
Gene Expression Dynamics of Sugar Metabolism and Accumulation During Fruit Ripening in Camellia drupifera




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
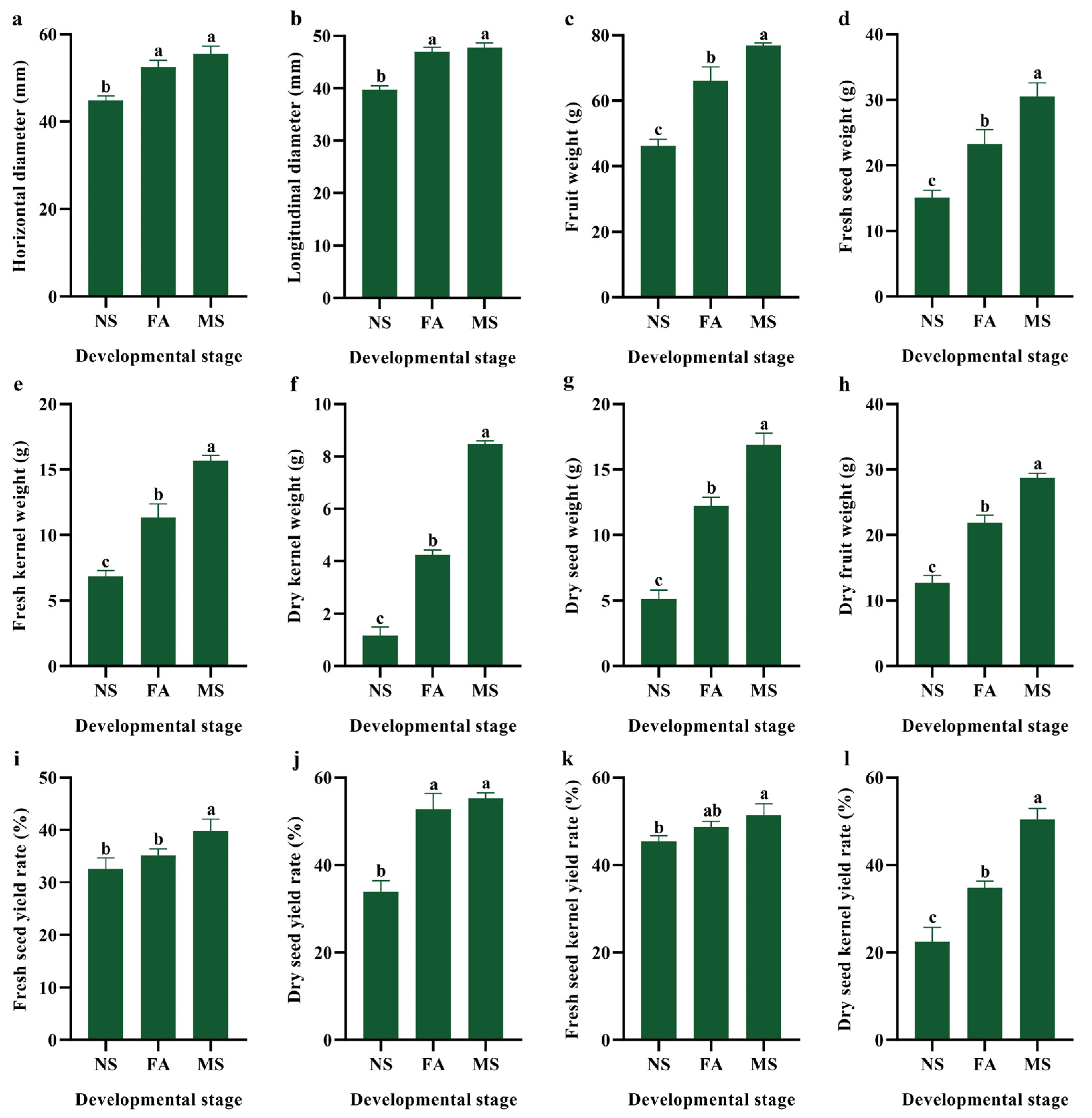
2.1. Developmental Changes in Phenotypic and Economic Traits
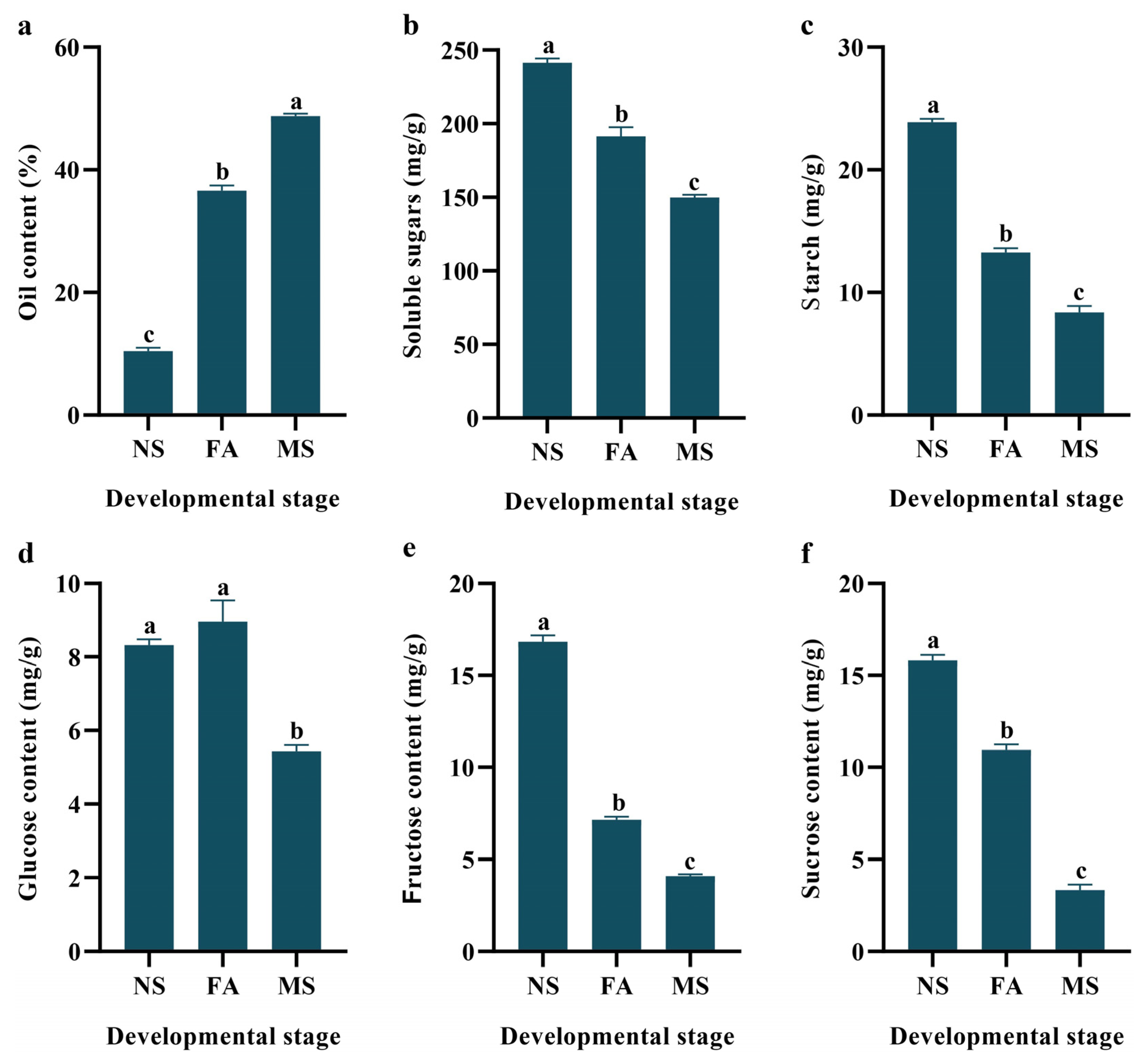
2.2. Physicochemical Analysis Changes
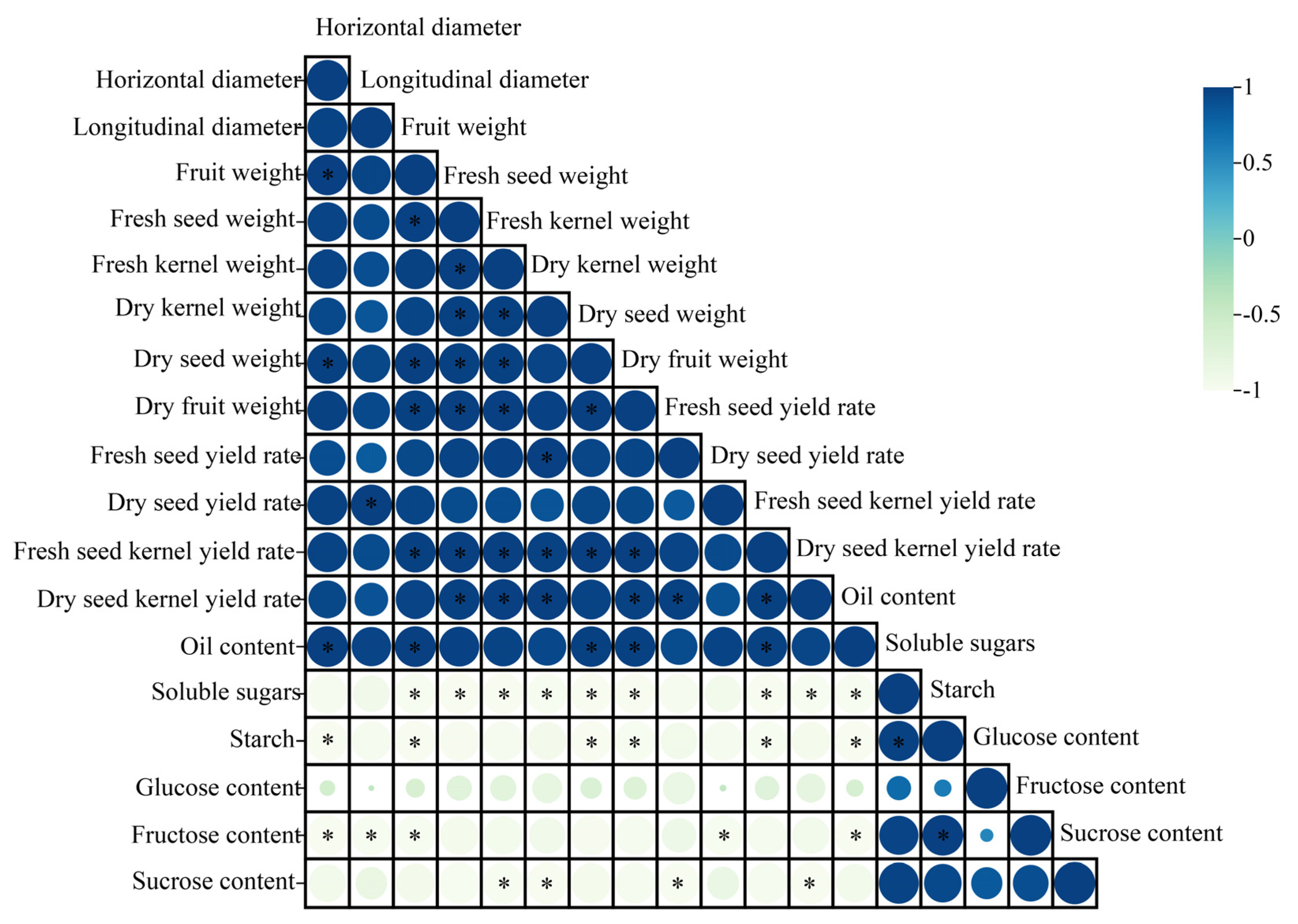
2.3. Correlation Analysis
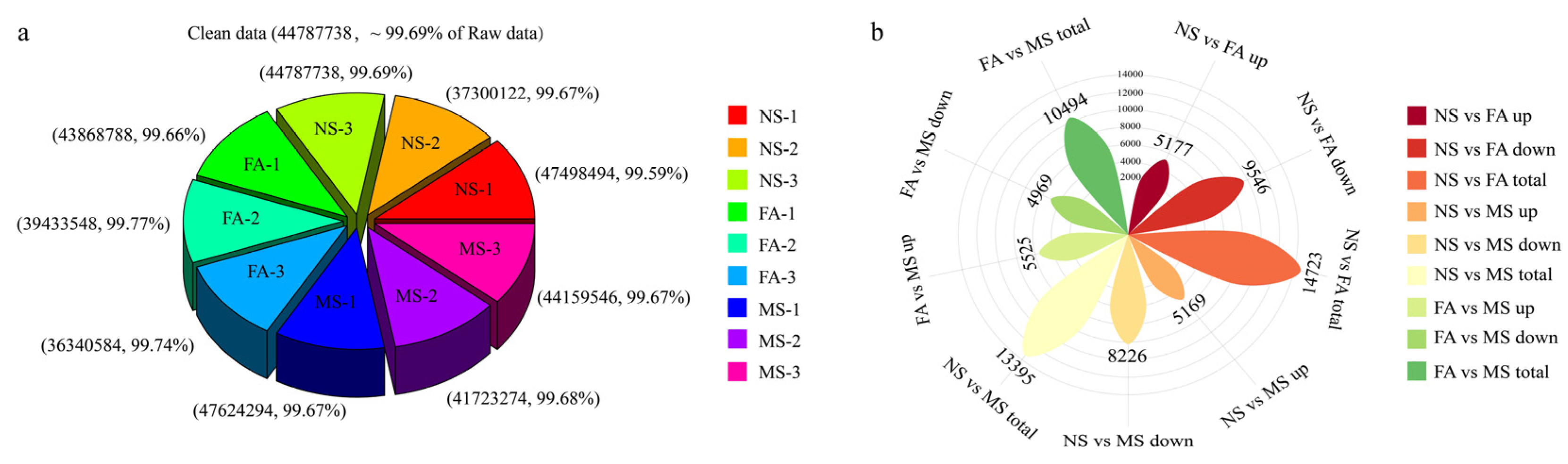
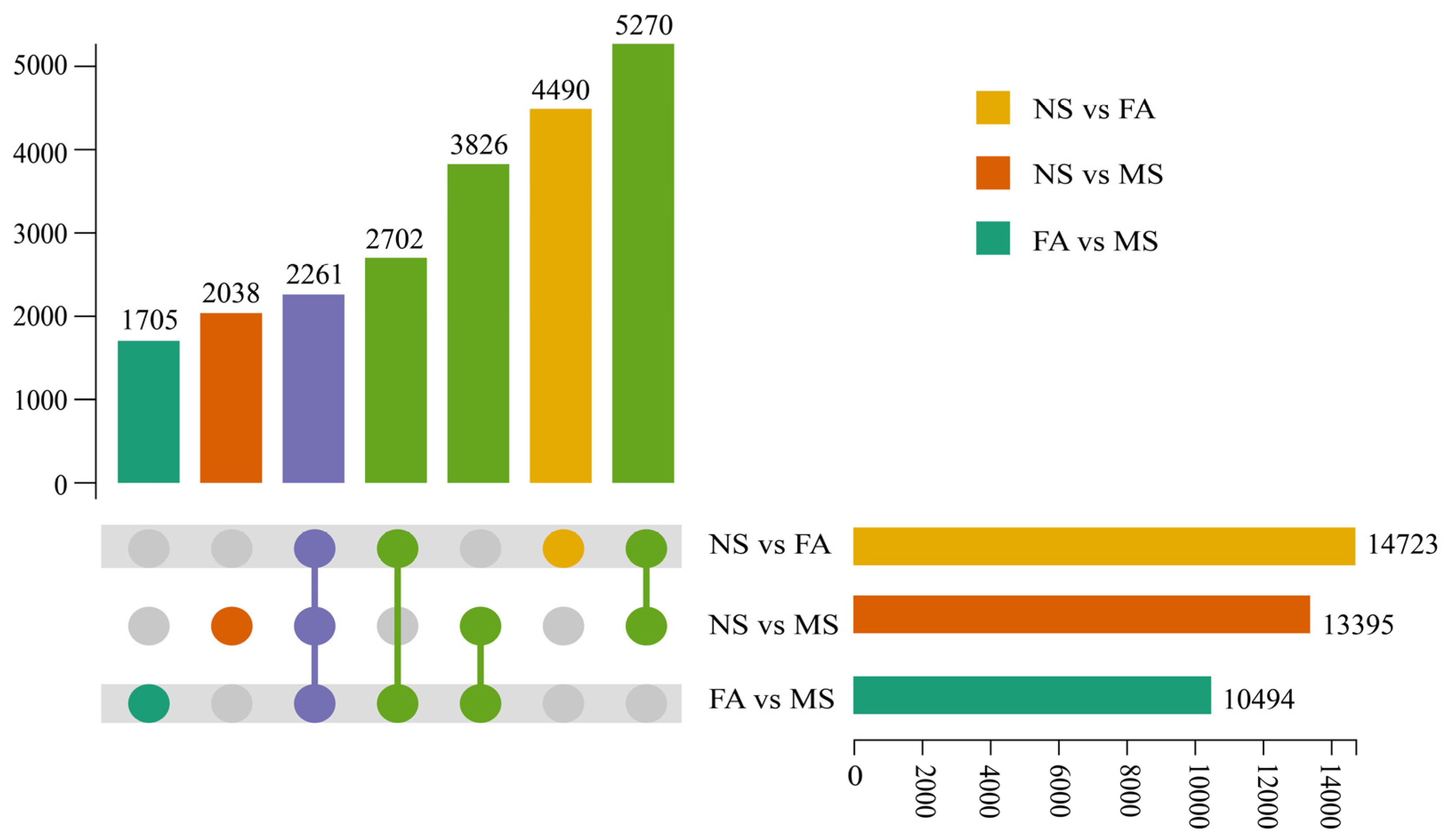
2.4. Transcriptome and Differential Unigene Analysis
2.5. Functional Annotation of DEGs
2.6. Co-Expression Network Analysis with WGCNA
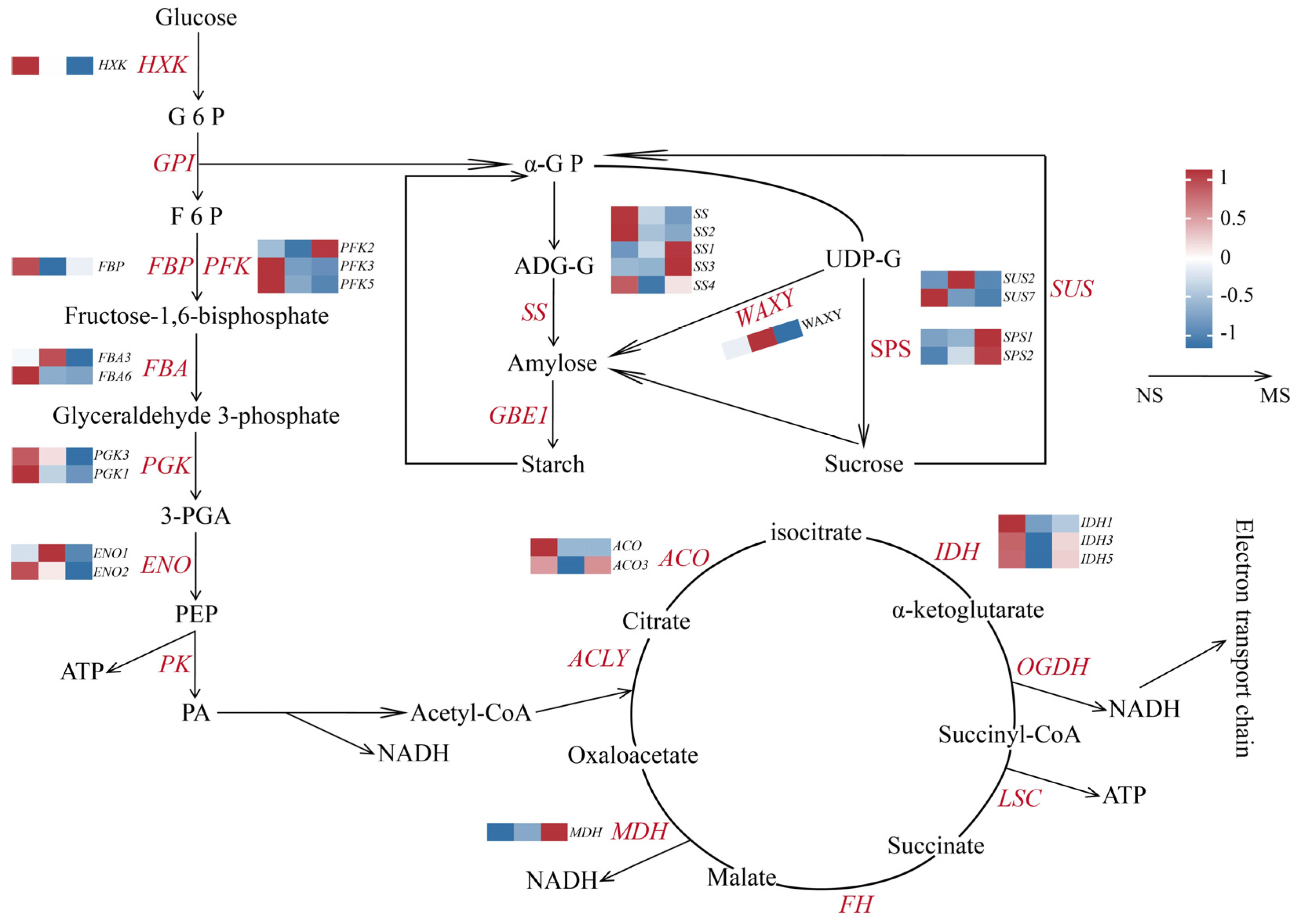
2.7. Analysis of Expression Patterns of DEGs
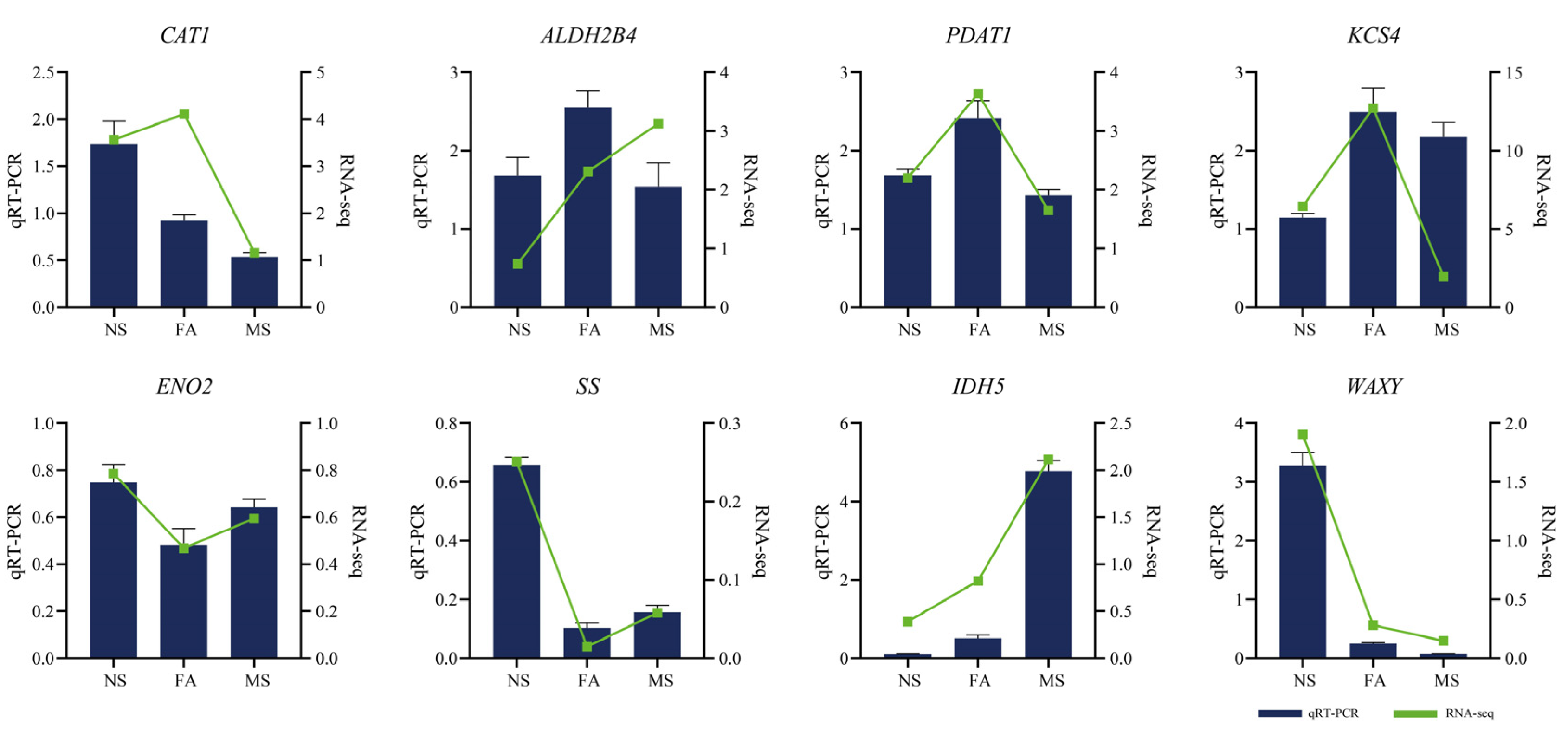
2.8. Quantitative Real-Time PCR Analysis of Selected Genes
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Fruit Economic Characteristics Determination
- Fresh seed yield (%) = (fresh seed weight/single fruit weight) × 100;
- Dry seed yield (%) = (dry seed weight/fresh seed weight) × 100;
- Fresh seed kernel yield (%) = (fresh kernel weight/fresh seed weight) × 100;
- Dry seed kernel yield (%) = (dry kernel weight/dry seed weight) × 100.
4.3. Physicochemical Parameters Measurement
4.4. RNA Extraction and Transcriptome Library Construction
4.5. Bioinformatics Analysis
4.6. WGCNA Used to Identify Relevant Gene Networks
4.7. Validation by qRT-PCR
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, F.; Feng, L.-Y.; Lin, P.-F.; Jia, J.-J.; Gao, L.-Z. Chromosome-scale genome assembly of oil-tea tree Camellia crapnelliana. Sci. Data 2024, 11, 599. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Shen, J.; Wei, J.; Chen, Y. A critical review of the bioactive ingredients and biological functions of camellia oleifera oil. Curr. Res. Food Sci. 2024, 8, 100753. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-X.; Luo, X.-F.; Luo, K.-X.; Liu, Y.-L.; Pan, T.; Li, Z.-Z.; Duns, G.J.; He, F.-L.; Qin, Z.-D. Small RNA sequencing reveals dynamic microRNA expression of important nutrient metabolism during development of Camellia oleifera fruit. Int. J. Biol. Sci. 2019, 15, 416. