jacq.) using somatic embryogenesis. Oil Crop Sci. 2024, 9, 111–120. [Google Scholar] [CrossRef]
- Sparjanbabu, D.S.; Prathapani, N.K.; Krishna, M.S.R.; Ramajayam, D.; Susanthi, B. Differential response of oil palm (Elaeis guineensis Jacq.) genotypes on somatic embryogenesis and plantlet regeneration from zygotic embryo. J. Appl. Biol. Biotechnol. 2023, 11, 139–143. [Google Scholar] [CrossRef]
- Hapsord, D.; Hamiranti, R.; Yusnita, Y. In vitro somatic embryogenesis of superior clones of robusta coffee from Lampung, Indonesia: Effect of genotypes and callus induction media. Biodiversitas 2020, 21, 3811–3817. [Google Scholar] [CrossRef]
- Etienne, H. Somatic embryogenesis protocol: Coffee (Coffea arabica L. and C. canephora P.). In Protocol for Somatic Embryogenesis in Woody Plants, 77th ed.; Jain, S., Gupta, P., Eds.; Springer: Dordrecht, The Netherlands, 2005; pp. 167–179. [Google Scholar] [CrossRef]
- Awada, R.; Campa, C.; Gibault, E.; Déchamp, E.; Georget, F.; Lepelley, M.; Abdallah, C.; Erban, A.; Martinez-Seidel, F.; Kopka, J.; et al. Unravelling the metabolic and hormonal machinery during key steps of somatic embryogenesis: A Case study in coffee. Int. J. Mol. Sci. 2019, 20, 4665. [Google Scholar] [CrossRef]
- Awada, R.; Lepelley, M.; Breton, D.; Charpagne, A.; Campa, C.; Berry, V.; Georget, F.; Breitler, J.C.; Léran, S.; Djerrab, D.; et al. Global transcriptome profiling reveals differential regulatory, metabolic and hormonal networks during somatic embryogenesis in Coffea arabica. BMC Genom. 2023, 24, 41. [Google Scholar] [CrossRef]
- Quiroz-Figueroa, F.R.; Monforte-González, M.; Galaz-Ávalos, R.M.; Loyola-Vargas, V.M. Direct somatic embryogenesis in Coffea canephora. In Plant Cell Culture Protocols; Loyola-Vargas, V.M., Vázquez-Flota, F.A., Eds.; Humana Press: Totowa, NJ, USA, 2006; pp. 111–117. [Google Scholar] [CrossRef]
- Quintana-Escobar, A.O.; Nic-Can, G.I.; Galaz-Ávalos, R.M.; Loyola-Vargas, V.M.; Góngora-Castillo, E. Transcriptome analysis of the induction of somatic embryogenesis in Coffea canephora and the participation of ARF and AUX/IAA genes. PeerJ 2019, 7, e7752. [Google Scholar] [CrossRef]
- Quintana-Escobar, A.O.; Bojórquez-Velázquez, E.; Ruiz-May, E.; Loyola-Vargas, V.M. Proteomic approach during the induction of somatic embryogenesis in Coffea canephora. Plants 2023, 12, 4095. [Google Scholar] [CrossRef]
- Quintana-Escobar, A.O.; Galaz-Ávalos, R.M.; Elizalde-Contreras, J.M.; Reyes-Soria, F.A.; Aguilar-Hernández, V.; Ruíz-May, E.; Loyola-Vargas, V.M. Differences in the abundance of auxin homeostasis proteins suggest their central roles for in vitro tissue differentiation in Coffea arabica. Plants 2021, 10, 2607. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Hernández, H.A.; Quintana-Escobar, A.O.; Uc-Chuc, M.Á.; Loyola-Vargas, V.M. Genome-wide analysis, modeling, and identification of amino acid binding motifs suggest the involvement of GH3 genes during somatic embryogenesis of Coffea canephora. Plants 2021, 10, 2034. [Google Scholar] [CrossRef]
- Ayil-Gutiérrez, B.A.; Galaz-Ávalos, R.M.; Peña-Cabrera, E.; Loyola-Vargas, V.M. Dynamics of the concentration of IAA and some of its conjugates during the induction of somatic embryogenesis in Coffea canephora. Plant Signal Behav. 2013, 8, e26998. [Google Scholar] [CrossRef]
