

Transcriptome Profiling of Spike Development Reveals Key Genes and Pathways Associated with Early Heading in Wheat–Psathyrstachys huashanica 7Ns Chromosome Addition Line
 , , , and
, , , and
Abstract

1. Introduction
2. Results
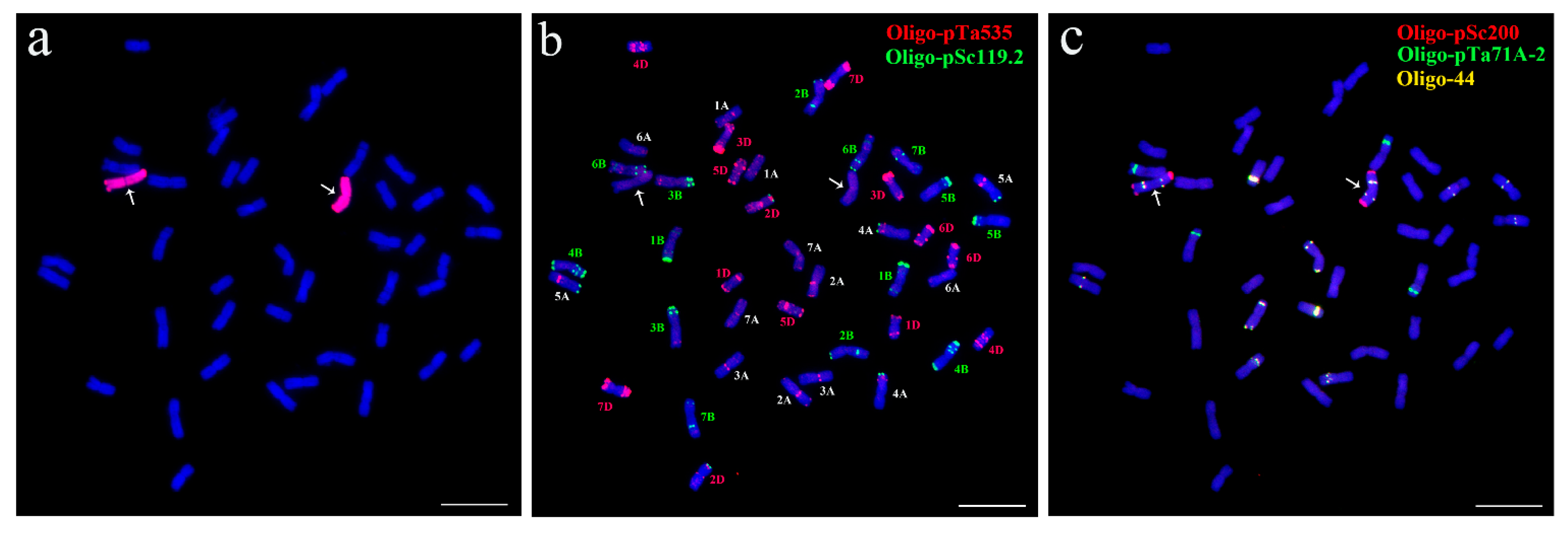
2.1. Cytological Identification of Wheat–P. huashanica 7Ns Addition Line
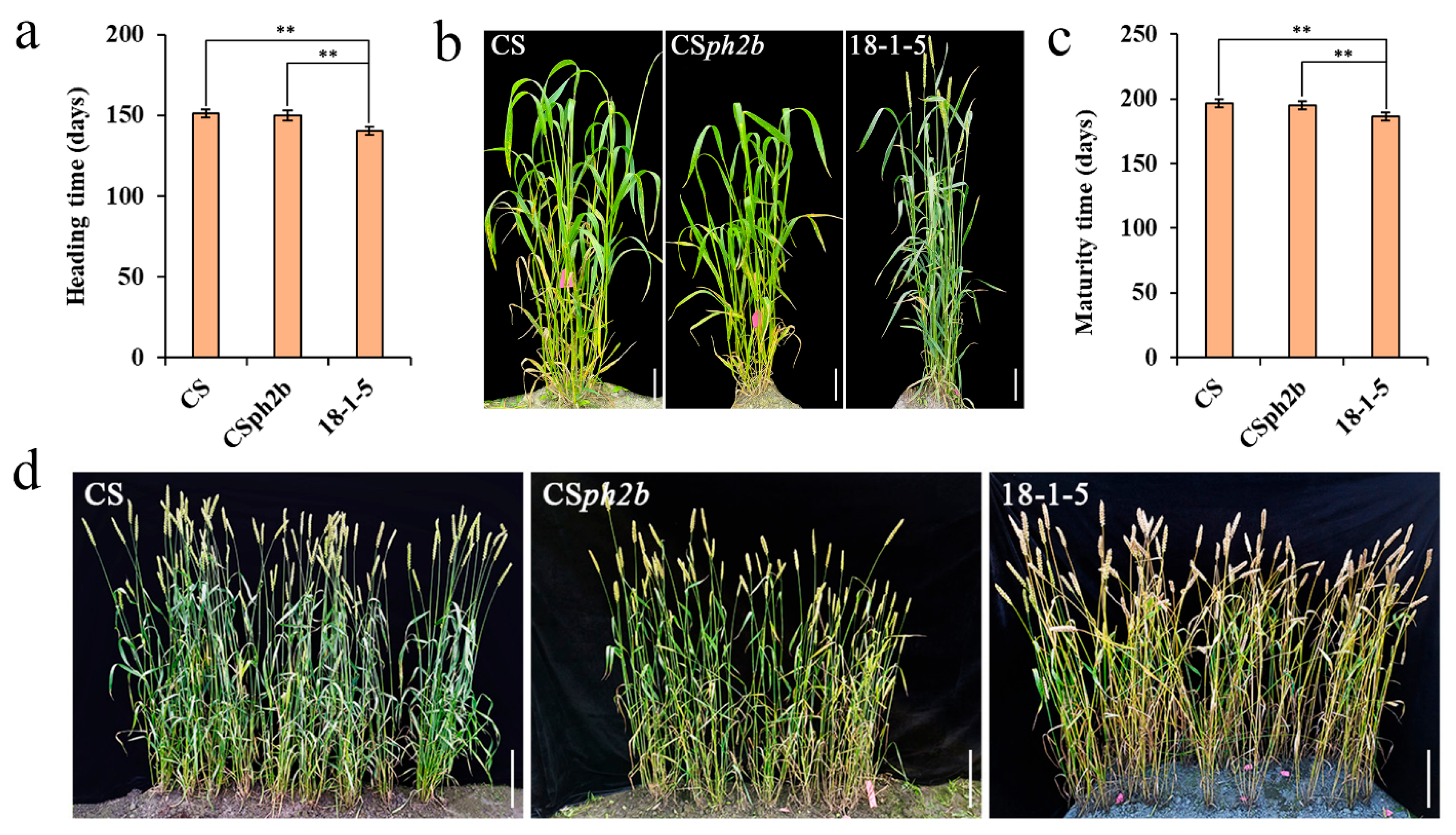
2.2. Investigation of Heading and Maturity Times for Wheat–P. huashanica 7Ns Addition Line
2.3. Observation of Spike Development in the Wheat–P. huashanica 7Ns Addition Line
2.4. Quality Analysis and Sequence Assembly of RNA-Seq Data
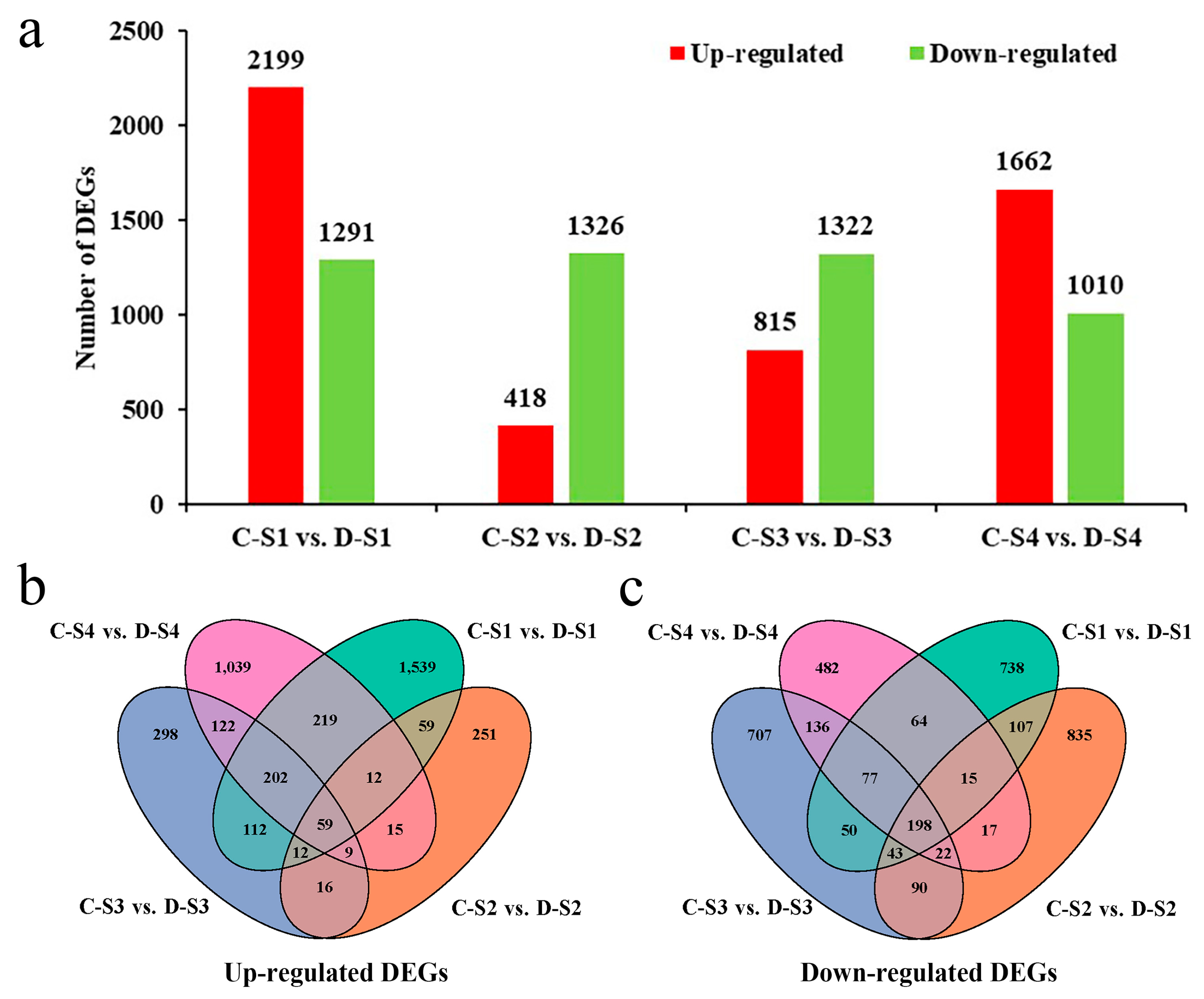
2.5. DEGs Obtained During Spike Development
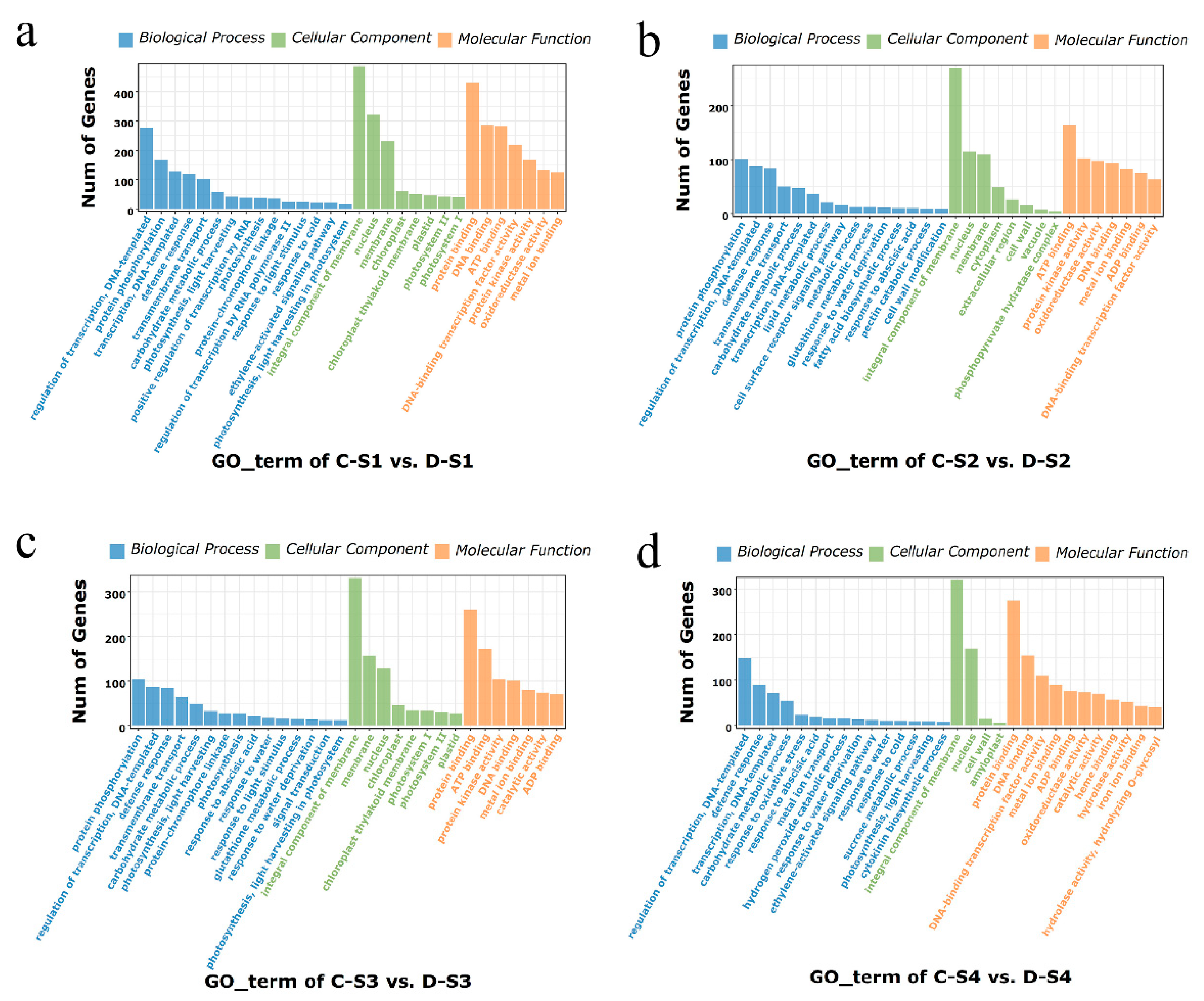
2.6. GO Enrichment Analysis of DEGs
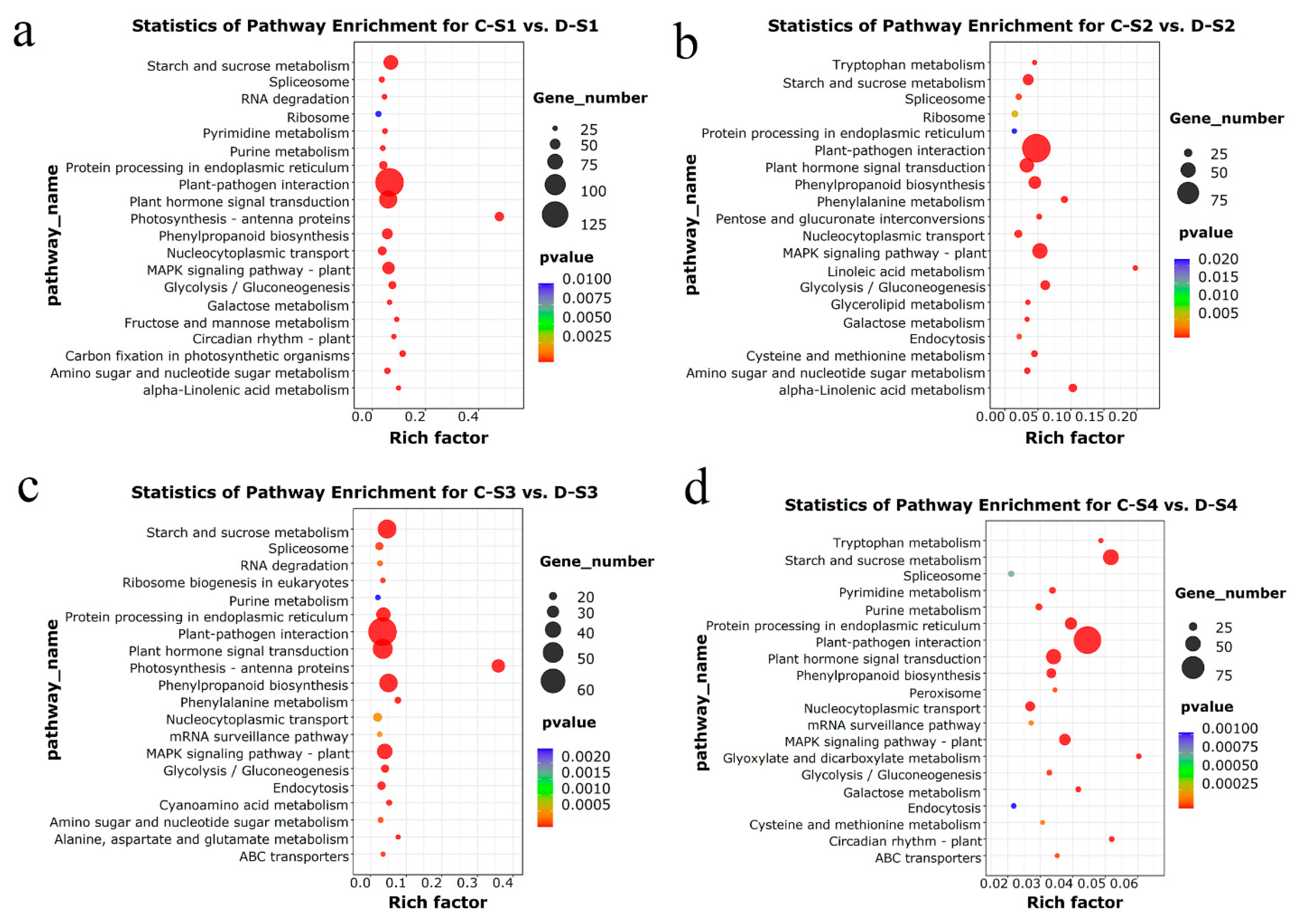
2.7. KEGG Enrichment Analysis of DEGs
2.8. Important Regulatory Pathways and DEGs Associated with Spike Development
2.9. TFs Associated with Spike Development
2.10. Quantitative Real-Time PCR Validation of Spike Development-Related DEGs
3. Discussion
3.1. Wheat–Alien Species Are Potentially Valuable Germplasm Resources for Improving Early Heading and Maturity
3.2. DEGs and Pathways Associated with Spike Development
3.3. Plant Hormone Signal Transduction and Starch and Sucrose Metabolism-Related Genes Involved in Spike Development
3.4. Photosynthetic Antenna Proteins and Circadian Rhythm-Related Genes Involved in Spike Development
3.5. TFs Involved in Spike Development
4. Materials and Methods
4.1. Plant Materials
4.2. GISH and FISH Analyses
4.3. Phenotypic Characterization
4.4. RNA Extraction, Library Construction, and Illumina Sequencing
4.5. Transcriptome Analysis
4.6. Transcription Factor Analysis
4.7. qRT-PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| CS | Chinese Spring |
| CSph2b | Chinese Spring ph2b mutant |
| GISH | Genomic in situ hybridization |
| FISH | Fluorescence in situ hybridization |
| DAPI | 4,6-diamidino-2-phenylindole |
| DEGs | Differentially expressed genes |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| TFs | Transcription factors |
| FPKM | Fragment per kilobase of transcript per million mapped reads |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
References
- Shiferaw, B.; Smale, M.; Braun, H.J.; Duveiller, E.; Reynolds, M.; Muricho, G. Crops that feed the world 10. Past successes and future challenges to the role played by wheat in global food security. Food Sec. 2013, 5, 291–317. [Google Scholar] [CrossRef]
- Fjellheim, S.; Boden, S.; Trevaskis, B. The role of seasonal flowering responses in adaptation of grasses to temperate climates. Front. Plant Sci. 2014, 5, 431. [Google Scholar] [CrossRef]
- Mizuno, N.; Matsunaka, H.; Yanaka, M.; Ishikawa, G.; Kobayashi, F.; Nakamura, K. Natural variations of wheat EARLY FLOWERING 3 highlight their contributions to local adaptation through fine-tuning of heading time. Theor. Appl. Genet. 2023, 136, 139. [Google Scholar] [CrossRef]
- Huang, M.; Mheni, N.; Brown-Guedira, G.; McKendry, A.; Griffey, C.; Van, S.D.; Costa, J.; Sneller, C. Genetic analysis of heading date in winter and spring wheat. Euphytica 2018, 214, 128. [Google Scholar] [CrossRef]
- Yan, L.; Fu, D.; Li, C.; Blechl, A.; Tranquilli, G.; Bonafede, M.; Sanchez, A.; Valarik, M.; Yasuda, S.; Dubcovsky, J. The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc. Natl. Acad. Sci. USA 2006, 103, 19581–19586. [Google Scholar] [CrossRef]
- Yan, L.; Loukoianov, A.; Tranquilli, G.; Helguera, M.; Fahima, T.; Dubcovsky, J. Positional cloning of the wheat vernalization gene VRN1. Proc. Natl. Acad. Sci. USA 2003, 100, 6263–6268. [Google Scholar] [CrossRef]
- Xie, L.; Zhang, Y.; Wang, K.; Luo, X.; Xu, D.; Tian, X.; Li, L.; Ye, X.; Xia, X.; Li, W.; et al. TaVrt2, an SVP-like gene, cooperates with TaVrn1 to regulate vernalization-induced flowering in wheat. New Phytol. 2021, 231, 834–848. [Google Scholar] [CrossRef]
