


Integrated Transcriptome and Proteome Analysis Reveals That Cell Wall Activity Affects Phelipanche aegyptiaca Parasitism

 and
and
Abstract
1. Introduction
2. Materials and Method
2.1. Plant Materials and Growing Conditions
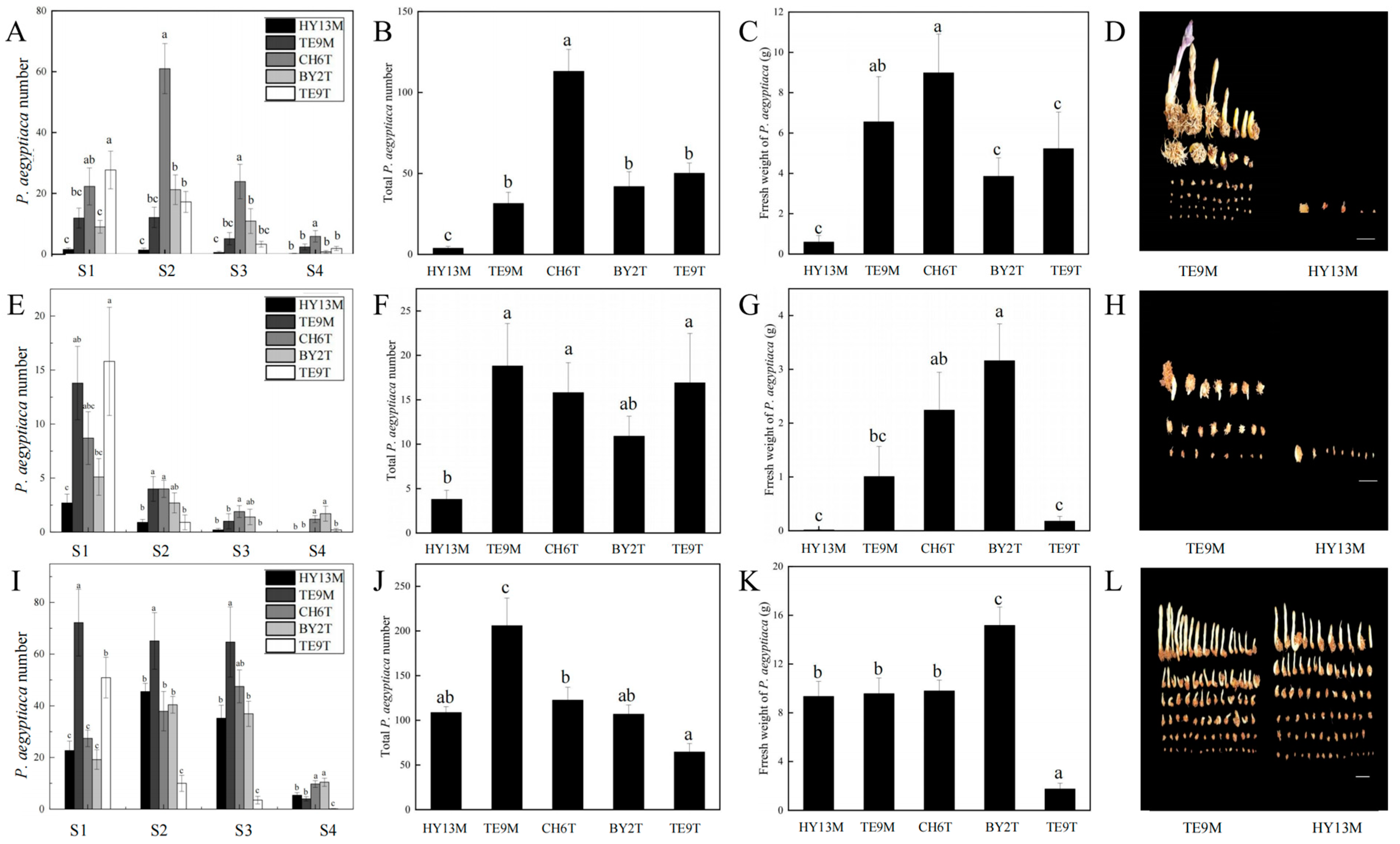
2.2. Determination of the Parasitic Ability of Different P. aegyptiaca Populations
2.3. Transcriptome Analysis of HY13M and TE9M Parasitizing C. lanatus
2.3.1. RNA Extraction, cDNA Library Construction, and Sequencing
2.3.2. Quantitative Real-Time PCR (qRT-PCR)
2.4. Proteome Analysis of HY13M and TE9M Parasitizing C. lanatus
2.4.1. Protein Extraction and Digestion
2.4.2. Liquid Chromatography–Mass Spectrometry Analysis
2.4.3. LC-MS Data Analysis
2.4.4. Quantitative Data Analysis
2.4.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
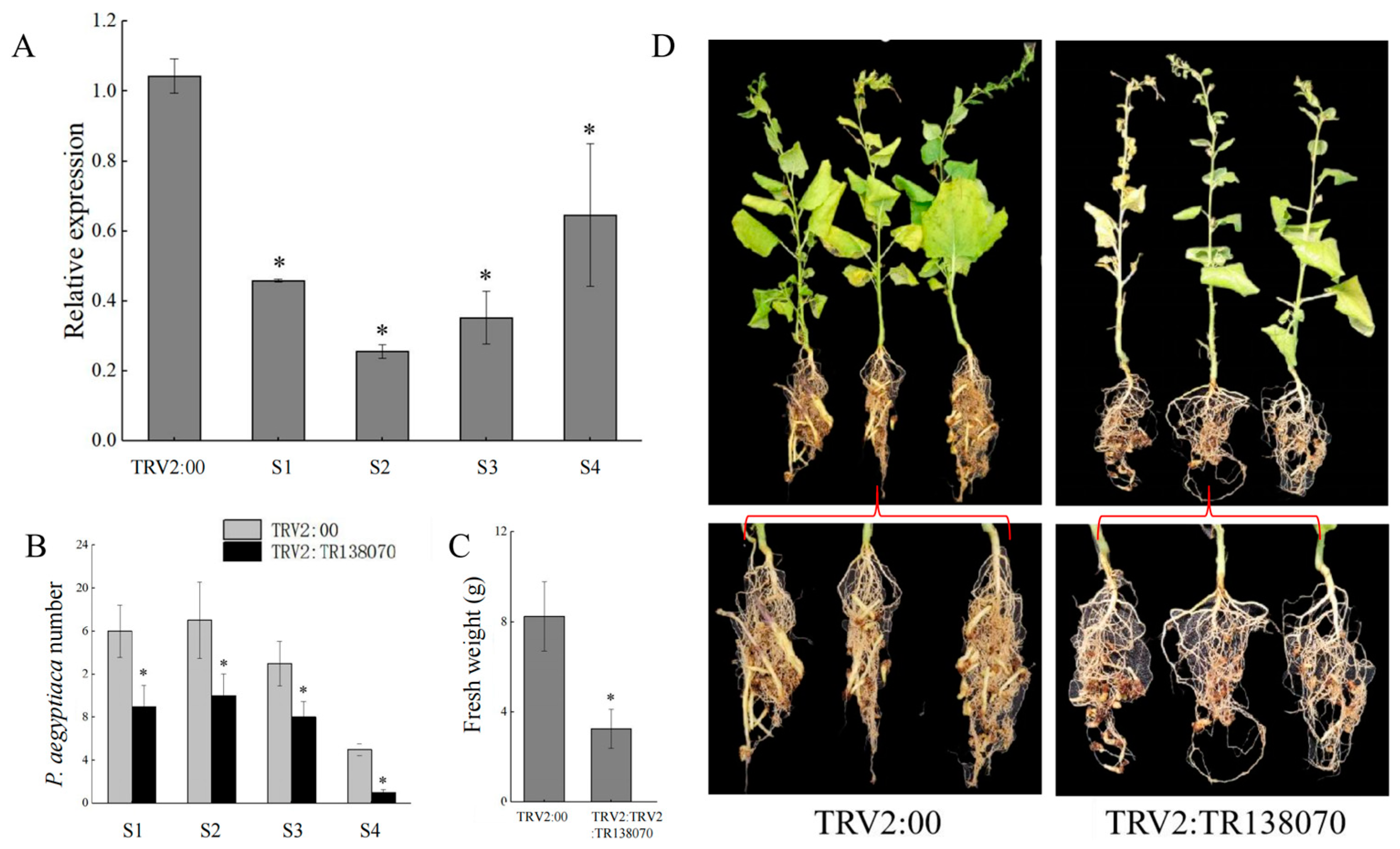
2.5. Verification of the Effect of the Pectinesterase Gene on Parasitism by Means of HIGS
3. Results
3.1. Parasitic Ability of Different P. aegyptiaca Populations
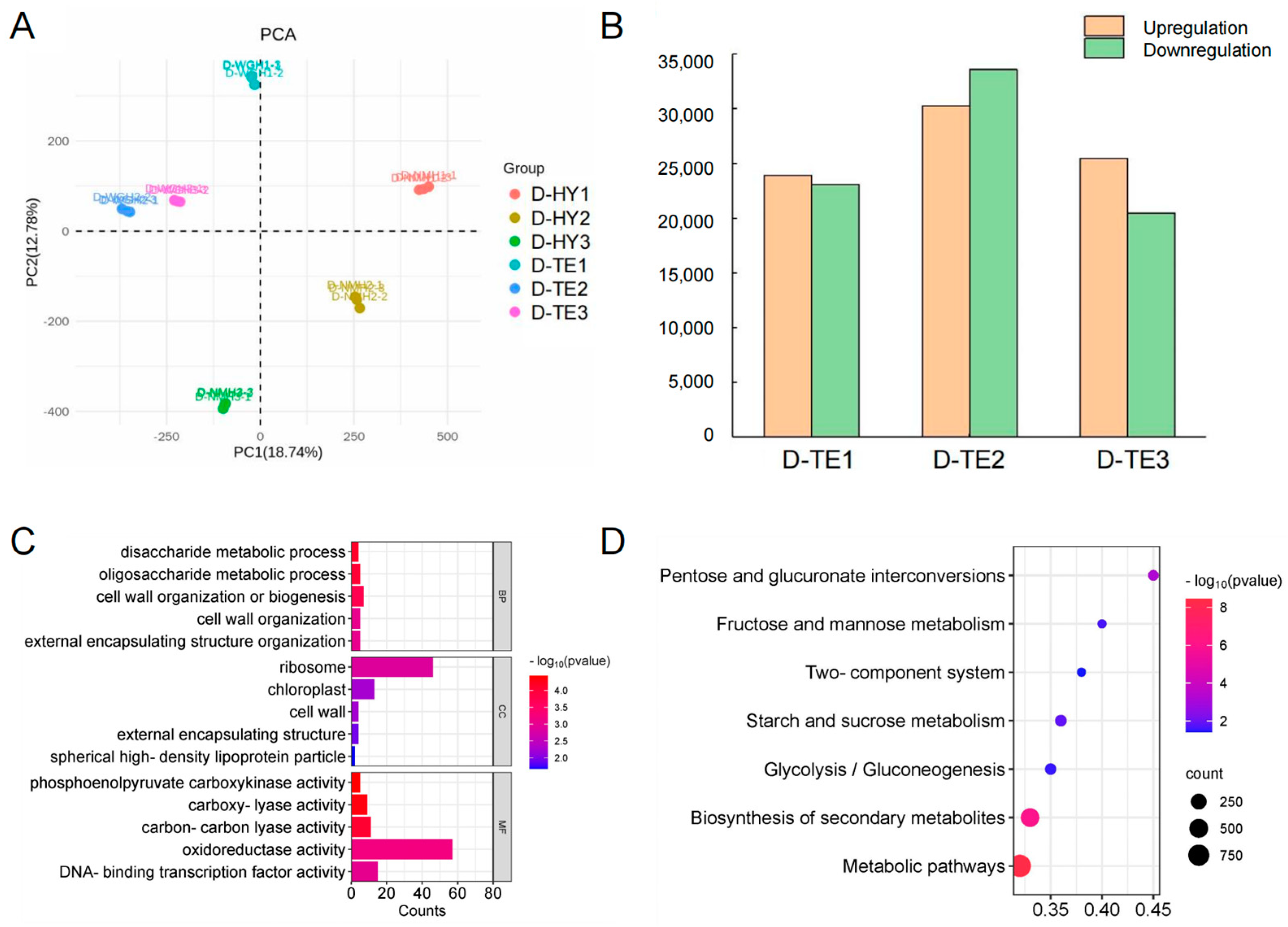
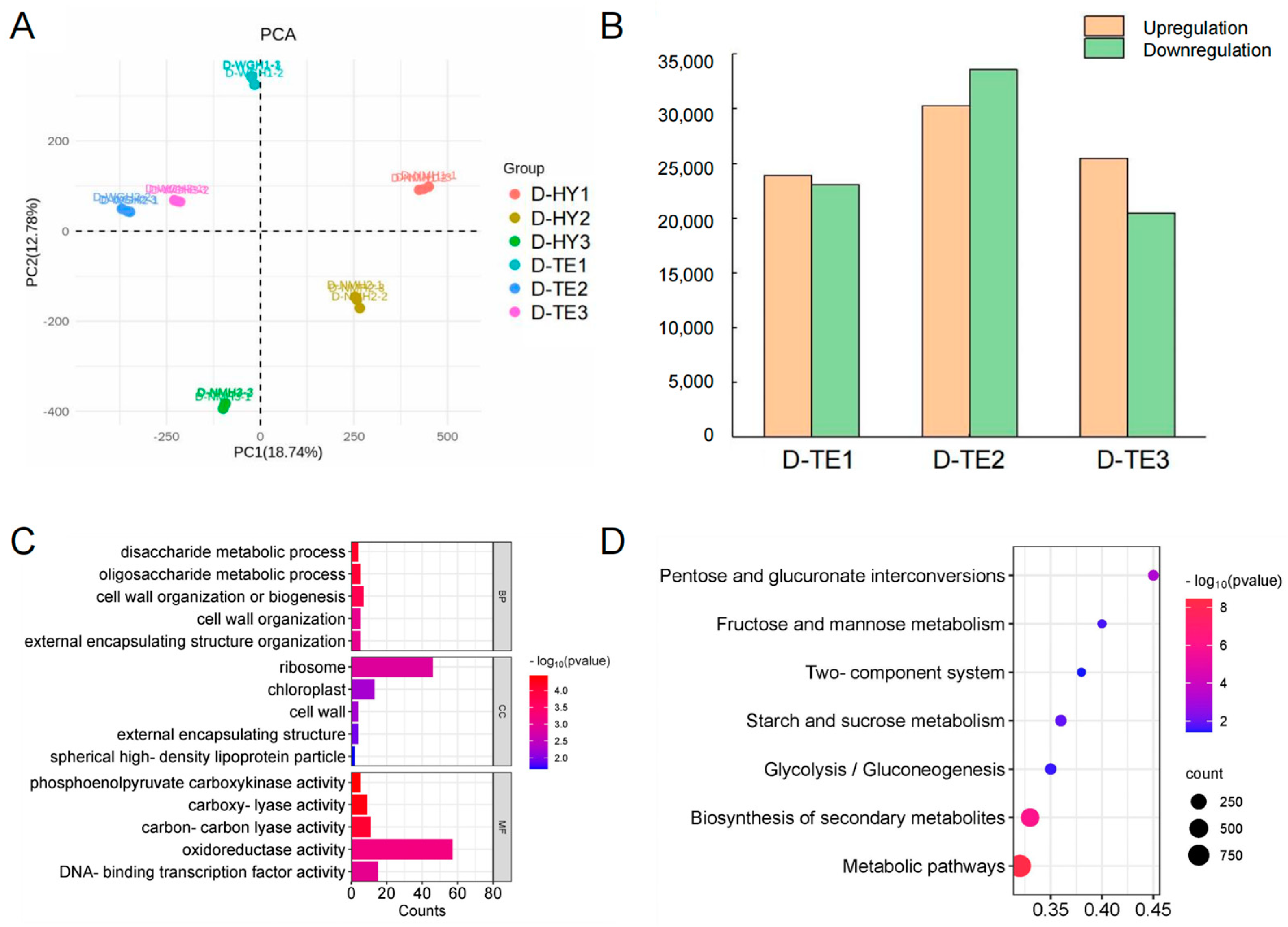
3.2. Transcriptomic Analysis and Function Analysis of DEGs
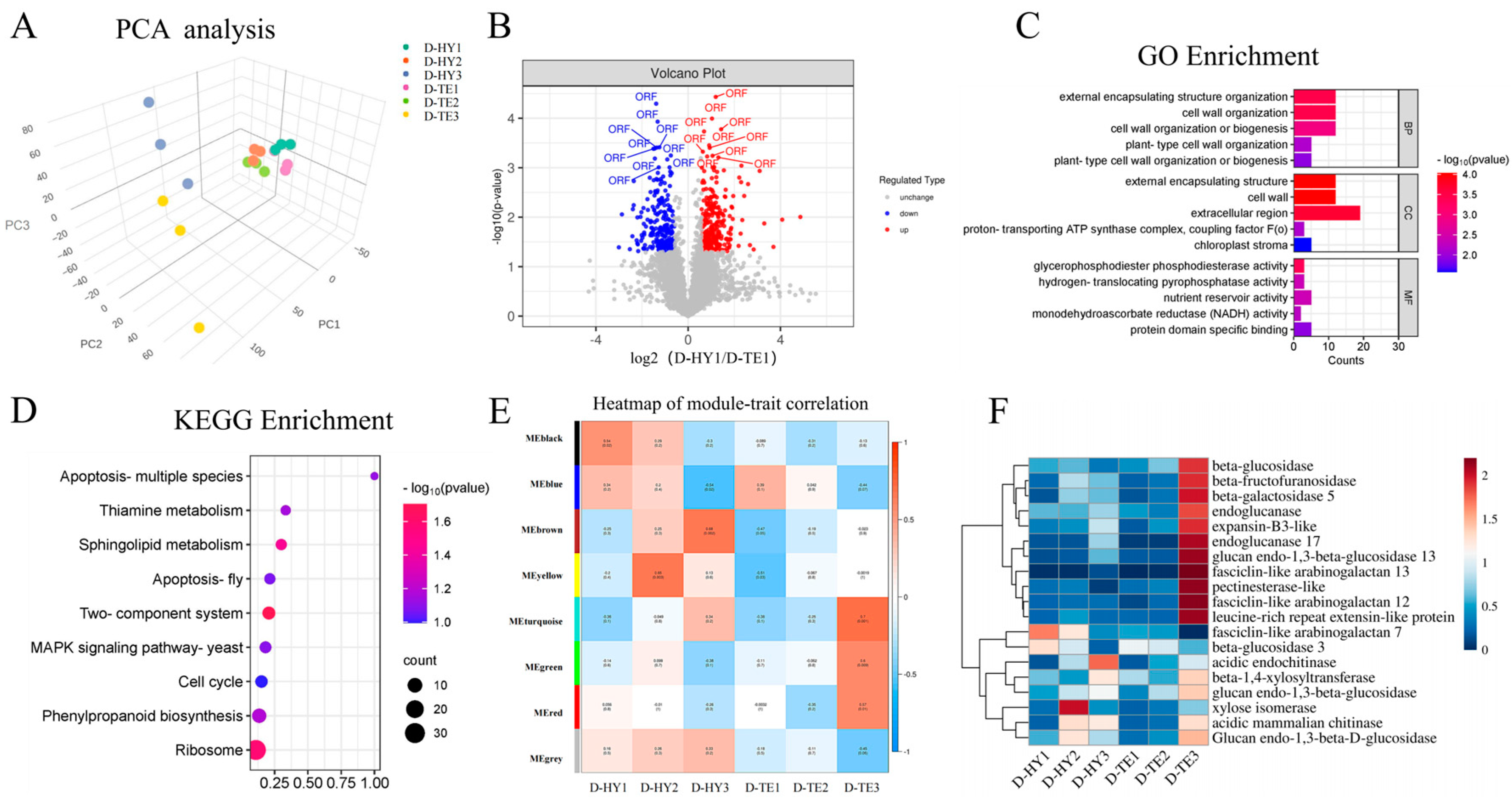
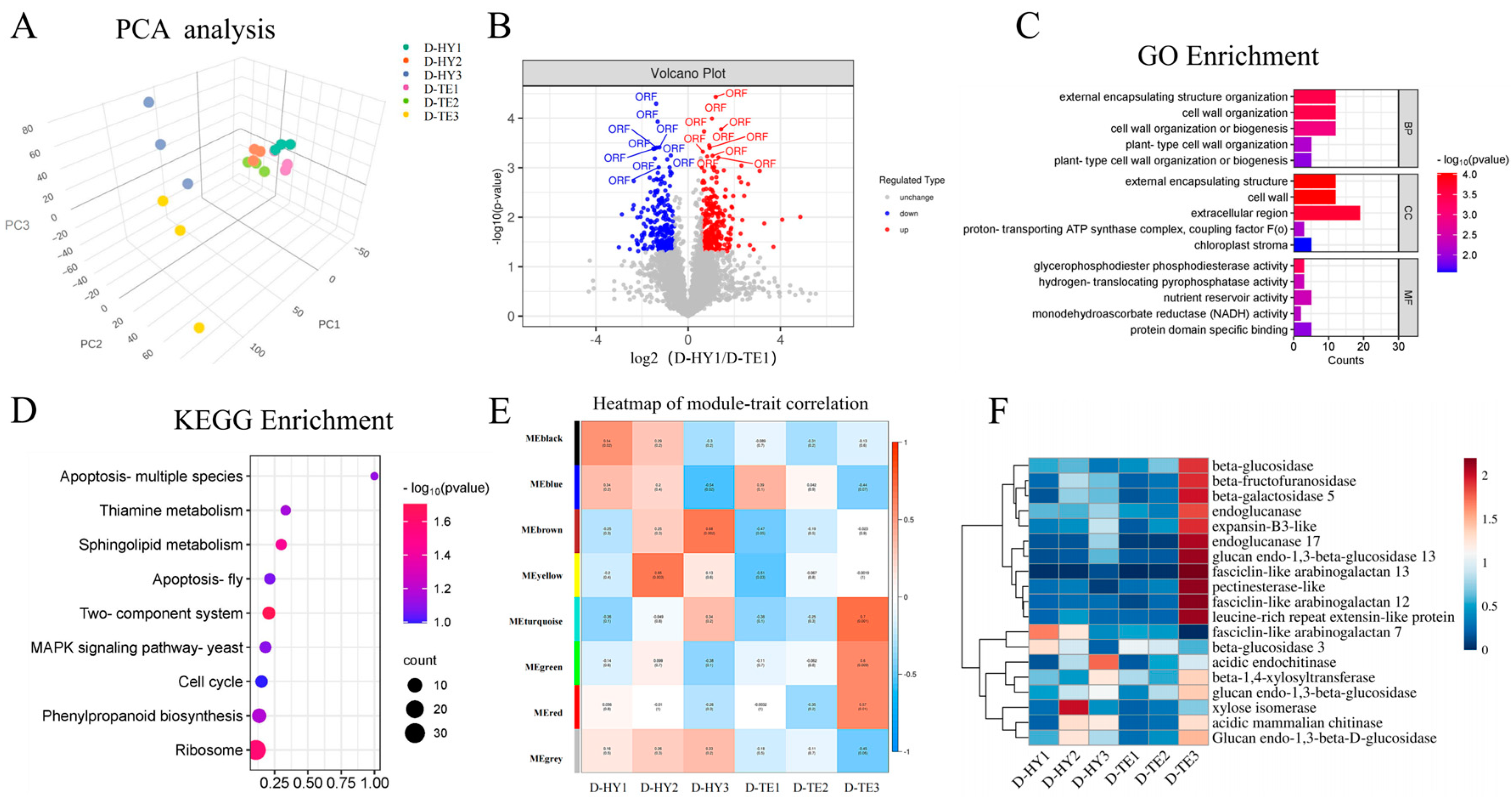
3.3. Proteomic Analysis of DEPs and Function Analysis
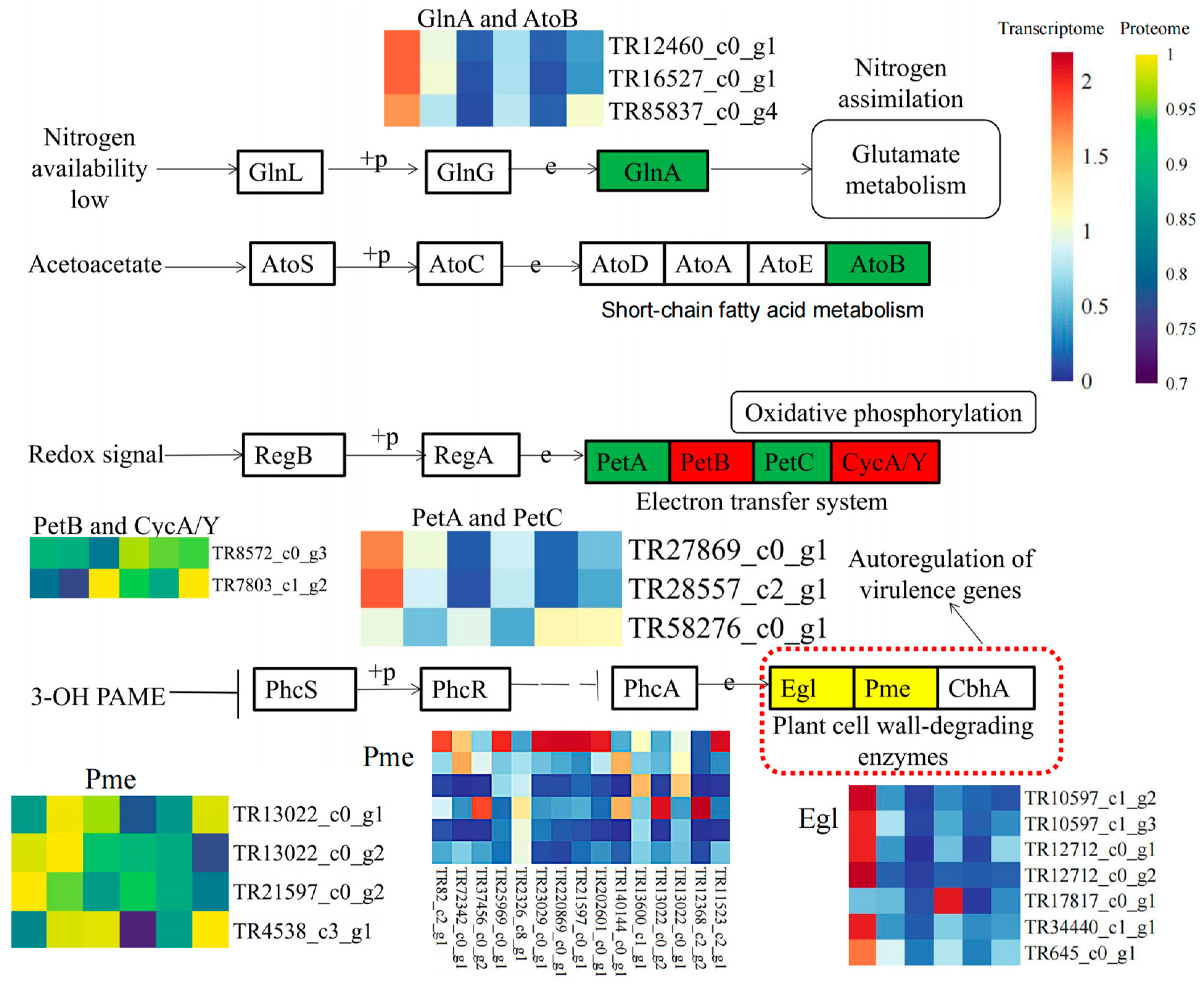
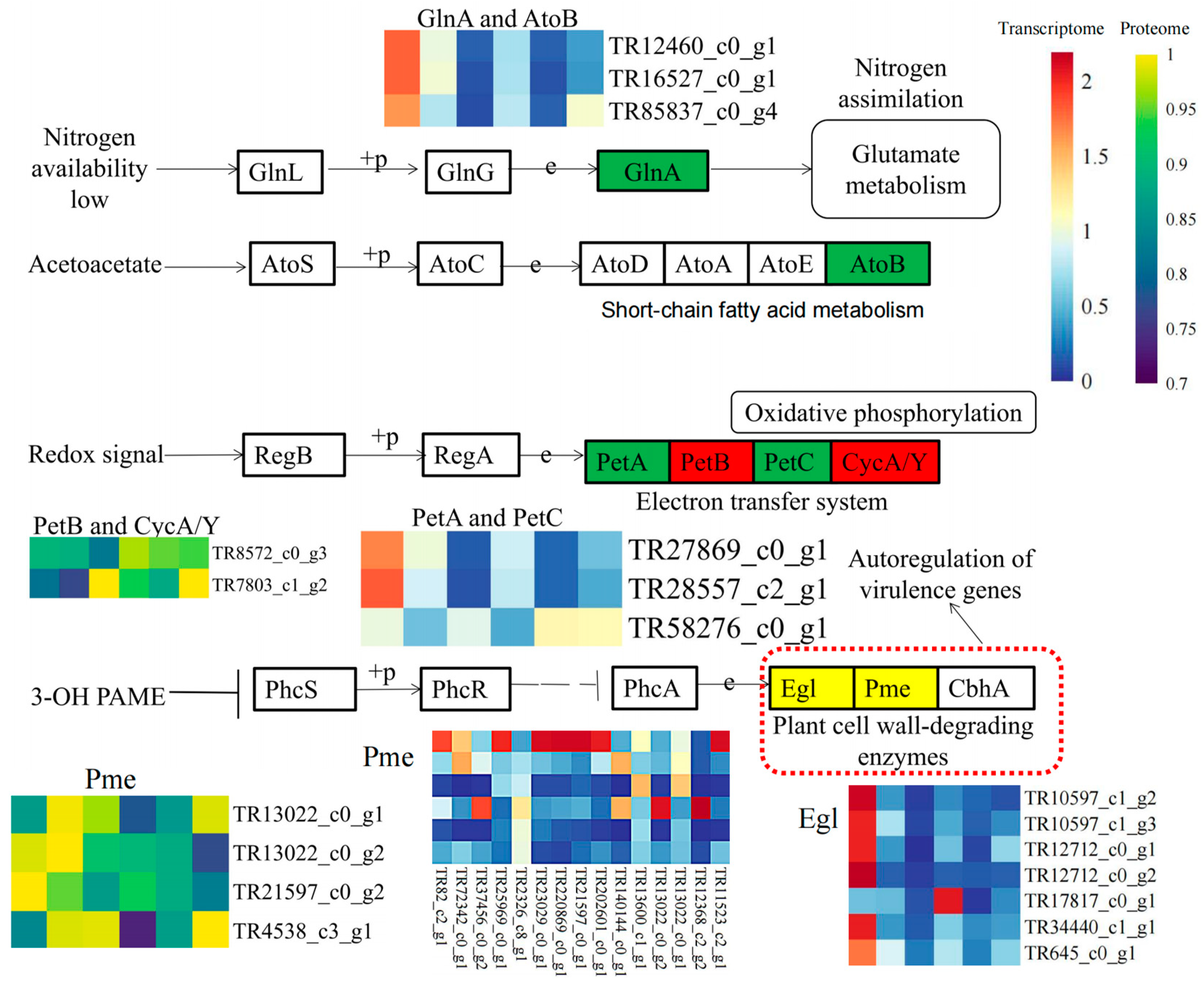
3.4. Combined Transcriptome and Proteome Analysis Revealed the Involvement of Cell Wall Metabolism Enzymes in P. aegyptiaca Parasitism
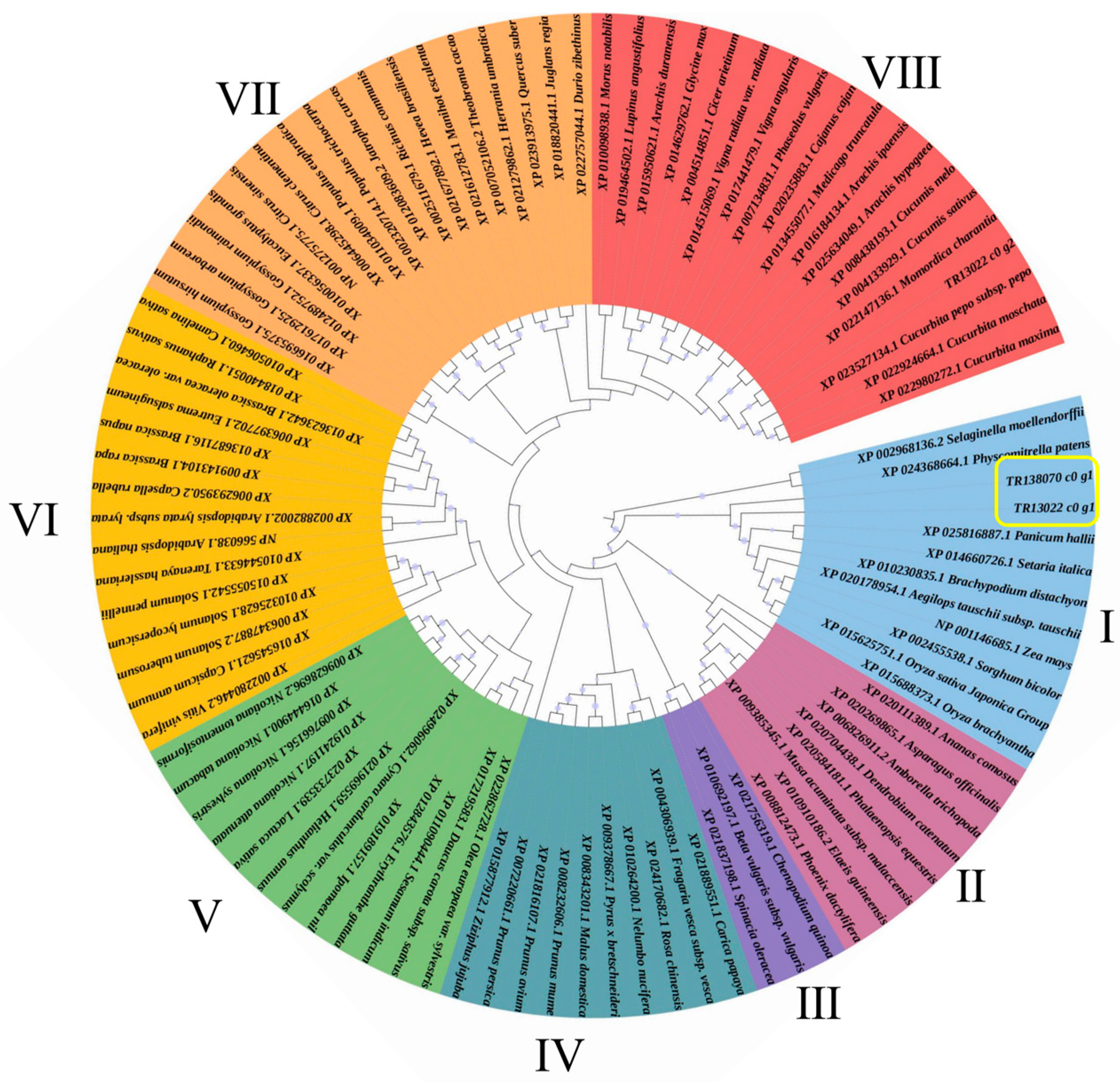
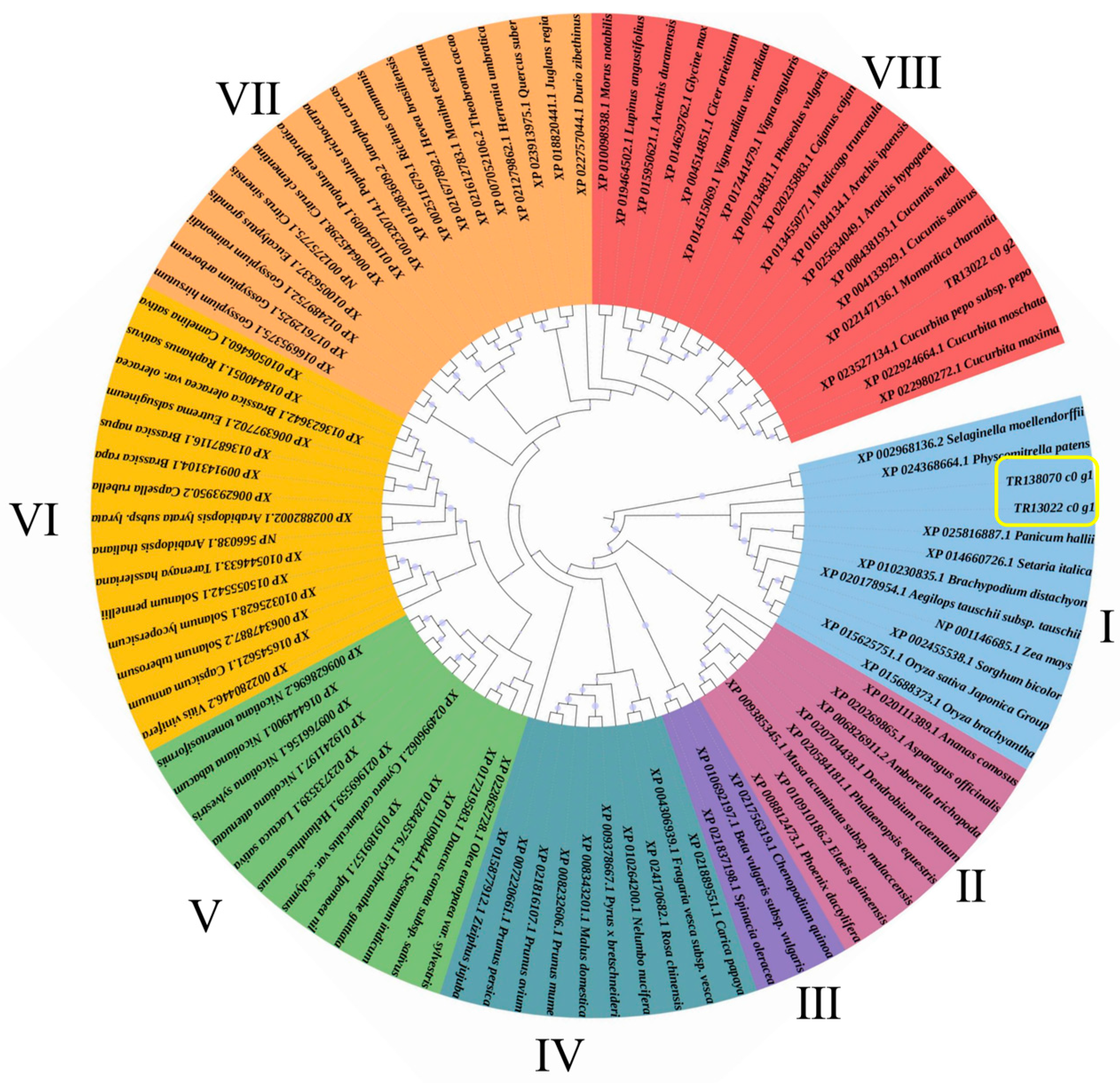
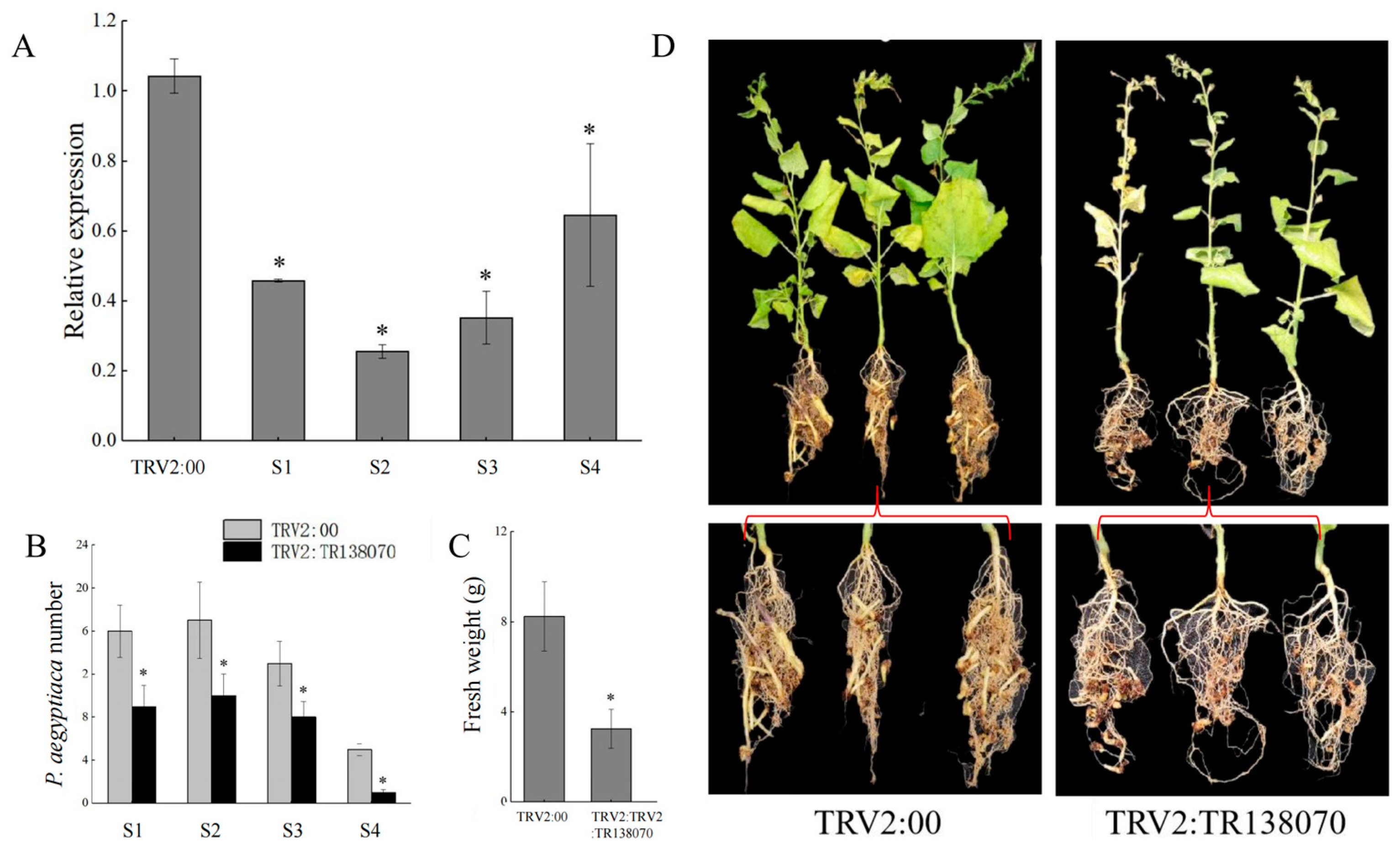
3.5. HIGS Confirmed Pectinesterase-like Gene Involved in P. aegyptiaca Parasitism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wallach, A.; Achdari, G.; Eizenberg, H. Good News for Cabbageheads: Controlling Phelipanche aegyptiaca infestation under hydroponic and field conditions. Plants 2022, 11, 1107. [Google Scholar] [CrossRef]
- Clarke, C.R.; Timko, M.P.; Yoder, J.I.; Axtell, M.J.; Westwood, J.H. Molecular dialog between parasitic plants and their hosts. Annu. Rev. Phytopathol. 2019, 57, 279–299. [Google Scholar] [CrossRef]