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, F.; Xu, Z.; Wang, G.; Hu, L.; Cheng, J.; Ge, X.; Liu, J.; Chen, W.; Li, Q. The complex hexaploid oil-Camellia genome traces back its phylogenomic history and multi-omics analysis of Camellia oil biosynthesis. Plant Biotechnol. J. 2024, 22, 2890–2906. [Google Scholar] [CrossRef]
- Yang, D.; Wang, R.; Lai, H.; He, Y.; Chen, Y.; Xun, C.; Zhang, Y.; He, Z. Comparative Transcriptomic and Lipidomic Analysis of Fatty Acid Accumulation in Three Camellia oleifera Varieties During Seed Maturing. J. Agric. Food Chem. 2024, 72, 18257–18270. [Google Scholar] [CrossRef]
- Zhao, S.; Rong, J. Single-cell RNA-seq reveals a link of ovule abortion and sugar transport in Camellia oleifera. Front. Plant Sci. 2024, 15, 1274013. [Google Scholar] [CrossRef]
- Ma, X.; Chen, S.; Li, J.; Ouyang, X. Transcriptome analysis unravels key pathways and hub genes related to immature fruit abscission in Camellia oleifera. Front. Plant Sci. 2024, 15, 1418358. [Google Scholar] [CrossRef]
- He, Y.; Chen, R.; Yang, Y.; Liang, G.; Zhang, H.; Deng, X.; Xi, R. Sugar metabolism and transcriptome analysis reveal key sugar transporters during Camellia oleifera fruit development. Int. J. Mol. Sci. 2022, 23, 822. [Google Scholar] [CrossRef]
- Li, Y.; Yan, H.; Zeeshan Ul Haq, M.; Liu, Y.; Wu, Y.; Yu, J.; Xia, P. Physiological and Transcriptional Analysis Provides Insights into Tea Saponin Biosynthesis and Regulation in Response to SA in Camellia vietnamensis Huang. Horticulturae 2023, 10, 8. [Google Scholar] [CrossRef]
- Lijun, C.; Wu, L.; Qi, S.; Yuqi, C.; CongYa, W.; Sheng, L. Long-term Camellia oleifera cultivation influences the assembly process of soil bacteria in different soil aggregate particles. Land Degrad. Dev. 2023, 34, 441–452. [Google Scholar] [CrossRef]
- Tan, Z.; Liu, T.; Ning, C.; Lin, X.; Liu, X.; Jiang, M.; Liu, S.; Yan, W. Effects of Transformation of Inefficient Camellia oleifera Plantation on Soil Quality and Fungal Communities. Forests 2024, 15, 603. [Google Scholar] [CrossRef]