- Yan, L.; Loukoianov, A.; Blechl, A.; Tranquilli, G.; Ramakrishna, W.; SanMiguel, P.; Bennetzen, J.L.; Echenique, V.; Dubcovsky, J. The wheat VRN2 gene is a flowering repressor down-regulated by vernalization. Science 2004, 303, 1640–1644. [Google Scholar] [CrossRef]
- Dubcovsky, J.; Loukoianov, A.; Fu, D.; Valarik, M.; Sanchez, A.; Yan, L. Effect of photoperiod on the regulation of wheat vernalization genes VRN1 and VRN2. Plant Mol. Biol. 2006, 60, 469–480. [Google Scholar] [CrossRef]
- Turner, A.; Beales, J.; Faure, S.; Dunford, R.P.; Laurie, D.A. The pseudo-response regulator Ppd-H1 provides adaptation to photoperiod in barley. Science 2005, 310, 1031–1034. [Google Scholar] [CrossRef]
- Boden, S.A.; Cavanagh, C.; Cullis, B.R.; Ramm, K.; Greenwood, J.; Jean Finnegan, E.; Trevaskis, B.; Swain, S.M. Ppd-1 is a key regulator of inflorescence architecture and paired spikelet development in wheat. Nat. Plants 2015, 1, 14016. [Google Scholar]
- Worland, A.J. The influence of flowering time genes on environmental adaptability in European wheats. Euphytica 1996, 89, 49–57. [Google Scholar] [CrossRef]
- Zikhali, M.; Wingen, L.U.; Griffiths, S. Delimitation of the Earliness per se D1 (Eps-D1) flowering gene to a subtelomeric chromosomal deletion in bread wheat (Triticum aestivum). J. Exp. Bot. 2016, 67, 287–299. [Google Scholar] [CrossRef]
- Balzan, S.; Johal, G.S.; Carraro, N. The role of auxin transporters in monocots development. Front. Plant Sci. 2014, 5, 393. [Google Scholar] [CrossRef]
- Domagalska, M.A.; Sarnowska, E.; Nagy, F.; Davis, S.J. Genetic analyses of interactions among gibberellin, abscisic acid, and brassinosteroids in the control of flowering time in Arabidopsis thaliana. PLoS ONE 2010, 5, e14012. [Google Scholar] [CrossRef]
- Yan, Z.; Deng, R.; Tang, H.; Zhang, H.; Zhu, S. Molecular basis of differential sensitivity to MeJA in floret opening between indica and japonica rice. Czech J. Genet. Plant Breed. 2024, 60, 136–148. [Google Scholar] [CrossRef]
- Bao, S.; Hua, C.; Shen, L.; Yu, H. New insights into gibberellin signaling in regulating flowering in Arabidopsis. J. Integr. Plant Biol. 2020, 62, 118–131. [Google Scholar] [CrossRef]
- Xu, F.; Li, T.; Xu, P.; Li, L.; Du, S.; Lian, H.; Yang, H. DELLA proteins physically interact with CONSTANS to regulate flowering under long days in Arabidopsis. FEBS Lett. 2016, 590, 541–549. [Google Scholar] [CrossRef]
- Moon, J.; Suh, S.S.; Lee, H.; Choi, K.R.; Hong, C.; Paek, N.C.; Kim, S.G.; Lee, I. The SOC1 MADS-box gene integrates vernalization and gibberellin signals for flowering in Arabidopsis. Plant J. 2003, 35, 613–623. [Google Scholar] [CrossRef]
- Pearce, S.; Vanzetti, L.S.; Dubcovsky, J. Exogenous gibberellins induce wheat spike development under short days only in the presence of VERNALIZATION1. Plant Physiol. 2013, 163, 1433–1445. [Google Scholar] [CrossRef]
- McMaster, G.S. Phytomers, phyllochrons, phenology and temperate cereal development. J. Agric. Sci. 2005, 143, 137–150. [Google Scholar] [CrossRef]
- Kamran, A.; Iqbal, M.; Spaner, D. Flowering time in wheat (Triticum aestivum L.): A key factor for global adaptability. Euphytica 2014, 197, 1–26. [Google Scholar] [CrossRef]
- Digel, B.; Pankin, A.; von Korff, M. Global transcriptome profiling of developing leaf and shoot apices reveals distinct genetic and environmental control of floral transition and inflorescence development in barley. Plant Cell 2015, 27, 2318–2334. [Google Scholar] [CrossRef]
- Li, Y.; Fu, X.; Zhao, M.; Zhang, W.; Li, B.; An, D.; Li, J.; Zhang, A.; Liu, R.; Liu, X. A Genome-wide view of transcriptome dynamics during early spike development in bread wheat. Sci. Rep. 2018, 8, 15338. [Google Scholar] [CrossRef]
- Liu, H.; Li, G.; Yang, X.; Kuijer, H.N.J.; Liang, W.; Zhang, D. Transcriptome profiling reveals phase-specific gene expression in the developing barley inflorescence. Crop J. 2020, 8, 71–86. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, X.; Wu, L.; Zhang, L.; Liu, G.; Xia, C.; Liu, X.; Kong, X. Transcriptome profiling of developing leaf and shoot apices to reveal the molecular mechanism and co-expression genes responsible for the wheat heading date. BMC Genom. 2021, 22, 468. [Google Scholar] [CrossRef]