- Yao, Z.; Tian, F.; Cao, X.; Xu, Y.; Chen, M.; Xiang, B.; Zhao, S. Global Transcriptomic analysis reveals the mechanism of Phelipanche aegyptiaca seed germination. Int. J. Mol. Sci. 2016, 17, 1139. [Google Scholar] [CrossRef]
- Bernhard, R.H.; Jensen, J.E.; Andreasen, C. Prediction of yield loss caused by Orobanche spp. in carrot and pea crops based on the soil seedbank. Weed Res. 2015, 38, 191–197. [Google Scholar] [CrossRef]
- Wiseglass, G.; Pri-Tal, O.; Mosquna, A. ABA signaling components in Phelipanche aegyptiaca. Sci. Rep. 2019, 9, 6476. [Google Scholar] [CrossRef]
- Eizenberg, H.; Goldwasser, Y. Control of egyptian broomrape in processing tomato: A summary of 20 years of research and successful implementation. Plant Dis. 2018, 102, 1477–1488. [Google Scholar] [CrossRef]
- Yang, N.; Wang, W.; Cao, C. Analysis on status and development trend of melon in China. Zhongguo Gua Cai 2019, 32, 50–54. (In Chinese) [Google Scholar]
- Zhang, Y.; Wu, M. Genetic diversity of melon landraces Cucumis melo L.) in the Xinjiang Uygur Autonomous region on the basis of simple sequence repeat marker. Zhongguo Gua Cai 2019, 32, 168–169. (In Chinese) [Google Scholar] [CrossRef]
- Zhang, X.; Yao, Z.; Zhao, S. Distribution, harmfulness and its assessment of Orobanche aegyptiaca in Xinjiang province. Plant Quar. 2012, 26, 31–33. (In Chinese) [Google Scholar]
- Babaei, S.; Alizadeh, H.; Jahansouz, M.R.; Mashhadi, H.R. Management of phelipanche aegyptiaca pomel. using trap crops in rotation on tomato (solanum lycopersicom L.). Aust. J. Crop. Sci. 2010, 4, 437–442. [Google Scholar]
- Ross, K.C.; Colquhoun, J.B.; Mallory-Smith, C.A. Small broomrape (Orobanche minor) germination and early development in response to plant species. Weed Sci. 2004, 52, 260–266. [Google Scholar] [CrossRef]
- Rubiales, D.; Ferna´ndez-Aparicio, M.; Wegmann, K.; Joel, D.M. Revisiting strategies for reducing the seedbank of Orobanche and Phelipanche spp. Weed Res. 2009, 49, 23–33. [Google Scholar] [CrossRef]