- Dong, H.; Li, F.; Xuan, X.; Ahiakpa, J.K.; Tao, J.; Zhang, X.; Ge, P.; Wang, Y.; Gai, W.; Zhang, Y. The genetic basis and improvement of photosynthesis in tomato. Hortic. Plant J. 2024, 11, 69–84. [Google Scholar] [CrossRef]
- Acharya, T.P.; Malladi, A.; Nambeesan, S.U. Sustained carbon import supports sugar accumulation and anthocyanin biosynthesis during fruit development and ripening in blueberry (Vaccinium ashei). Sci. Rep. 2024, 14, 24964. [Google Scholar] [CrossRef]
- Farcuh, M.; Li, B.; Rivero, R.M.; Shlizerman, L.; Sadka, A.; Blumwald, E. Sugar metabolism reprogramming in a non-climacteric bud mutant of a climacteric plum fruit during development on the tree. J. Exp. Bot. 2017, 68, 5813–5828. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.P.; Dean, L.L. Peanut composition, flavor and nutrition. In Peanuts; Elsevier: Amsterdam, The Netherlands, 2016; pp. 289–345. [Google Scholar]
- Hu, L.; Wu, G.; Hao, C.; Yu, H.; Tan, L. Transcriptome and selected metabolite analyses reveal points of sugar metabolism in jackfruit (Artocarpus heterophyllus Lam.). Plant Sci. 2016, 248, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Wingler, A.; Henriques, R. Sugars and the speed of life—Metabolic signals that determine plant growth, development and death. Physiol. Plant. 2022, 174, e13656. [Google Scholar] [CrossRef]
- Hartmann, H.; Trumbore, S. Understanding the roles of nonstructural carbohydrates in forest trees–from what we can measure to what we want to know. New Phytol. 2016, 211, 386–403. [Google Scholar] [CrossRef]
- Lu, W.; Hao, W.; Liu, K.; Liu, J.; Yin, C.; Su, Y.; Hang, Z.; Peng, B.; Liu, H.; Xiong, B. Analysis of sugar components and identification of SPS genes in citrus fruit development. Front. Plant Sci. 2024, 15, 1372809. [Google Scholar] [CrossRef]
- Dai, N.; Cohen, S.; Portnoy, V.; Tzuri, G.; Harel-Beja, R.; Pompan-Lotan, M.; Carmi, N.; Zhang, G.; Diber, A.; Pollock, S. Metabolism of soluble sugars in developing melon fruit: A global transcriptional view of the metabolic transition to sucrose accumulation. Plant Mol. Biol. 2011, 76, 1–18. [Google Scholar] [CrossRef]