- Márquez-López, R.E.; Pérez-Hernández, C.A.; Kú-González, Á.; Galaz-Ávalos, R.M.; Loyola-Vargas, V.M. Localization and transport of indole-3-acetic acid during somatic embryogenesis in Coffea canephora. Protoplasma 2018, 255, 695–708. [Google Scholar] [CrossRef]
- Avilez-Montalvo, J.; Quintana-Escobar, A.O.; Méndez-Hernández, H.A.; Uc-Chuc, M.Á.; Brito-Argáez, L.; Aguilar-Hernández, V.; Galaz-Ávalos, R.M.; Loyola-Vargas, V.M. Auxin-cytokinin cross talk in somatic embryogenesis of Coffea canephora. Plants 2022, 11, 2013. [Google Scholar] [CrossRef] [PubMed]
- Nic-Can, G.I.; López-Torres, A.; Barredo-Pool, F.A.; Wrobel, K.; Loyola-Vargas, V.M.; Rojas-Herrera, R.; De-la-Peña, C. New insights into somatic embryogenesis: LEAFY COTYLEDON1, BABY BOOM1 and WUSCHEL-RELATED HOMEOBOX4 are epigenetically regulated in Coffea canephora. PLoS ONE 2013, 8, e72160. [Google Scholar] [CrossRef]
- Carrillo-Bermejo, E.A.; Brito-Argáez, L.; Galaz-Ávalos, R.M.; Barredo-Pool, F.; Loyola-Vargas, V.M.; Aguilar-Hernández, V. Protein profile changes during priming explants to embryogenic response in Coffea canephora: Identification of the RPN12 proteasome subunit involved in the protein degradation. PeerJ 2024, 12, e18372. [Google Scholar] [CrossRef]
- Yasuda, T.; Fujii, Y.; Yamaguchi, T. Embryogenic callus induction from Coffea arabica leaf explants by benzyladenine. Plant Cell Physiol. 1985, 26, 595–597. [Google Scholar] [CrossRef]
- Goldberg, R.B.; De Paiva, G.; Yadegari, R. Plant embryogenesis: Zygote to seed. Science 1994, 266, 605–614. [Google Scholar] [CrossRef]
- Ljung, K.; Bhalerao, R.P.; Sandberg, G. Sites and homeostatic control of auxin biosynthesis in Arabidopsis during vegetative growth. Plant J. 2001, 28, 465–474. [Google Scholar] [CrossRef]
- Suzuki, M.; Yamazaki, C.; Mitsui, M.; Kakei, Y.; Mitani, Y.; Nakamura, A.; Ishii, T.; Soeno, K.; Shimada, Y. Transcriptional feedback regulation of YUCCA genes in response to auxin levels in Arabidopsis. Plant Cell Rep. 2015, 34, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Kalve, S.; Sizani, B.L.; Markakis, M.N.; Helsmoortel, C.; Vandeweyer, G.; Laukens, K.; Sommen, M.; Naulaerts, S.; Vissenberg, K.; Prinsen, E.; et al. Osmotic stress inhibits leaf growth of Arabidopsis thaliana by enhancing ARF-mediated auxin responses. New Phytol. 2020, 226, 1766–1780. [Google Scholar] [CrossRef]
- Han, J.; Li, Y.; Zhao, Y.; Sun, Y.; Li, Y.; Peng, Z. How does light regulate plant regeneration? Front. Plant Sci. 2025, 15, 1474431. [Google Scholar] [CrossRef] [PubMed]
- Khadem, A.; Moshtaghi, N.; Bagheri, A. Regulatory networks of hormone-involved transcription factors and their downstream pathways during somatic embryogenesis of Arabidopsis thaliana. 3 Biotech 2023, 13, 132. [Google Scholar] [CrossRef]
- Soy, J.; Leivar, P.; González-Schain, N.; Sentandreu, M.; Prat, S.; Quail, P.H.; Monte, E. Phytochrome-imposed oscillations in PIF3 protein abundance regulate hypocotyl growth under diurnal light/dark conditions in Arabidopsis. Plant J. 2012, 71, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Soy, J.; Leivar, P.; Monte, E. PIF1 promotes phytochrome-regulated growth under photoperiodic conditions in Arabidopsis together with PIF3, PIF4, and PIF5. J. Exp. Bot. 2014, 65, 2925–2936. [Google Scholar] [CrossRef]