- Kannan, C.; Aditi, P.; Zwanenburg, B. Quenching the action of germination stimulants using borax and thiourea, a new method for controlling parasitic weeds: A proof of concept. Crop. Prot. 2015, 70, 92–98. [Google Scholar] [CrossRef]
- Pérez-de-Luque, A.; Eizenberg, H.; Grenz, J.H.; Sillero, J.C.; Ávila, C.; Sauerborn, J.; Rubiales, D. Broomrape management in faba bean. Field Crop. Res. 2010, 115, 319–328. [Google Scholar] [CrossRef]
- Dor, E.; Galili, S.; Smirnov, E.; Hacham, Y.; Amir, R.; Hershenhorn, J. The effects of herbicides targeting aromatic and branched chain amino acid biosynthesis support the presence of functional pathways in broomrape. Front. Plant Sci. 2017, 8, 707. [Google Scholar] [CrossRef]
- Cao, X.; Xiao, L.; Zhang, L.; Chen, M.; Bian, P.; Ma, Q.; Chen, S.; He, Q.; Ma, X.; Yao, Z.; et al. Phenotypic and histological analyses on the resistance of melon to Phelipanche aegyptiaca. Front. Plant Sci. 2023, 14, 1070319. [Google Scholar] [CrossRef]
- Fernández-Aparicio, M.; Reboud, X.; Gibot-Leclerc, S. Broomrape weeds. Underground mechanisms of parasitism and associated strategies for their control: A review. Front. Plant Sci. 2016, 7, 135. [Google Scholar] [CrossRef]
- Nelson, D.C. The mechanism of host-induced germination in root parasitic plants. Plant Physiol. 2021, 185, 1353–1373. [Google Scholar] [CrossRef]
- Velasco, L.; Goffman, F.D.; Pujadas-Salvà, A.J. Fatty acids and tocochromanols in seeds of Orobanche. Phytochemistry 2000, 54, 295–300. [Google Scholar] [CrossRef]
- Rubiales, D.; Pérez-de-Luque, A.; Fernández-Aparico, M.; Sillero, J.C.; Román, B.; Kharrat, M.; Khalil, S.; Joel, D.M.; Riches, C. Screening techniques and sources of resistance against parasitic weeds in grain legumes. Euphytica 2006, 147, 187–199. [Google Scholar] [CrossRef]
- Pérez-de-Luque, A.; Fondevilla, S.; Pérez-Vich, B.; Aly, R.; Thoiron, S.; Simier, P.; Castillejo, M.A.; Fernander-Martinez, J.M.; Jorrin, J.; Rubiales, D.; et al. Understanding Orobanche and Phelipanche-host plant interactions and developing resistance. Weed Res. 2009, 49, 8–22. [Google Scholar] [CrossRef]