- Saladié, M.; Cañizares, J.; Phillips, M.A.; Rodriguez-Concepcion, M.; Larrigaudière, C.; Gibon, Y.; Stitt, M.; Lunn, J.E.; Garcia-Mas, J. Comparative transcriptional profiling analysis of developing melon (Cucumis melo L.) fruit from climacteric and non-climacteric varieties. BMC Genom. 2015, 16, 440. [Google Scholar] [CrossRef]
- Schemberger, M.O.; Stroka, M.A.; Reis, L.; de Souza Los, K.K.; de Araujo, G.A.T.; Sfeir, M.Z.T.; Galvão, C.W.; Etto, R.M.; Baptistão, A.R.G.; Ayub, R.A. Transcriptome profiling of non-climacteric ‘yellow’melon during ripening: Insights on sugar metabolism. BMC Genom. 2020, 21, 262. [Google Scholar] [CrossRef] [PubMed]
- Stroka, M.A.; Reis, L.; de Souza Los, K.K.; Pinto, C.A.; Gustani, F.M.; Forney, C.F.; Etto, R.M.; Galvão, C.W.; Ayub, R.A. The maturation profile triggers differential expression of sugar metabolism genes in melon fruits. Plant Physiol. Biochem. 2024, 207, 108418. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.M.; Deng, L.; Wang, X.; Wang, Y.; Pan, L.; Liu, H.; Niu, L.; Lu, Z.; Cui, G.; Zeng, W. Expression patterns of genes involved in sugar metabolism and accumulation during peach fruit development and ripening. Sci. Hortic. 2019, 257, 108633. [Google Scholar] [CrossRef]
- Yu, X.; Ali, M.M.; Li, B.; Fang, T.; Chen, F. Transcriptome data-based identification of candidate genes involved in metabolism and accumulation of soluble sugars during fruit development in ‘Huangguan’plum. J. Food Biochem. 2021, 45, e13878. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Y.; Wang, M.; Han, H.; Luo, Y.; Ding, W.; Xu, W.; Zhong, Y.; Huang, H.; Qu, S. Soluble sugars accumulation and related gene expression during fruit development in Cucurbita maxima Duchesne. Sci. Hortic. 2020, 272, 109520. [Google Scholar] [CrossRef]
- Wyatt, L.E.; Strickler, S.R.; Mueller, L.A.; Mazourek, M. Comparative analysis of Cucurbita pepo metabolism throughout fruit development in acorn squash and oilseed pumpkin. Hortic. Res. 2016, 3, 16045. [Google Scholar] [CrossRef]