- Zhang, Y.; Mayba, O.; Pfeiffer, A.; Shi, H.; Tepperman, J.M.; Speed, T.P.; Quail, P.H. A quartet of PIF bHLH factors provides a transcriptionally centered signaling hub that regulates seedling morphogenesis through differential expression-patterning of shared target genes in Arabidopsis. PLoS Genet. 2013, 9, e1003244. [Google Scholar] [CrossRef]
- Mira, M.M.; Day, S.; Ibrahim, S.; Hill, R.D.; Stasolla, C. The Arabidopsis Phytoglobin 2 mediates phytochrome B (phyB) light signaling responses during somatic embryogenesis. Planta 2023, 257, 88. [Google Scholar] [CrossRef]
- Wang, X.-D.; Nolan, K.E.; Irwanto, R.R.; Sheahan, M.B.; Rose, R.J. Ontogeny of embryogenic callus in Medicago truncatula: The fate of the pluripotent and totipotent stem cells. Ann. Bot. 2011, 107, 599–609. [Google Scholar] [CrossRef]
- Rose, R.J. Somatic embryogenesis in the Medicago truncatula model: Cellular and molecular mechanisms. Front. Plant Sci. 2019, 10, 267. [Google Scholar] [CrossRef]
- Soares, N.C.; Francisco, R.; Vielba, J.M.; Ricardo, C.P.; Jackson, P.A. Associating wound-related changes in the apoplast proteome of Medicago with early steps in the ROS signal-transduction pathway. J. Proteome Res. 2009, 8, 2298–2309. [Google Scholar] [CrossRef] [PubMed]
- Tiew, T.W.-Y.; Sheahan, M.B.; Rose, R.J. Peroxisomes contribute to reactive oxygen species homeostasis and cell division induction in Arabidopsis protoplasts. Front. Plant Sci. 2015, 6, 658. [Google Scholar] [CrossRef]
- Imin, N.; Nizamidin, M.; Daniher, D.; Nolan, K.E.; Rose, R.J.; Rolfe, B.G. Proteomic analysis of somatic embryogenesis in Medicago truncatula. Explant cultures grown under 6-benzylaminopurine and 1-naphthaleneacetic acid treatments. Plant Physiol. 2005, 137, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Yang, X.; Guo, K.; Deng, J.; Xu, J.; Gao, W.; Lindsey, K.; Zhang, X. ROS homeostasis regulates somatic embryogenesis via the regulation of auxin signaling in cotton. Mol. Cell. Proteom. 2016, 15, 2108–2124. [Google Scholar]
- Ding, M.; Dong, H.; Xue, Y.; Su, S.; Wu, Y.; Li, S.; Liu, H.; Li, H.; Han, J.; Shan, X.; et al. Transcriptomic analysis reveals somatic embryogenesis-associated signaling pathways and gene expression regulation in maize (Zea mays L.). Plant Mol. Biol. 2020, 104, 647–663. [Google Scholar] [CrossRef]
- Wickramasuriya, A.M.; Dunwell, J.M. Global scale transcriptome analysis of Arabidopsis embryogenesis in vitro. BMC Genom. 2015, 16, 301. [Google Scholar] [CrossRef]
- Elhiti, M.; Stasolla, C. Transduction of signals during somatic embryogenesis. Plants 2022, 11, 178. [Google Scholar] [CrossRef]
- Chen, R.; Chen, X.; Huo, W.; Zheng, S.; Lin, Y.; Lai, Z. Transcriptome analysis of azacitidine (5-AzaC)-treatment affecting the development of early somatic embryogenesis in longan. J. Hortic. Sci. Biotechnol. 2020, 96, 311–323. [Google Scholar] [CrossRef]
- Awon, V.K.; Dutta, D.; Banerjee, S.; Pal, S.; Gangopadhyay, G. Integrated metabolomics and transcriptomics analysis highlight key pathways involved in the somatic embryogenesis of Darjeeling tea. BMC Genom. 2024, 25, 207. [Google Scholar] [CrossRef]