- VanGessel, C.; Hamilton, J.; Tabbita, F.; Dubcovsky, J.; Pearce, S. Transcriptional signatures of wheat inflorescence development. Sci. Rep. 2022, 12, 17224. [Google Scholar] [CrossRef]
- Benaouda, S.; Stöcker, T.; Schoof, H.; Léon, J.; Ballvora, A. Transcriptome profiling at the transition to the reproductive stage uncovers stage and tissue-specific genes in wheat. BMC Plant Biol. 2023, 23, 25. [Google Scholar] [CrossRef]
- Gauley, A.; Pasquariello, M.; Yoshikawa, G.V.; Alabdullah, A.K.; Hayta, S.; Smedley, M.A.; Dixon, L.E.; Boden, S.A. Photoperiod-1 regulates the wheat inflorescence transcriptome to influence spikelet architecture and flowering time. Curr. Biol. 2024, 34, 2330–2343. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, A.; Fu, J. The hybridization between Triticum aestivum and Psathyrostachys huashanica Keng. Acta Genet. Sin. 1991, 18, 508–512. [Google Scholar]
- Fu, J.; Wang, M.; Zhao, J.; Chen, S.; Hou, W.; Yang, Q. Studies on cytogenetics and utilization of wheat-Psathyrostachys huashanica medium material H8911 with resistance to wheat take-all fungus. Acta Bot. Boreal. Occident. Sin. 2003, 23, 2157–2162. [Google Scholar]
- Kang, H.; Zhang, H.; Fan, X.; Zhou, Y. Morphological and cytogenetic studies on the hybrid between bread wheat and Psathyrostachys huashanica Keng ex Kuo. Euphytica 2008, 162, 441–448. [Google Scholar] [CrossRef]
- Li, J.; Zhao, L.; Cheng, X.; Bai, G.; Li, M.; Wu, J.; Yang, Q.; Chen, X.; Yang, Z.; Zhao, J. Molecular cytogenetic characterization of a novel wheat-Psathyrostachys huashanica Keng T3DS-5NsL•5NsS and T5DL-3DS•3DL dual translocation line with powdery mildew resistance. BMC Plant Biol. 2020, 20, 163. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Huang, C.; Wang, Y.; Wen, X.; Deng, P.; Li, T.; Wang, C.; Liu, X.; Chen, C.; Zhao, J.; et al. Molecular cytological analysis and specific marker development in wheat-Psathyrostachys huashanica keng 3Ns additional line with elongated glume. Int. J. Mol. Sci. 2023, 24, 6726. [Google Scholar] [CrossRef]
- Tan, B.; Wang, M.; Cai, L.; Li, S.; Zhu, W.; Xu, L.; Wang, Y.; Zeng, J.; Fan, X.; Sha, L.; et al. Cytogenetic and molecular marker analyses of a novel wheat-Psathyrostachys huashanica 7Ns disomic addition line with powdery mildew resistance. Int. J. Mol. Sci. 2022, 23, 10285. [Google Scholar] [CrossRef]
- Tang, Z.; Yang, Z.; Fu, S. Oligonucleotides replacing the roles of repetitive sequences pAs1, pSc119.2, pTa-535, pTa71, CCS1, and pAWRC.1 for FISH analysis. J. Appl. Genet. 2014, 55, 313–318. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, F.; Zeng, C.; Zhu, W.; Xu, L.; Wang, Y.; Zeng, J.; Fan, X.; Sha, L.; Wu, D.; et al. Development and application of specific FISH probes for karyotyping Psathyrostachys huashanica chromosomes. BMC Genom. 2022, 23, 309. [Google Scholar] [CrossRef]
- Zhao, H.; Huang, X.; Yang, Z.; Li, F.; Ge, X. Synergistic optimization of crops by combining early maturation with other agronomic traits. Trends Plant Sci. 2023, 28, 1178–1191. [Google Scholar] [CrossRef]
- Johansson, E.; Lan, Y.; Olalekan, O.; Kuktaite, R.; Chawade, A.; Rahmatov, M. Alien introgression to wheat for food security: Functional and nutritional quality for novel products under climate change. Front. Nutr. 2024, 11, 1393357. [Google Scholar] [CrossRef]
- Mujeeb-Kazi, A.; Kazi, A.G.; Dundas, I.; Rasheed, A.; Ogbonnaya, F.; Kishii, M.; Bonnett, D.; Wang, R.C.; Xu, S.; Chen, P.; et al. Genetic diversity for wheat improvement as a conduit to food security. Adv. Agron. 2013, 122, 179–257. [Google Scholar]
- Kong, L.; Song, X.; Xiao, J.; Sun, H.; Dai, K.; Lan, C.; Singh, P.; Yuan, C.; Zhang, S.; Singh, R.; et al. Development and characterization of a complete set of Triticum aestivum-Roegneria ciliaris disomic addition lines. Theor. Appl. Genet. 2018, 131, 1793–1806. [Google Scholar] [CrossRef]