- Yang, Z.; Wafula, E.K.; Honaas, L.A.; Zhang, H.; Das, M.; Fernandez-Aparicio, M.; Huang, K.; Bandaranayake, P.C.; Wu, B.; Der, J.P.; et al. Comparative transcriptome analyses reveal core parasitism genes and suggest gene duplication and repurposing as sources of structural novelty. Mol. Biol. Evol. 2015, 32, 767–790. [Google Scholar] [CrossRef]
- Duca, M.; Boicu, A.; Clapco, S.; Port, A. Comparative analysis of two Orobanche cumana wallr. accessions with a different virulence. Acta Physiol. Plant. 2020, 42, 170. [Google Scholar] [CrossRef]
- Véronési, C.; Bonnin, E.; Benharrat, H.; Fer, A.; Thalouarn, P. Opinion: Are pectinolytic activities of orobanche cumana seedlings related to virulence towards sunflower? Isr. J. Plant Sci. 2005, 53, 19–27. [Google Scholar] [CrossRef]
- Qiu, S.; Bradley, J.M.; Zhang, P.; Chaudhuri, R.; Blaxter, M.; Butlin, R.K.; Scholes, J.D. Genome-enabled discovery of candidate virulence loci in Striga hermonthica, a devastating parasite of African cereal crops. New Phytol. 2022, 236, 622–628. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, Y.; Wafula, E.K.; Honaas, L.A.; Ralph, P.E.; Jones, S.; Clarke, C.R.; Liu, S.; Su, C.; Zhang, H.; et al. Horizontal gene transfer is more frequent with increased heterotrophy and contributes to parasite adaptation. Proc. Natl. Acad. Sci. USA 2016, 113, E7010–E7019. [Google Scholar] [CrossRef]
- Veronesi, C.; Bonnin, E.; Calvez, S.; Thalouarn, P.; Simier, P. Activity of secreted cell wall-modifying enzymes and expression of peroxidase-encoding gene following germination of Orobanche ramosa. Biol. Plant 2007, 51, 391–394. [Google Scholar] [CrossRef]
- Yang, C.; Fu, F.; Zhang, N.; Wang, J.; Hu, L.; Islam, F.; Bai, Q.; Yun, X.; Zhou, W. Transcriptional profiling of underground interaction of two contrasting sunflower cultivars with the root parasitic weed orobanche cumana. Plant Soil 2020, 450, 303–321. [Google Scholar] [CrossRef]
- Brotman, Y.; Briff, E.; Viterbo, A.; Chet, I. Role of swollenin, an expansin-like protein from Trichoderma, in plant root colonization. Plant Physiol. 2008, 147, 779–789. [Google Scholar] [CrossRef]