- Apriyanto, A.; Compart, J.; Zimmermann, V.; Alseekh, S.; Fernie, A.R.; Fettke, J. Indication that starch and sucrose are biomarkers for oil yield in oil palm (Elaeis guineensis Jacq.). Food Chem. 2022, 393, 133361. [Google Scholar] [CrossRef]
- Nazari, M.; Kordrostami, M.; Ghasemi-Soloklui, A.A.; Eaton-Rye, J.J.; Pashkovskiy, P.; Kuznetsov, V.; Allakhverdiev, S.I. Enhancing Photosynthesis and Plant Productivity through Genetic Modification. Cells 2024, 13, 1319. [Google Scholar] [CrossRef]
- Yang, L.; Gao, C.; Xie, J.; Qiu, J.; Deng, Q.; Zhou, Y.; Liao, D.; Deng, C. Fruit economic characteristics and yields of 40 superior Camellia oleifera Abel plants in the low-hot valley area of Guizhou Province, China. Sci. Rep. 2022, 12, 7068. [Google Scholar] [CrossRef]
- Hu, Z.-Y.; Hua, W.; Zhang, L.; Deng, L.-B.; Wang, X.-F.; Liu, G.-H.; Hao, W.-J.; Wang, H.-Z. Seed structure characteristics to form ultrahigh oil content in rapeseed. PLoS ONE 2013, 8, e62099. [Google Scholar] [CrossRef]
- Yuan, X.; Jiang, X.; Zhang, M.; Wang, L.; Jiao, W.; Chen, H.; Mao, J.; Ye, W.; Song, Q. Integrative omics analysis elucidates the genetic basis underlying seed weight and oil content in soybean. Plant Cell 2024, 36, 2160–2175. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, H.; Baaghideh, M.; Dadashi-Roudbari, A. Climate change impacts on pistachio cultivation areas in Iran: A simulation analysis based on CORDEX-MENA multi-model ensembles. Theor. Appl. Climatol. 2021, 145, 109–120. [Google Scholar] [CrossRef]
- Adaskaveg, J.; Lee, C.; Wei, Y.; Wang, F.; Grilo, F.S.; Mesquida-Pesci, S.D.; Davis, M.; Wang, S.C.; Marino, G.; Ferguson, L. In a nutshell: Pistachio genome and kernel development. bioRxiv 2024. [Google Scholar] [CrossRef]
- Ye, C.; He, Z.; Peng, J.; Wang, R.; Wang, X.; Fu, M.; Zhang, Y.; Wang, A.; Liu, Z.; Jia, G. Genomic and genetic advances of oiltea-camellia (Camellia oleifera). Front. Plant Sci. 2023, 14, 1101766. [Google Scholar] [CrossRef]