- Efremova, T.T.; Laĭkova, L.I.; Arbuzova, V.S.; Popova, O.M. Effect of the 5R(5A) alien chromosome substitution on the growth habit and winter hardiness of wheat. Russ. J. Genet. 2004, 40, 810–812. [Google Scholar] [CrossRef]
- Liu, S.; Chen, X.; Cai, Z.; Wu, J.; Zhao, J.; Yang, Q. Selection and identification of early mature derive lines from common wheat× cultivated barley hybrid progenies. J. Shanxi Agric. Univ. 2011, 31, 142–145. [Google Scholar]
- Farkas, A.; Molnár, I.; Kiss, T.; Karsai, I.; Molnár-Láng, M. Effect of added barley chromosomes on the flowering time of new wheat/winter barley addition lines in various environments. Euphytica 2014, 195, 45–55. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Du, W.; Jing, F.; Wang, Z.; Wu, J.; Chen, X. Anatomy and cytogenetic identification of a wheat-Psathyrostachys huashanica keng line with early maturation. PLoS ONE 2015, 10, e0131841. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Zhao, L.; Li, L.; Zhang, H.; Zhu, W.; Xu, L.; Wang, Y.; Zeng, J.; Fan, X.; Sha, L.; et al. Identification of a wheat-Psathyrostachys huashanica 7Ns ditelosomic addition line conferring early maturation by cytological analysis and newly developed molecular and FISH markers. Front. Plant Sci. 2021, 12, 784001. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Zhao, M.; Guo, L.; Guo, X.; Zhao, D.; Batool, A.; Dong, B.; Xu, H.; Cui, S.; et al. Wheat FRIZZY PANICLE activates VERNALIZATION1-A and HOMEOBOX4-A to regulate spike development in wheat. Plant Biotechnol. J. 2021, 19, 1141–1154. [Google Scholar] [CrossRef]
- Feng, N.; Song, G.; Guan, J.; Chen, K.; Jia, M.; Huang, D.; Wu, J.; Zhang, L.; Kong, X.; Geng, S.; et al. Transcriptome profiling of wheat inflorescence development from spikelet initiation to floral patterning identified stage-specific regulatory genes. Plant Physiol. 2017, 174, 1779–1794. [Google Scholar] [CrossRef]
- Pearce, S.; Kippes, N.; Chen, A.; Debernardi, J.M.; Dubcovsky, J. RNA-seq studies using wheat PHYTOCHROME B and PHYTOCHROME C mutants reveal shared and specific functions in the regulation of flowering and shade-avoidance pathways. BMC Plant Biol. 2016, 16, 141. [Google Scholar] [CrossRef]
- Sun, F.; Niu, Y.; Song, T.; Han, B.; Liu, Z.; You, W.; Wang, P.; Su, P. Comparative transcriptome analysis reveals genetic mechanism for flowering response in two wheat (Triticum aestivum L.) cultivars. Russ. J. Genet. 2023, 59, 9–18. [Google Scholar] [CrossRef]
- Dong, X.; Li, Y.; Guan, Y.; Wang, S.; Luo, H.; Li, X.; Li, H.; Zhang, Z. Auxin-induced AUXIN RESPONSE FACTOR4 activates APETALA1 and FRUITFULL to promote flowering in woodland strawberry. Hortic. Res. 2021, 8, 115. [Google Scholar] [CrossRef]
- Su, P.; Sui, C.; Wang, S.; Liu, X.; Zhang, G.; Sun, H.; Wan, K.; Yan, J.; Guo, S. Genome-wide evolutionary analysis of AUX/IAA gene family in wheat identifies a novel gene TaIAA15-1A regulating flowering time by interacting with ARF. Int. J. Biol. Macromol. 2023, 227, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, T.; Yue, J.; Pu, N.; Liu, J.; Luo, L.; Huang, M.; Guo, T.; Xiao, W. Small Auxin Up RNA 56 (SAUR56) regulates heading date in rice. Mol. Breed. 2023, 43, 62. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Gao, Y.; Liu, Y.; Zhang, X.; Gu, X.; Ma, D.; Zhao, Z.; Yuan, Z.; Xue, H.; Liu, H. BES1-regulated BEE1 controls photoperiodic flowering downstream of blue light signaling pathway in Arabidopsis. New Phytol. 2019, 223, 1407–1419. [Google Scholar] [CrossRef]
- Fukazawa, J.; Ohashi, Y.; Takahashi, R.; Nakai, K.; Takahashi, Y. DELLA degradation by gibberellin promotes flowering via GAF1-TPR-dependent repression of floral repressors in Arabidopsis. Plant Cell 2021, 33, 2258–2272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Jian, M.; Li, W.; Yao, X.; Tan, C.; Qian, Q.; Hu, Y.; Liu, X.; Hou, X. Gibberellin signaling modulates flowering via the DELLA-BRAHMA-NF-YC module in Arabidopsis. Plant Cell 2023, 35, 3470–3484. [Google Scholar] [CrossRef]