- Qin, L.; Kudla, U.; Roze, E.H.; Goverse, A.; Popeijus, H.; Nieuwland, J.; Overmars, H.; Jones, J.T.; Schots, A.; Smant, G.; et al. Plant degradation: A nematode expansin acting on plants. Nature 2004, 427, 30. [Google Scholar] [CrossRef]
- Honaas, L.A.; Wafula, E.K.; Yang, Z.; Der, J.P.; Wickett, N.J.; Altman, N.S.; Taylor, C.G.; Yoder, J.I.; Timko, M.P.; Westwood, J.H.; et al. Functional genomics of a generalist parasitic plant: Laser microdissection of host-parasite interface reveals host-specific patterns of parasite gene expression. BMC. Plant Biol. 2013, 13, 9. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Fan, K.T.; Wang, K.H.; Chang, W.H.; Yang, J.C.; Yeh, C.F.; Cheng, K.T.; Hung, S.C.; Chen, Y.R. Application of data-independent acquisition approach to study the proteome change from early to later phases of tomato pathogenesis responses. Int. J. Mol. Sci. 2019, 20, 863. [Google Scholar] [CrossRef]
- Mertins, P.; Tang, L.C.; Krug, K.; Clark, D.J.; Gritsenko, M.A.; Chen, L.; Clauser, K.R.; Clauss, T.R.; Shah, P.; Gillette, M.A.; et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 2018, 13, 1632–1661. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, W.Y.; Tseng, M.C.; Chen, Y.R. Tunnel frit: A nonmetallic in-capillary frit for nanoflow ultra high-performance liquid chromatography-mass spectrometryapplications. Anal. Chem. 2012, 84, 297–303. [Google Scholar] [CrossRef]
- Scheltema, R.A.; Hauschild, J.P.; Lange, O.; Hornburg, D.; Denisov, E.; Damoc, E.; Kuehn, A.; Makarov, A.; Mann, M. The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol. Cell. Proteom. 2014, 13, 3698–3708. [Google Scholar] [CrossRef]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Xuan, Y.; Sondermann, J.; Schmidt, M.; Gomez-Varela, D.; Reiter, L. Optimization of experimental parameters in data-independent mass spectrometry significantly increases depth and reproducibility of results. Mol. Cell. Proteom. 2017, 16, 2296–2309. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Eigengene networks for studying the relationships between co-expression modules. BMC. Syst. Biol. 2007, 1, 54. [Google Scholar] [CrossRef]