- Zhang, F.; Li, Z.; Zhou, J.; Gu, Y.; Tan, X. Comparative study on fruit development and oil synthesis in two cultivars of Camellia oleifera. BMC Plant Biol. 2021, 21, 348. [Google Scholar] [CrossRef]
- Du, B.; Cao, Y.; Zhou, J.; Chen, Y.; Ye, Z.; Huang, Y.; Zhao, X.; Zou, X.; Zhang, L. Sugar import mediated by sugar transporters and cell wall invertases for seed development in Camellia oleifera. Hortic. Res. 2024, 11, uhae133. [Google Scholar] [CrossRef]
- Akagić, A.; Oras, A.V.; Oručević Žuljević, S.; Spaho, N.; Drkenda, P.; Bijedić, A.; Memić, S.; Hudina, M. Geographic variability of sugars and organic acids in selected wild fruit species. Foods 2020, 9, 462. [Google Scholar] [CrossRef]
- Zhi, Y.; Chuanjiang, Z.; Xinfang, Y.; Mengyi, D.; Zhenlei, W.; Fenfen, Y.; Cuiyun, W.; Jiurui, W.; Mengjun, L.; Minjuan, L. Genetic analysis of mixed models of fruit sugar–acid fractions in a cross between jujube (Ziziphus jujuba Mill.) and wild jujube (Z. acido jujuba). Front. Plant Sci. 2023, 14, 1181903. [Google Scholar] [CrossRef]
- Ren, Y.; Liao, S.; Xu, Y. An update on sugar allocation and accumulation in fruits. Plant Physiol. 2023, 193, 888–899. [Google Scholar] [CrossRef]
- Zhai, Z.; Keereetaweep, J.; Liu, H.; Xu, C.; Shanklin, J. The role of sugar signaling in regulating plant fatty acid synthesis. Front. Plant Sci. 2021, 12, 643843. [Google Scholar] [CrossRef]
- Ahmed, M.; Iqbal, A.; Latif, A.; Din, S.U.; Sarwar, M.B.; Wang, X.; Rao, A.Q.; Husnain, T.; Ali Shahid, A. Overexpression of a sucrose synthase gene indirectly improves cotton fiber quality through sucrose cleavage. Front. Plant Sci. 2020, 11, 476251. [Google Scholar] [CrossRef] [PubMed]
- Durán-Soria, S.; Pott, D.M.; Osorio, S.; Vallarino, J.G. Sugar signaling during fruit ripening. Front. Plant Sci. 2020, 11, 564917. [Google Scholar] [CrossRef] [PubMed]
- Koch, K. Sucrose metabolism: Regulatory mechanisms and pivotal roles in sugar sensing and plant development. Curr. Opin. Plant Biol. 2004, 7, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.Z.; Hu, X.-M.; Jin, L.-F.; Liu, Y.-Z.; Peng, S.-A. Genome-wide identification and expression profile analysis of citrus sucrose synthase genes: Investigation of possible roles in the regulation of sugar accumulation. PLoS ONE 2014, 9, e113623. [Google Scholar] [CrossRef]
