
Polyploids of Brassicaceae: Genomic Insights and Assembly Strategies


Abstract
1. Introduction
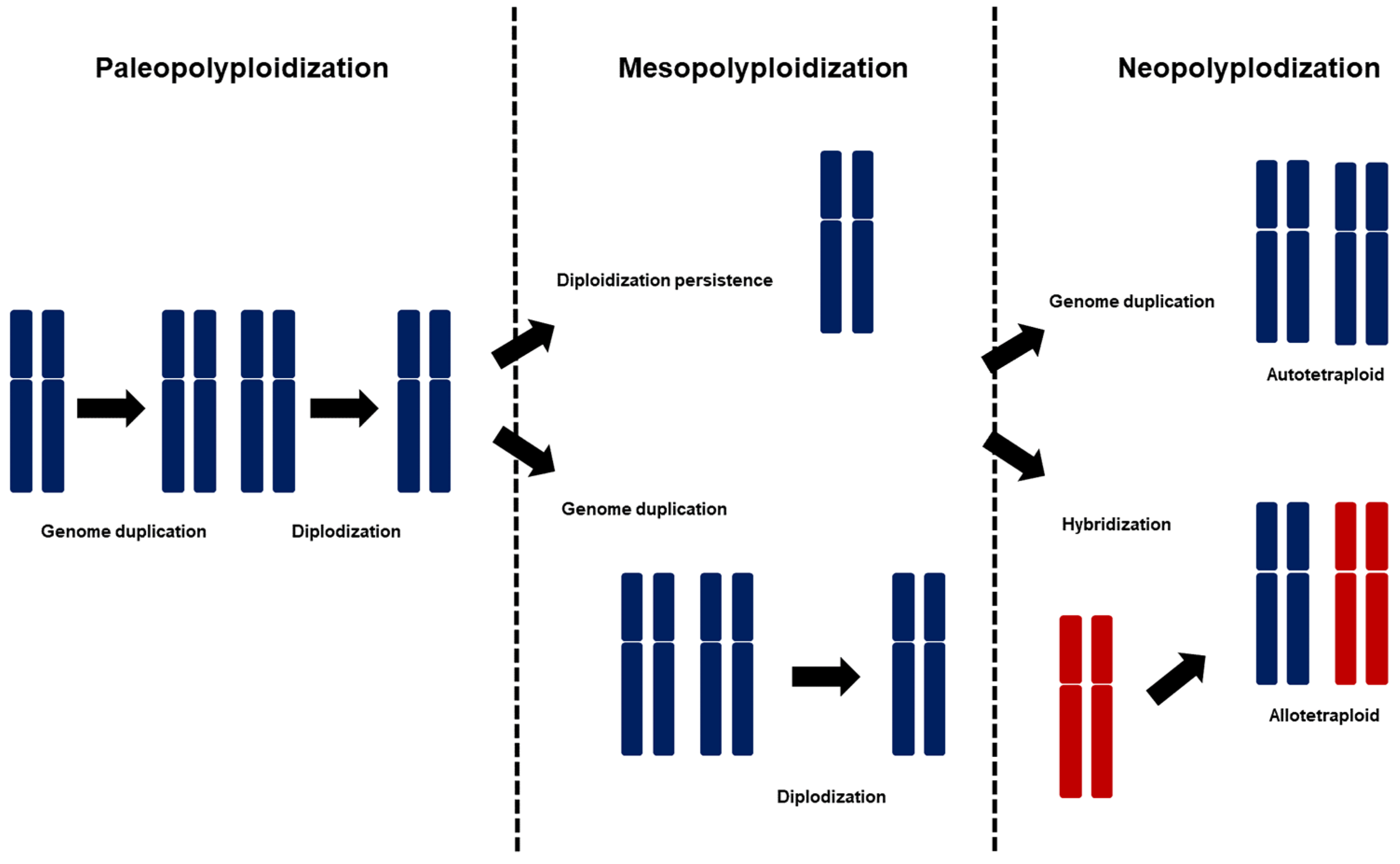
2. Polyploidization of the Brassicaceae Family
2.1. Diversification of Brassicaceae
2.2. Evolution of the Biosynthetic Pathways to Produce GSLs
2.3. Polyploidy and Its Role in the Evolution and Domestication of Brassica Species for Agricultural Trait Development
3. Challenges and Solutions in Sequencing and Assembling Polyploidy Plant Genomes
3.1. Assessing Ploidy Levels in Plants
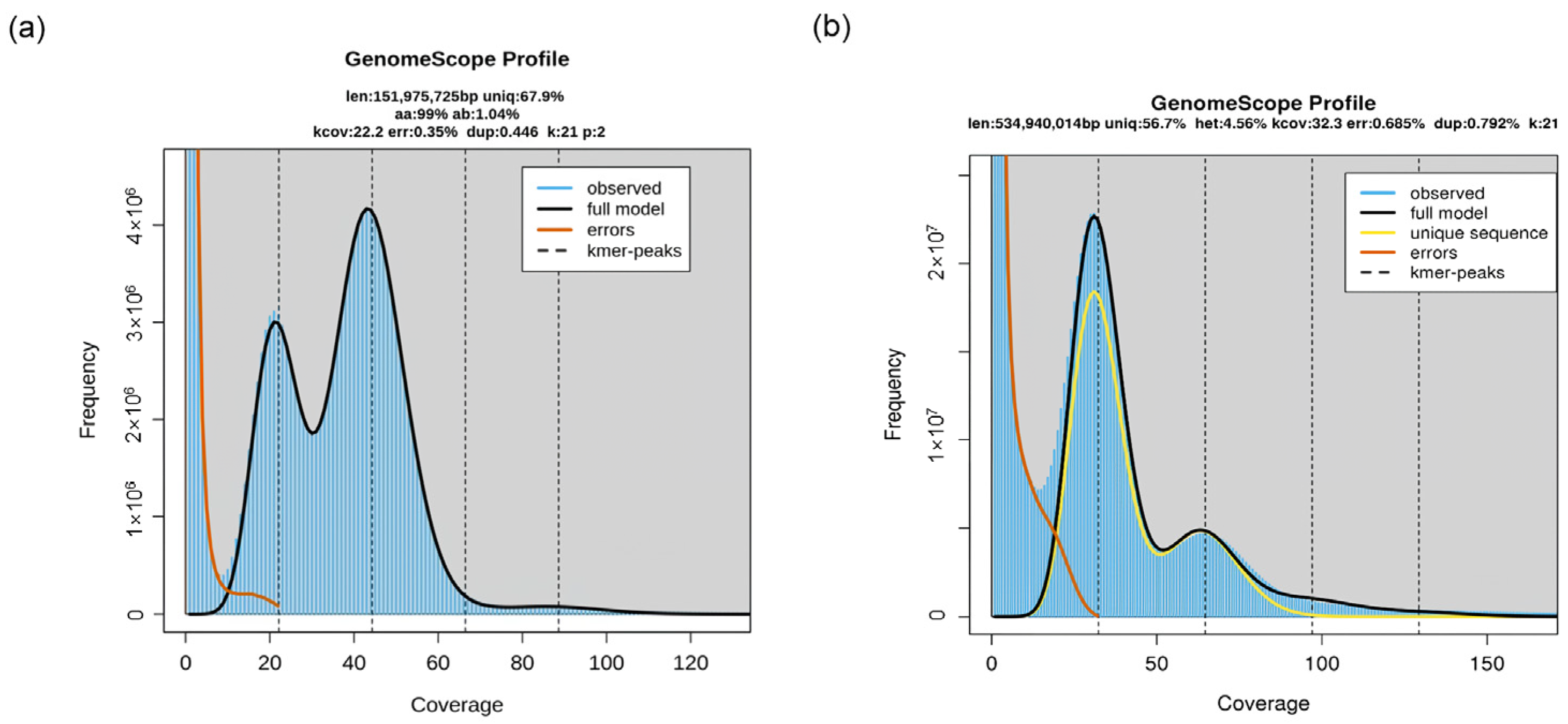
3.2. Genomic Characteristics of Polyploid Plants
3.3. Sequencing Technology Advancements and Limitations in Polyploids
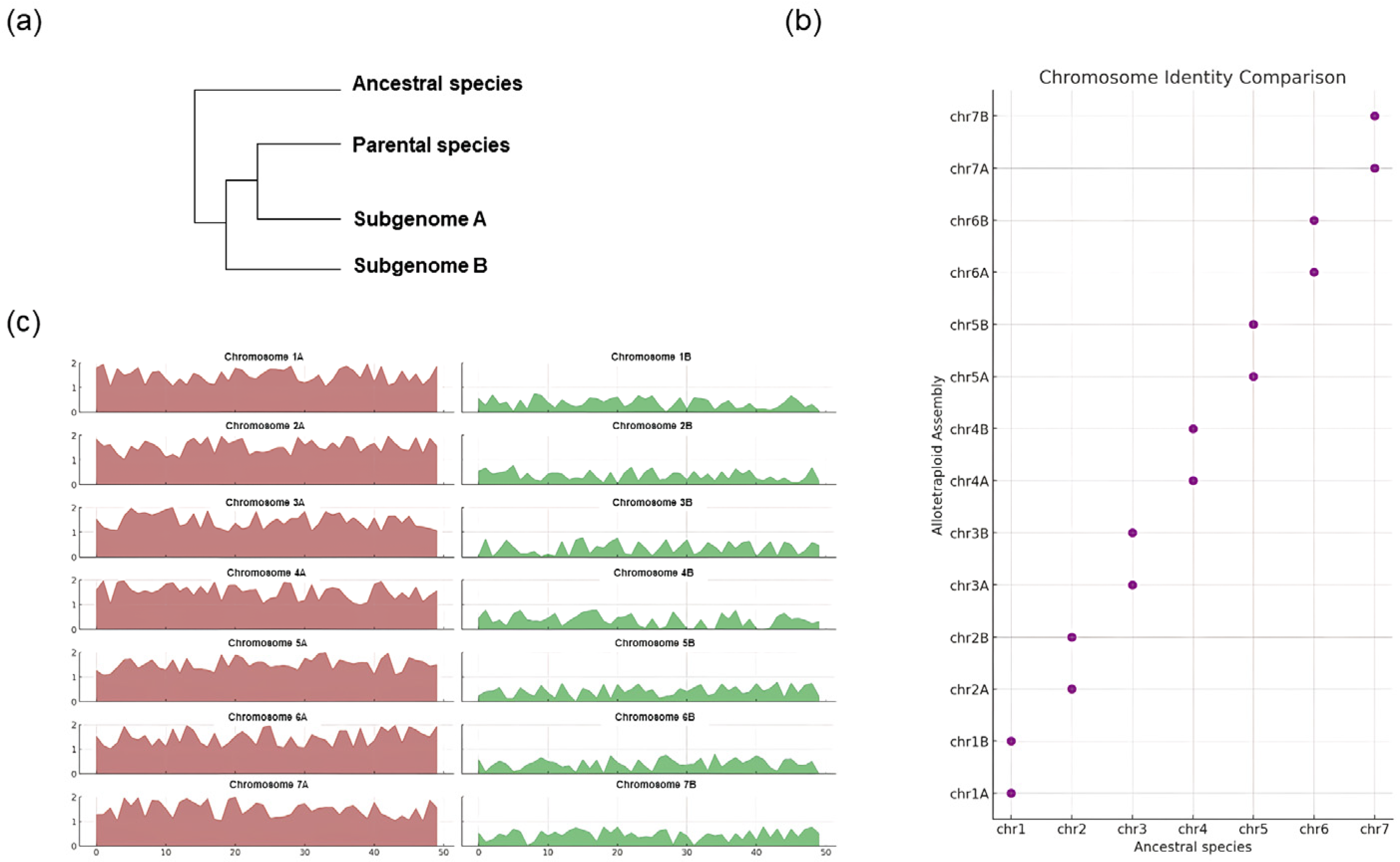
3.4. Sub-Genome Discrimination in Allopolyploids
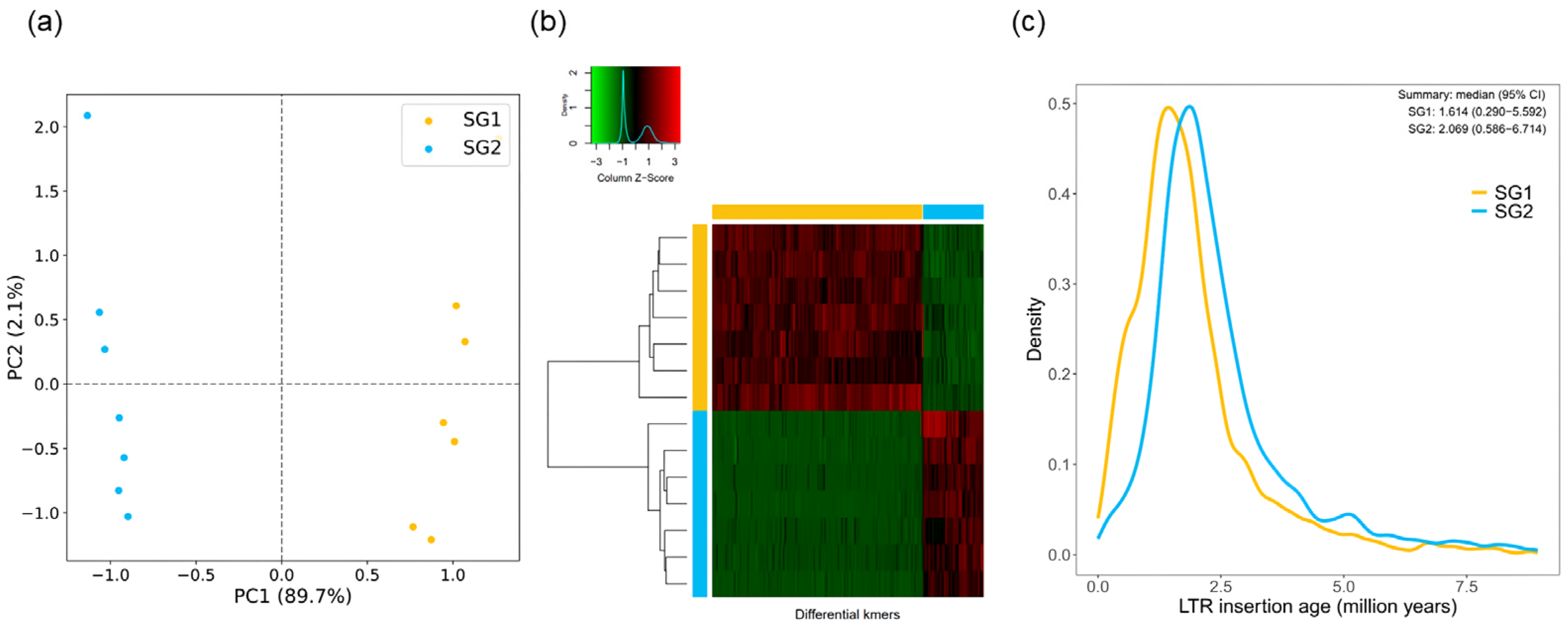
3.5. Genome Assembly Strategies of Autopolyploids in the Brassicaceae