- Ogawara, T.; Higashi, K.; Kamada, H.; Ezura, H. Ethylene advances the transition from vegetative growth to flowering in Arabidopsis thaliana. J. Plant Physiol. 2003, 160, 1335–1340. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Q.; Huang, X.; Liu, S.; Zhang, H.; Zhang, Z.; Sun, G. Molecular cloning and characterization of four genes encoding ethylene receptors associated with pineapple (Ananas comosus L.) flowering. Front. Plant Sci. 2016, 7, 710. [Google Scholar] [CrossRef]
- Bodson, M. Changes in the carbohydrate content of the leaf and the apical bud of Sinapis during transition to flowering. Planta 1977, 135, 19–23. [Google Scholar] [CrossRef]
- Serrano, G.; Herrera-Palau, R.; Romero, J.M.; Serrano, A.; Coupland, G.; Valverde, F. Chlamydomonas CONSTANS and the evolution of plant photoperiodic signaling. Curr. Biol. 2009, 19, 359–368. [Google Scholar] [CrossRef]
- Wahl, V.; Ponnu, J.; Schlereth, A.; Arrivault, S.; Langenecker, T.; Franke, A.; Feil, R.; Lunn, J.E.; Stitt, M.; Schmid, M. Regulation of flowering by trehalose-6-phosphate signaling in Arabidopsis thaliana. Science 2013, 339, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Miao, F.; Jia, H.; Li, G.; Powers, C.; Nagarajan, R.; Alderman, P.D.; Carver, B.F.; Ma, Z.; Yan, L. O-linked N-acetylglucosamine transferase is involved in fine regulation of flowering time in winter wheat. Nat. Commun. 2021, 12, 2303. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Blankenship, R.E. On the interface of light-harvesting antenna complexes and reaction centers in oxygenic photosynthesis. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 148079. [Google Scholar] [CrossRef]
- Peng, H.; Gao, J.; Song, X. Transcriptome analyses reveal photosynthesis-related genes involved in chloroplast development of the EMS-induced maize mutant. Plant Biotechnol. Rep. 2022, 16, 565–578. [Google Scholar] [CrossRef]
- Novoderezhkin, V.I.; Croce, R. The location of the low-energy states in Lhca1 favors excitation energy transfer to the core in the plant PSI-LHCI supercomplex. Photosynth. Res. 2023, 156, 59–74. [Google Scholar] [CrossRef]
- Li, H.; He, X.; Gao, Y.; Liu, W.; Song, J.; Zhang, J. Integrative analysis of transcriptome, proteome, and phosphoproteome reveals potential roles of photosynthesis antenna proteins in response to brassinosteroids signaling in maize. Plants 2023, 12, 1290. [Google Scholar] [CrossRef]
- Murphy, R.L.; Klein, R.R.; Morishige, D.T.; Brady, J.A.; Rooney, W.L.; Miller, F.R.; Dugas, D.V.; Klein, P.E.; Mullet, J.E. Coincident light and clock regulation of pseudoresponse regulator protein 37 (PRR37) controls photoperiodic flowering in sorghum. Proc. Natl. Acad. Sci. USA 2011, 108, 16469–16474. [Google Scholar] [CrossRef] [PubMed]
- Nasution, K.; Satyawan, D.; Yunus, M.; Dewi, A.; Melati, P.; Maryono, M.; Dwimahyani, I.; Enggarini, W.; Sobrizal, S. Detection of genomic loci associated with days to heading in tropical japonica rice through QTL-seq. Czech J. Genet. Plant Breed. 2025, 61, 23–30. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, L.; Xie, Y.; Hao, L.; Wang, Z.; Yi, C.; Guo, H.; Gan, Y.; Xiang, G.; Yan, Z.; et al. QTL mapping for heading date and plant height using a RIL population in rice in different photoperiod environments. Czech J. Genet. Plant Breed. 2024, 60, 119–125. [Google Scholar] [CrossRef]
- Kojima, S.; Takahashi, Y.; Kobayashi, Y.; Monna, L.; Sasaki, T.; Araki, T.; Yano, M. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol. 2002, 43, 1096–1105. [Google Scholar] [CrossRef]