- Yu, J.; Liu, X.; Wang, W.; Zhang, L.; Wang, C.; Zhang, Q.; Wang, J.; Du, M.; Sheng, L.; Hu, D. MdCIbHLH1 modulates sugar metabolism and accumulation in apple fruits by coordinating carbohydrate synthesis and allocation. Hortic. Plant J. 2024, 11, 578–592. [Google Scholar] [CrossRef]
- Jiang, M.; Li, S.; Zhao, C.; Zhao, M.; Xu, S.; Wen, G. Identification and analysis of sucrose synthase gene family associated with polysaccharide biosynthesis in Dendrobium catenatum by transcriptomic analysis. PeerJ 2022, 10, e13222. [Google Scholar] [CrossRef]
- Zhao, C.; Hua, L.-N.; Liu, X.-F.; Li, Y.-Z.; Shen, Y.-Y.; Guo, J.-X. Sucrose synthase FaSS1 plays an important role in the regulation of strawberry fruit ripening. Plant Growth Regul. 2017, 81, 175–181. [Google Scholar] [CrossRef]
- Li, J.; Baroja-Fernández, E.; Bahaji, A.; Muñoz, F.J.; Ovecka, M.; Montero, M.; Sesma, M.T.; Alonso-Casajús, N.; Almagro, G.; Sánchez-López, A.M. Enhancing sucrose synthase activity results in increased levels of starch and ADP-glucose in maize (Zea mays L.) seed endosperms. Plant Cell Physiol. 2013, 54, 282–294. [Google Scholar] [CrossRef]
- Fan, C.; Wang, G.; Wang, Y.; Zhang, R.; Wang, Y.; Feng, S.; Luo, K.; Peng, L. Sucrose synthase enhances hull size and grain weight by regulating cell division and starch accumulation in transgenic rice. Int. J. Mol. Sci. 2019, 20, 4971. [Google Scholar] [CrossRef]
- Xu, S.-M.; Brill, E.; Llewellyn, D.J.; Furbank, R.T.; Ruan, Y.-L. Overexpression of a potato sucrose synthase gene in cotton accelerates leaf expansion, reduces seed abortion, and enhances fiber production. Mol. Plant 2012, 5, 430–441. [Google Scholar] [CrossRef]