- Yang, J.; Wang, L.; Ji, X.; Feng, Y.; Li, X.; Zou, C.; Xu, J.; Ren, Y.; Mi, Q.; Wu, J.; et al. Genomic and proteomic analyses of the fungus arthrobotrys oligospora provide insights into nematode-trap formation. PLoS Pathog. 2011, 7, e1002179. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, S.; Lin, H.; Lu, W.; Wang, H.; Chen, Y.; Lin, Y.; Fan, Z. The role of cell wall polysaccharides disassembly in Lasiodiplodia theobromae-induced disease occurrence and softening of fresh longan fruit. Food Chem. 2021, 351, 129294. [Google Scholar] [CrossRef]
- Li, W.; Jiang, Y.; Hu, C.; Liu, G.; Li, Y.; Wang, S. Identification, pathogenic mechanism and control of Rhizopus oryzae causing postharvest fruit rot in pumpkin. Postharvest. Biol. Technol. 2023, 204, 112460. [Google Scholar] [CrossRef]
- Nishimura, M. Cell wall reorganization during infection in fungal plant pathogens. Physiol. Mol. Plant Pathol. 2016, 95, 14–19. [Google Scholar] [CrossRef]
- Mu, B.; Teng, Z.; Tang, R.; Lu, M.; Chen, J.; Xu, X.; Wen, Y.Q. An effector of Erysiphe necator translocates to chloroplasts and plasma membrane to suppress host immunity in grapevine. Hortic. Res. 2023, 10, uhad163. [Google Scholar] [CrossRef]
- Prado, S.B.R.; Beukema, M.; Jermendi, E.; Schols, H.A.; de Vos, P.; Fabi, J.P. Pectin Interaction with immune receptors is modulated by ripening process in papayas. Sci. Rep. 2020, 10, 1690. [Google Scholar] [CrossRef]
- Frías, M.; González, M.; González, C.; Brito, N. BcIEB1, a Botrytis cinerea secreted protein, elicits a defense response in plants. Plant Sci. 2016, 250, 115–124. [Google Scholar] [CrossRef]
- Ashwin, N.M.R.; Barnabas, L.; Ramesh Sundar, A.; Malathi, P.; Viswanathan, R.; Masi, A.; Agrawal, G.K.; Rakwal, R. CfPDIP1, a novel secreted protein of Colletotrichum falcatum, elicits defense responses in sugarcane and triggers hypersensitive response in tobacco. Appl. Microbiol. Biotechnol. 2018, 102, 6001–6021. [Google Scholar] [CrossRef]