- Zhu, Y.; Fan, Y.; Wang, K.; Huang, D.; Liu, W.; Ying, J.; Zhuang, J. Rice Flowering Locus T 1 plays an important role in heading date influencing yield traits in rice. Sci. Rep. 2017, 7, 4918. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhang, Y.; Zhang, M.; Zhan, X.; Shen, X.; Yu, P.; Chen, D.; Liu, Q.; Sinumporn, S.; Hussain, K.; et al. The rice CONSTANS-like protein OsCOL15 suppresses flowering by promoting Ghd7 and repressing RID1. Biochem. Biophys. Res. Commun. 2018, 495, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gu, F.; Dong, S.; Liu, W.; Wang, H.; Chen, Z.; Wang, J. CONSTANS-like 9 (COL9) delays the flowering time in Oryza sativa by repressing the Ehd1 pathway. Biochem. Biophys. Res. Commun. 2016, 479, 173–178. [Google Scholar] [CrossRef]
- Ridge, S.; Sussmilch, F.C.; Hecht, V.; Vander Schoor, J.K.; Lee, R.; Aubert, G.; Burstin, J.; Macknight, R.C.; Weller, J.L. Identification of LATE BLOOMER2 as a CYCLING DOF FACTOR Homolog reveals conserved and divergent features of the flowering response to photoperiod in Pea. Plant Cell 2016, 28, 2545–2559. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N. Role of bHLH93 in Controlling Flowering Time in Arabidopsis thaliana. Ph.D. Thesis, The University of Texas at Austin, Austin, TX, USA, 2011. [Google Scholar]
- Sharma, N.; Xin, R.; Kim, D.H.; Sung, S.; Lange, T.; Huq, E. NO FLOWERING IN SHORT DAY (NFL) is a bHLH transcription factor that promotes flowering specifically under short-day conditions in Arabidopsis. Development 2016, 143, 682–690. [Google Scholar]
- Bhagat, P.K.; Verma, D.; Sharma, D.; Sinha, A.K. HY5 and ABI5 transcription factors physically interact to fine tune light and ABA signaling in Arabidopsis. Plant Mol. Biol. 2021, 107, 117–127. [Google Scholar] [CrossRef]
- Becker, A.; Theissen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Nam, J.; DePamphilis, C.W.; Ma, H.; Nei, M. Antiquity and evolution of the MADS-box gene family controlling flower development in plants. Mol. Biol. Evol. 2003, 20, 1435–1447. [Google Scholar] [CrossRef]
- Chen, L.; Yan, Y.; Ke, H.; Zhang, Z.; Meng, C.; Ma, L.; Sun, Z.; Chen, B.; Liu, Z.; Wang, G.; et al. SEP-like genes of Gossypium hirsutum promote flowering via targeting different loci in a concentration-dependent manner. Front. Plant Sci. 2022, 13, 990221. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Cai, Q.; Hu, Y.; Jin, Z.; Zhao, X.; Fan, W.; Huang, Q.; Luo, Z.; Chen, M.; et al. OsMADS32 interacts with PI-like proteins and regulates rice flower development. J. Integr. Plant Biol. 2015, 57, 504–513. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, J.; Adrian, J.; Gissot, L.; Coupland, G.; Yu, D.; Turck, F. Elevated levels of MYB30 in the phloem accelerate flowering in Arabidopsis through the regulation of FLOWERING LOCUS T. PLoS ONE 2014, 9, e89799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, G.; Jia, J.; Zhao, G.; Xia, C.; Zhang, L.; Li, F.; Zhang, Q.; Dong, C.; Gao, S.; et al. The wheat MYB-related transcription factor TaMYB72 promotes flowering in rice. J. Integr. Plant Biol. 2016, 58, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xie, Z.; Li, D.; Chen, Y.; Xia, C.; Kong, X.; Liu, X.; Zhang, L. TaMYB72 directly activates the expression of TaFT to promote heading and enhance grain yield traits in wheat (Triticum aestivum L.). J. Integr. Plant Biol. 2024, 66, 1266–1269. [Google Scholar] [CrossRef]
- Zhang, H.; Cui, X.; Guo, Y.; Luo, C.; Zhang, L. Picea wilsonii transcription factor NAC2 enhanced plant tolerance to abiotic stress and participated in RFCP1-regulated flowering time. Plant Mol. Biol. 2018, 98, 471–493. [Google Scholar] [CrossRef]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Wu, M.; Yang, L.; Wu, G.; Poethig, R.S.; Wagner, D. The microRNA-regulated SBP-Box transcription factor SPL3 is a direct upstream activator of LEAFY, FRUITFULL, and APETALA1. Dev. Cell 2009, 17, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, Z.; Wang, L.; Kim, S.G.; Seo, P.J.; Qiao, M.; Wang, N.; Li, S.; Cao, X.; Park, C.M.; et al. WRKY71 accelerates flowering via the direct activation of FLOWERING LOCUS T and LEAFY in Arabidopsis thaliana. Plant J. 2016, 85, 96–106. [Google Scholar] [CrossRef]