- Li, B.; Zhu, L.; Yang, N.; Qu, S.; Cao, W.; Ma, W.; Wei, X.; Ma, B.; Ma, F.; Fu, A. Transcriptional landscape and dynamics involved in sugar and acid accumulation during apple fruit development. Plant Physiol. 2024, 195, kiae273. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jiang, L.; Chen, Y.; Li, C.; Li, P.; Yang, Y.; Chen, J.; Liu, Q. Transcriptome analysis unveiled the intricate interplay between sugar metabolism and lipid biosynthesis in Symplocos paniculate fruit. Plants 2023, 12, 2703. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Upadhyay, S.; Verma, P.C.; Solomon, S.; Singh, S.B. Functional analysis of sucrose phosphate synthase (SPS) and sucrose synthase (SS) in sugarcane (Saccharum) cultivars. Plant Biol. 2011, 13, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Meng, J.; Cui, Y.; Tian, M.; Shi, Z.; Wang, J. Transcriptome and targeted hormone metabolome reveal the molecular mechanisms of flower abscission in camellia. Front. Plant Sci. 2022, 13, 1076037. [Google Scholar] [CrossRef]
- Song, Q.; Ji, K.; Mo, W.; Wang, L.; Chen, L.; Gao, L.; Gong, W.; Yuan, D. Dynamics of sugars, endogenous hormones, and oil content during the development of Camellia oleifera fruit. Botany 2021, 99, 515–529. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Zeeshan Ul Haq, M.; Liu, Y.; Yang, D.; Yang, H.; Wu, Y. Gene Expression Dynamics of Sugar Metabolism and Accumulation During Fruit Ripening in Camellia drupifera. Plants 2025, 14, 817. https://doi.org/10.3390/plants14050817
Sun X, Zeeshan Ul Haq M, Liu Y, Yang D, Yang H, Wu Y. Gene Expression Dynamics of Sugar Metabolism and Accumulation During Fruit Ripening in Camellia drupifera. Plants. 2025; 14(5):817. https://doi.org/10.3390/plants14050817
Chicago/Turabian StyleSun, Xue, Muhammad Zeeshan Ul Haq, Ya Liu, Dongmei Yang, Huageng Yang, and Yougen Wu. 2025. "Gene Expression Dynamics of Sugar Metabolism and Accumulation During Fruit Ripening in Camellia drupifera" Plants 14, no. 5: 817. https://doi.org/10.3390/plants14050817
APA StyleSun, X., Zeeshan Ul Haq, M., Liu, Y., Yang, D., Yang, H., & Wu, Y. (2025). Gene Expression Dynamics of Sugar Metabolism and Accumulation During Fruit Ripening in Camellia drupifera. Plants, 14(5), 817. https://doi.org/10.3390/plants14050817