- Cosgrove, D.J. Loosening of plant cell walls by expansins. Nature 2020, 407, 321–326. [Google Scholar] [CrossRef]
- Vieira, P.; Nemchinov, L.G. An Expansin-Like candidate effector protein from Pratylenchus penetrans modulates immune responses in Nicotiana benthamiana. Phytopathology 2020, 110, 684–693. [Google Scholar] [CrossRef]
- Culbertson, A.T.; Chou, Y.H.; Smith, A.L.; Young, Z.T.; Tietze, A.A.; Cottaz, S.; Fauré, R.; Zabotina, O.A. Enzymatic activity of xyloglucan xylosyltransferase 5. Plant Physiol. 2016, 171, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Yan, J.Y.; Ren, J.Y.; Sun, L.; Xu, C.; Li, G.X.; Ding, Z.J.; Zheng, S.J. A WRKY transcription factor confers aluminum tolerance via regulation of cell wall modifying genes. Integr. Plant. Biol. 2020, 62, 1176–1192. [Google Scholar] [CrossRef] [PubMed]
- Wakatake, T.; Yoshida, S.; Shirasu, K. Induced cell fate transitions at multiple cell layers configure haustorium development in parasitic plants. Development 2018, 145, dev164848. [Google Scholar] [CrossRef]
- Olivier, A.; Benhamou, N.; Leroux, G. Cell surface interactions between sorghum roots and the parasitic weed Striga hermonthica: Cytochemical aspects of cellulose distribution in resistant and susceptible host tissues. Can. J. Bot. 1991, 69, 1679–1690. [Google Scholar] [CrossRef]
- Losner-goshen, D. Pectolytic activity by the haustorium of the parasitic plant Orobanche L. (Orobanchaceae) in host roots. Ann. Bot. 1998, 2, 319–326. [Google Scholar] [CrossRef]
- Kurotani, K.I.; Wakatake, T.; Ichihashi, Y.; Okayasu, K.; Sawai, Y.; Ogawa, S.; Cui, S.; Suzuki, T.; Shirasu, K.; Notaguchi, M. Host-parasite tissue adhesion by a secreted type of β-1,4-glucanase in the parasitic plant Phtheirospermum japonicum. Commun. Biol. 2020, 3, 407. [Google Scholar] [CrossRef]
- Ranjan, A.; Ichihashi, Y.; Farhi, M.; Zumstein, K.; Townsley, B.; David-Schwartz, R.; Sinha, N.R. De novo assembly and characterization of the transcriptome of the parasitic weed dodder identifies genes associated with plant parasitism. Plant Physiol. 2014, 166, 1186–1199. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population Name | Collection Time | Collection Location | Host | Longitude, Latitude |