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- German, D.A.; Hendriks, K.P.; Koch, M.A.; Lens, F.; Lysak, M.A.; Bailey, C.D.; Mummenhoff, K.; Al-Shehbaz, I.A. An updated classification of the Brassicaceae (Cruciferae). Phytokeys 2023, 220, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, K.P.; Kiefer, C.; Al-Shehbaz, I.A.; Bailey, C.D.; van Huysduynen, A.H.; Nikolov, L.A.; Nauheimer, L.; Zuntini, A.R.; German, D.A.; Franzke, A.; et al. Global Brassicaceae phylogeny based on filtering of 1000-gene dataset. Curr. Biol. 2023, 33, 4052–4068. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, P.; Pislewska-Bednarek, M.; Svatos, A.; Schneider, B.; Doubsky, J.; Mansurova, M.; Humphry, M.; Consonni, C.; Panstruga, R.; Sanchez-Vallet, A.; et al. A Glucosinolate Metabolism Pathway in Living Plant Cells Mediates Broad-Spectrum Antifungal Defense. Science 2009, 323, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, Y.; Koshiba-Takeuchi, K. Significance of whole-genome duplications on the emergence of evolutionary novelties. Brief. Funct. Genom. 2018, 17, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.E.; Takebayashi, N.; Barker, M.S.; Mayrose, I.; Greenspoon, P.B.; Rieseberg, L.H. The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. USA 2009, 106, 13875–13879. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.E.; Chapman, B.A.; Rong, J.; Paterson, A.H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 2003, 422, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Kagale, S.; Robinson, S.J.; Nixon, J.; Xiao, R.; Huebert, T.; Condie, J.; Kessler, D.; Clarke, W.E.; Edger, P.P.; Links, M.G.; et al. Polyploid Evolution of the Brassicaceae during the Cenozoic Era. Plant Cell 2014, 26, 2777–2791. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.C.; German, D.A.; Koch, M.A. Temporal patterns of diversification in Brassicaceae demonstrate decoupling of rate shifts and mesopolyploidization events. Ann. Bot. 2020, 125, 29–47. [Google Scholar] [CrossRef] [PubMed]