- Suo, J.; Zhou, C.; Zeng, Z.; Li, X.; Bian, H.; Wang, J.; Zhu, M.; Han, N. Identification of regulatory factors promoting embryogenic callus formation in barley through transcriptome analysis. BMC Plant Biol. 2021, 21, 145. [Google Scholar] [CrossRef]
- Ikeuchi, M.; Iwase, A.; Rymen, B.; Lambolez, A.; Kojima, M.; Takebayashi, Y.; Heyman, J.; Watanabe, S.; Seo, M.; De Veylder, L. Wounding triggers callus formation via dynamic hormonal and transcriptional changes. Plant Physiol. 2017, 175, 1158–1174. [Google Scholar] [PubMed]
- Liu, X.; Bie, X.M.; Lin, X.; Li, M.; Wang, H.; Zhang, X.; Yang, Y.; Zhang, C.; Zhang, X.S.; Xiao, J. Uncovering the transcriptional regulatory network involved in boosting wheat regeneration and transformation. Nat. Plants 2023, 9, 908–925. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhuang, S.; Zhang, W. Advances in plant auxin biology: Synthesis, metabolism, signaling, interaction with other hormones, and roles under abiotic stress. Plants 2024, 13, 2523. [Google Scholar] [CrossRef]
- Wang, B.; Chu, J.; Yu, T.; Xu, Q.; Sun, X.; Yuan, J.; Xiong, G.; Wang, G.; Wang, Y.; Li, J. Tryptophan-independent auxin biosynthesis contributes to early embryogenesis in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 4821–4826. [Google Scholar] [CrossRef]
- Casanova-Sáez, R.; Mateo-Bonmatí, E.; Ljung, K. Auxin metabolism in plants. Cold Spring Harbor Perpect. Biol. 2021, 13, a039867. [Google Scholar] [CrossRef]
- LeClere, S.; Tellez, R.; Rampey, R.A.; Matsuda, S.P.T.; Bartel, B. Characterization of a family of IAA-amino acid conjugate hydrolases from Arabidopsis. J. Biol. Chem. 2002, 277, 20446–20452. [Google Scholar]
- Hammes, U.Z.; Pedersen, B.P. Structure and function of auxin transporters. Annu. Rev. Plant Biol. 2024, 75, 185–209. [Google Scholar] [CrossRef]
- Weijers, D.; Wagner, D. Transcriptional responses to the auxin hormone. Annu. Rev. Plant Biol. 2016, 67, 539–574. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, H. Five unaddressed questions about cytokinin biosynthesis. J. Exp. Bot. 2024, erae348. [Google Scholar] [CrossRef]
- Tokunaga, H.; Kojima, M.; Kuroha, T.; Ishida, T.; Sugimoto, K.; Kiba, T.; Sakakibara, H. Arabidopsis lonely guy (LOG) multiple mutants reveal a central role of the LOG-dependent pathway in cytokinin activation. Plant J. 2012, 69, 355–365. [Google Scholar] [CrossRef]
- Schmülling, T.; Werner, T.; Riefler, M.; Krupková, E.; Manns, I.B. Structure and function of cytokinin oxidase/dehydrogenase genes of maize, rice, Arabidopsis and other species. J. Plant Res. 2003, 116, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Zhao, Y.; Zhang, K. Cytokinin transporters: Multisite players in cytokinin homeostasis and signal distribution. Front. Plant Sci. 2019, 10, 693. [Google Scholar] [CrossRef]
- Kieber, J.J.; Schaller, G.E. Cytokinin signaling in plant development. Development 2018, 145, dev149344. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Kurdyukov, S.; Kereszt, A.; Wang, X.; Gresshoff, P.; Rose, R. The association of hoemeobox gene expression with stem cell formation and morphogenesis in cultured Medicago truncatula. Planta 2009, 230, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.H.; Zhao, X.Y.; Liu, Y.B.; Zhang, C.L.; O’Neill, S.D.; Zhang, X.S. Auxin-induced WUS expression is essential for embryonic stem cell renewal during somatic embryogenesis in Arabidopsis. Plant J. 2009, 59, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.Y.; Smet, W.; Brunoud, G.; Yoshida, S.; Vernoux, T.; Weijers, D. Reporters for sensitive and quantitative measurement of auxin response. Nat. Methods 2015, 12, 207–210. [Google Scholar] [CrossRef]