- Hwang, Y.H.; Kim, S.K.; Lee, K.C.; Chung, Y.S.; Lee, J.H.; Kim, J.K. Functional conservation of rice OsNF-YB/YC and Arabidopsis AtNF-YB/YC proteins in the regulation of flowering time. Plant Cell Rep. 2016, 35, 857–865. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, L.; Zhang, Y.; Fan, J.; Li, L. Genome-wide analysis of poplar NF-YB gene family and identified PtNF-YB1 important in regulate flowering timing in transgenic plants. BMC Plant Biol. 2019, 19, 251. [Google Scholar] [CrossRef]
- Han, F.; Lamb, J.; Birchler, J.A. High frequency of centromere inactivation resulting in stable dicentric chromosomes of maize. Proc. Natl. Acad. Sci. USA 2006, 103, 3238–3243. [Google Scholar] [CrossRef]
- Cui, J.; Wang, Y.; Wang, H. Differentiation and formation of winter wheat spikes. In The Spike of Wheat; Cui, J., Guo, T., Eds.; China Agriculture Press: Beijing, China, 2008; pp. 22–33. [Google Scholar]
- Guo, W.; Fan, G. Wheat. In Treatise on Crop Cultivation; Yang, W., Tu, N., Eds.; China Agriculture Press: Beijing, China, 2011; pp. 77–78. [Google Scholar]
- Liu, Z.; Niu, F.; Yuan, S.; Feng, S.; Li, Y.; Lu, F.; Zhang, T.; Bai, J.; Zhao, C.; Zhang, L. Comparative transcriptome analysis reveals key insights into fertility conversion in the thermo-sensitive cytoplasmic male sterile wheat. Int. J. Mol. Sci. 2022, 23, 14354. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiao, C.; Sun, H.; Rosli, H.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Yang, X.; Jiang, Y.; Yu, X.; Zhang, H.; Wang, Y.; Guan, F.; Long, L.; Li, H.; Li, W.; Jiang, Q.; et al. Fine mapping and transcriptome sequencing reveal candidate genes conferring all-stage resistance to stripe rust on chromosome arm 1AL in Chinese wheat landrace AS1676. Crop J. 2023, 11, 1501–1511. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KEGG Pathways | Gene ID | Functional Annotation | C1 vs. D1 | C2 vs. D2 | C3 vs. D3 | C4 vs. D4 |
|---|---|---|---|---|---|---|
| Plant hormone signal transduction | TraesCS3A02G233000 | DELLA protein GAI | 3.04 ↑ | - | - | - |
| TraesCS3D02G220100 | DELLA protein GAI | 3.21 ↑ | - | - | - | |
| Starch and sucrose metabolism | TraesCS4A02G446700 | Sucrose synthase 1 (SUS1) | 6.33 ↑ | 5.62 ↑ | 3.53 ↑ | 3.27 ↑ |
| TraesCS2A02G161000 | Trehalose 6-phosphate phosphatase RA3 | 10.63 ↑ | - | - | - | |
| TraesCS2A02G161100 | Trehalose 6-phosphate phosphatase RA3 | 22.77 ↑ | - | - | - | |
| TraesCS2A02G161200 | Trehalose 6-phosphate phosphatase RA3 | 7.00 ↑ | - | 2.26 ↓ | - | |
| TraesCS2B02G187100 | Trehalose 6-phosphate phosphatase RA3 | 3.17 ↑ | - | - | - | |
| TraesCS2B02G187200 | Trehalose 6-phosphate phosphatase RA3 | 5.71 ↑ | - | - | - | |
| TraesCS2D02G168100 | Trehalose 6-phosphate phosphatase RA3 | 10.49 ↑ | - | - | 2.29 ↓ | |
| TraesCS2D02G168200 | Trehalose 6-phosphate phosphatase RA3 | 3.66 ↑ | - | - | - | |
| Photosynthetic antenna proteins | TraesCS7A02G227100 | Chlorophyll a/b-binding protein 1B-21, chloroplastic | 3.90 ↑ | - | - | - |
| TraesCS7B02G192500 | Chlorophyll a/b-binding protein 1B-21, chloroplastic | 5.49 ↑ | - | - | - | |
| TraesCS7D02G227300 | Chlorophyll a/b-binding protein 1B-21, chloroplastic | 4.81 ↑ | - | - | - | |
| Circadian rhythm—plant | TraesCSU02G199500 | Two-component response regulator-like PRR37 | 37.05 ↑ | 8.16 ↑ | 69.31 ↑ | 4.75 ↑ |
| TraesCSU02G221500 | Two-component response regulator-like PRR37 | 47.40 ↑ | 7.29 ↑ | 73.61 ↑ | 7.61 ↑ | |
| TraesCS3A02G143100 | Protein HEADING DATE 3A (Hd3a) | 4.77 ↑ | - | - | - | |
| TraesCS3B02G162000 | Protein HEADING DATE 3A (Hd3a) | 13.85 ↑ | - | - | - | |
| TraesCS3D02G144500 | Protein HEADING DATE 3A (Hd3a) | 8.87 ↑ | - | - | - | |
| TraesCS2B02G365300 | Protein FLOWERING LOCUS T | 4.98 ↓ | - | - | - | |
| TraesCS5A02G297300 | Protein FLOWERING LOCUS T | 5.41 ↓ | - | - | - | |
| TraesCS6A02G286400 | Zinc finger protein CONSTANS-LIKE 10 | 2.73 ↑ | - | - | - | |
| TraesCS4B02G045700 | Zinc finger protein CONSTANS-LIKE 16 | 7.03 ↑ | - | - | - | |