|---|---|---|---|---|
| TE9M | 2017 | 4th company, 163rd regiment, 9th division, Emin County, Tacheng, Xinjiang | Melon | 82°55′10″ E, 46°47′59″ N, |
| CH6T | 2017 | 3rd company, Junhu farm, 6th division, Hutubi County, Changji Hui Autonomous Prefecture, Xinjiang | Processing tomato | 87°0′19″ E, 44°1′27″ N, |
| BY2T | 2017 | 7th company, 21st regiment, 2nd division, Yanqi County, Bayingolin Mongolian Autonomous Prefecture, Xinjiang | Processing tomato | 86°18′54″ E, 42°9′36″ N, |
| TE9T | 2017 | 4th company, 163rd regiment, 9th division, Emin County, Tacheng, Xinjiang | Processing tomato | 82°55′10″ E, 46°47′59″ N, |
| HY13M | 2017 | Naomaohu farm, 13th division, Yiwu County Hami, Xinjiang | Melon | 94°58′56″ E, 43°55′52″ N, |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.; Zhang, L.; Yao, Z.; Cao, X.; Ma, Q.; Chen, S.; Zhang, X.; Zhao, S. Integrated Transcriptome and Proteome Analysis Reveals That Cell Wall Activity Affects Phelipanche aegyptiaca Parasitism. Plants 2024, 13, 869. https://doi.org/10.3390/plants13060869
Chen M, Zhang L, Yao Z, Cao X, Ma Q, Chen S, Zhang X, Zhao S. Integrated Transcriptome and Proteome Analysis Reveals That Cell Wall Activity Affects Phelipanche aegyptiaca Parasitism. Plants. 2024; 13(6):869. https://doi.org/10.3390/plants13060869
Chicago/Turabian StyleChen, Meixiu, Lu Zhang, Zhaoqun Yao, Xiaolei Cao, Qianqian Ma, Siyu Chen, Xuekun Zhang, and Sifeng Zhao. 2024. "Integrated Transcriptome and Proteome Analysis Reveals That Cell Wall Activity Affects Phelipanche aegyptiaca Parasitism" Plants 13, no. 6: 869. https://doi.org/10.3390/plants13060869
APA StyleChen, M., Zhang, L., Yao, Z., Cao, X., Ma, Q., Chen, S., Zhang, X., & Zhao, S. (2024). Integrated Transcriptome and Proteome Analysis Reveals That Cell Wall Activity Affects Phelipanche aegyptiaca Parasitism. Plants, 13(6), 869. https://doi.org/10.3390/plants13060869