- Zhao, Z.; Andersen, S.U.; Ljung, K.; Dolezal, K.; Miotk, A.; Schultheiss, S.J.; Lohmann, J.U. Hormonal control of the shoot stem-cell niche. Nature 2010, 465, 1089–1092. [Google Scholar] [CrossRef]
- Pierdonati, E.; Unterholzner, S.J.; Salvi, E.; Svolacchia, N.; Bertolotti, G.; Dello Ioio, R.; Sabatini, S.; Di Mambro, R. Cytokinin-dependent control of GH3 group II family genes in the Arabidopsis root. Plants 2019, 8, 94. [Google Scholar] [CrossRef]
- To, J.P.; Haberer, G.; Ferreira, F.J.; Deruére, J.; Mason, M.G.; Schaller, G.E.; Alonso, J.M.; Ecker, J.R.; Kieber, J.J. Type-A Arabidopsis response regulators are partially redundant negative regulators of cytokinin signaling. Plant Cell 2004, 16, 658–671. [Google Scholar] [CrossRef]
- Rashotte, A.M.; Carson, S.D.B.; To, J.P.C.; Kieber, J.J. Expression profiling of cytokinin action in Arabidopsis. Plant Physiol. 2003, 132, 1998–2011. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. In Babraham Bioinformatics; Institute Babraham: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Denoeud, F.; Carretero-Paulet, L.; Dereeper, A.; Droc, G.; Guyot, R.; Pietrella, M.; Zheng, C.; Alberti, A.; Anthony, F.; Aprea, G.; et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 2014, 345, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 51, D638–D646. [Google Scholar] [CrossRef]
- Salojärvi, J.; Rambani, A.; Yu, Z.; Guyot, R.; Strickler, S.; Lepelley, M.; Wang, C.; Rajaraman, S.; Rastas, P.; Zheng, C.; et al. The genome and population genomics of allopolyploid Coffea arabica reveal the diversification history of modern coffee cultivars. Nat. Genet. 2024, 56, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Couoh-Cauich, M.-D.; Méndez-Hernández, H.A.; Galaz-Ávalos, R.M.; Quintana-Escobar, A.O.; Ibarra-Laclette, E.; Loyola-Vargas, V.M. Transcriptional Dynamics Underlying Somatic Embryogenesis in Coffea canephora. Plants 2025, 14, 1108. https://doi.org/10.3390/plants14071108
Couoh-Cauich M-D, Méndez-Hernández HA, Galaz-Ávalos RM, Quintana-Escobar AO, Ibarra-Laclette E, Loyola-Vargas VM. Transcriptional Dynamics Underlying Somatic Embryogenesis in Coffea canephora. Plants. 2025; 14(7):1108. https://doi.org/10.3390/plants14071108
Chicago/Turabian StyleCouoh-Cauich, Marcos-David, Hugo A. Méndez-Hernández, Rosa M. Galaz-Ávalos, Ana Odetth Quintana-Escobar, Enrique Ibarra-Laclette, and Víctor M. Loyola-Vargas. 2025. "Transcriptional Dynamics Underlying Somatic Embryogenesis in Coffea canephora" Plants 14, no. 7: 1108. https://doi.org/10.3390/plants14071108
APA StyleCouoh-Cauich, M.-D., Méndez-Hernández, H. A., Galaz-Ávalos, R. M., Quintana-Escobar, A. O., Ibarra-Laclette, E., & Loyola-Vargas, V. M. (2025). Transcriptional Dynamics Underlying Somatic Embryogenesis in Coffea canephora. Plants, 14(7), 1108. https://doi.org/10.3390/plants14071108