| TraesCS7B02G113400 | Zinc finger protein CONSTANS-LIKE 16 | 2.23 ↑ | - | - | - | |
| TraesCS7D02G209000 | Zinc finger protein CONSTANS-LIKE 16 | - | - | - | 3.42 ↓ | |
| TraesCS6D02G274100 | Zinc finger protein CONSTANS-LIKE 16 | - | - | 2.31 ↓ | - | |
| TraesCS2D02G351900 | Zinc finger protein CONSTANS-LIKE 5 | - | - | 5.67 ↓ | - | |
| TraesCS3D02G185500 | Cycling DOF factor 2 | 2.52 ↓ | - | - | - | |
| TraesCS3A02G180600 | Cycling DOF factor 2 | - | - | 2.08 ↓ | - | |
| TraesCS3B02G210300 | Cycling DOF factor 2 | - | - | - | 2.27 ↓ |
| TFs Family | Gene Name | Gene ID | C1 vs. D1 | C2 vs. D2 | C3 vs. D3 | C4 vs. D4 |
|---|---|---|---|---|---|---|
| bHLH | TabHLH25-5B | TraesCS5B02G518400 | 3.23 ↑ | - | - | - |
| TabHLH25 | TraesCSU02G075200 | 3.99 ↑ | 2.97 ↓ | - | - | |
| TabHLH93 | TraesCS7A02G543300 | 32.90 ↓ | 10.54 ↓ | 50.51 ↓ | 111.88 ↓ | |
| bZIP | TaHY5 | TraesCS3A02G128900 | 2.07 ↑ | 2.57 ↑ | - | - |
| TabZIP44 | TraesCS6B02G124700 | - | 3.29 ↓ | - | 4.02 ↑ | |
| MADS-box | TaSEP6-7A | TraesCS7A02G122000 | 31.03 ↑ | - | - | - |
| TaSEP6-7B | TraesCS7B02G020800 | 17.46 ↑ | - | - | - | |
| TaSEP6-7D | TraesCS7D02G120500 | 8.30 ↑ | - | - | - | |
| TaMADS32-3A | TraesCS3A02G284400 | 2.18 ↑ | - | - | - | |
| TaMADS32-3B | TraesCS3B02G318300 | 2.36 ↑ | - | - | - | |
| TaMADS32-3D | TraesCS3D02G284200 | 2.61 ↑ | - | - | - | |
| MYB | TaMYB94 | TraesCS2A02G157600 | - | - | 2.53 ↑ | 2.76 ↑ |
| TaMYB30 | TraesCS2B02G183100 | - | - | 2.16 ↑ | 2.42 ↑ | |
| TaMYB44 | TraesCS6B02G201700 | - | - | 3.15 ↑ | 3.63 ↑ | |
| NAC | TaNAC2-5A | TraesCS5A02G468300 | 6.48 ↑ | - | - | - |
| TaNAC2-5B | TraesCS5B02G480900 | 10.83 ↑ | - | - | - | |
| TaNAC8 | TraesCS2D02G336500 | 2.66 ↑ | 1.52 ↑ | 1.05 ↑ | 3.66 ↑ | |
| SBP | TaSPL17-7A | TraesCS7A02G246500 | - | 2.48 ↑ | - | - |
| TaSPL17-7B | TraesCS7B02G144900 | - | 2.51 ↑ | - | - | |
| TaSPL17-7D | TraesCS7D02G245200 | - | 2.83 ↑ | - | - | |
| WRKY | TaWRKY71-6A | TraesCS6A02G146900 | 4.28 ↑ | - | - | - |
| TaWRKY71-6B | TraesCS6B02G175100 | 5.86 ↑ | - | - | - | |
| TaWRKY71-6D | TraesCS6D02G136200 | 5.94 ↑ | - | - | - | |
| TaWRKY46 | TraesCS5B02G183800 | 2.80 ↑ | - | - | - | |
| TaWRKY11 | TraesCS2D02G431000 | - | 2.31 ↑ | - | - | |
| TaWRKY24 | TraesCS3B02G379200 | - | - | - | 2.49 ↑ | |
| NF-Y | TaNF-YB5-3A | TraesCS3A02G457100 | 8.09 ↑ | - | - | - |
| TaNF-YB5-3D | TraesCS3D02G450100 | 24.10 ↑ | - | - | - | |
| TaNF-YC6 | TraesCS5D02G265000 | - | - | - | 2.96 ↑ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, B.; Xie, Y.; Peng, H.; Wang, M.; Zhu, W.; Xu, L.; Cheng, Y.; Wang, Y.; Zeng, J.; Fan, X.; et al. Transcriptome Profiling of Spike Development Reveals Key Genes and Pathways Associated with Early Heading in Wheat–Psathyrstachys huashanica 7Ns Chromosome Addition Line. Plants 2025, 14, 2077. https://doi.org/10.3390/plants14132077
Tan B, Xie Y, Peng H, Wang M, Zhu W, Xu L, Cheng Y, Wang Y, Zeng J, Fan X, et al. Transcriptome Profiling of Spike Development Reveals Key Genes and Pathways Associated with Early Heading in Wheat–Psathyrstachys huashanica 7Ns Chromosome Addition Line. Plants. 2025; 14(13):2077. https://doi.org/10.3390/plants14132077
Chicago/Turabian StyleTan, Binwen, Yangqiu Xie, Hang Peng, Miaomiao Wang, Wei Zhu, Lili Xu, Yiran Cheng, Yi Wang, Jian Zeng, Xing Fan, and et al. 2025. "Transcriptome Profiling of Spike Development Reveals Key Genes and Pathways Associated with Early Heading in Wheat–Psathyrstachys huashanica 7Ns Chromosome Addition Line" Plants 14, no. 13: 2077. https://doi.org/10.3390/plants14132077
APA StyleTan, B., Xie, Y., Peng, H., Wang, M., Zhu, W., Xu, L., Cheng, Y., Wang, Y., Zeng, J., Fan, X., Sha, L., Zhang, H., Qin, P., Zhou, Y., Wu, D., Li, Y., & Kang, H. (2025). Transcriptome Profiling of Spike Development Reveals Key Genes and Pathways Associated with Early Heading in Wheat–Psathyrstachys huashanica 7Ns Chromosome Addition Line. Plants, 14(13), 2077. https://doi.org/10.3390/plants14132077