- Franzke, A.; Lysak, M.A.; Al-Shehbaz, I.A.; Koch, M.A.; Mummenhoff, K. Cabbage family affairs: The evolutionary history of Brassicaceae. Trends Plant Sci. 2011, 16, 108–116. [Google Scholar] [CrossRef]
- Paritosh, K.; Yadava, S.K.; Singh, P.; Bhayana, L.; Mukhopadhyay, A.; Gupta, V.; Bisht, N.C.; Zhang, J.; Kudrna, D.A.; Copetti, D.; et al. A chromosome-scale assembly of allotetraploid Brassica juncea (AABB) elucidates comparative architecture of the A and B genomes. Plant Biotechnol. J. 2021, 19, 602–614. [Google Scholar] [CrossRef]
- Sun, H.Q.; Jiao, W.B.; Campoy, J.A.; Krause, K.; Goel, M.; Folz-Donahue, K.; Kukat, C.; Huettel, B.; Schneeberger, K. Chromosome-scale and haplotype-resolved genome assembly of a tetraploid potato cultivar. Nat. Genet. 2022, 54, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Xu, S.X.; Shen, Q.; Bi, C.W.; Lysak, M.A. The allotetraploid horseradish genome provides insights into subgenome diversification and formation of critical traits. Nat. Commun. 2023, 14, 4102. [Google Scholar] [CrossRef] [PubMed]
- Van De Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Ashman, T.L.; Soltis, P.S.; Soltis, D.E. Polyploidy: An evolutionary and ecological force in stressful times. Plant Cell 2021, 33, 11–26. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542. [Google Scholar] [CrossRef]
- Kyriakidou, M.; Tai, H.H.; Anglin, N.L.; Ellis, D.; Strömvik, M.V. Current Strategies of Polyploid Plant Genome Sequence Assembly. Front. Plant Sci. 2018, 9, 1660. [Google Scholar] [CrossRef]
- Amborella Genome, P. The Amborella genome and the evolution of flowering plants. Science 2013, 342, 1241089. [Google Scholar] [CrossRef] [PubMed]
- Walden, N.; Nguyen, T.-P.; Mandáková, T.; Lysak, M.A.; Schranz, M.E. Genomic blocks in Aethionema arabicum support Arabideae as next diverging clade in Brassicaceae. Front. Plant Sci. 2020, 11, 719. [Google Scholar] [CrossRef]
- Mandakova, T.; Li, Z.; Barker, M.S.; Lysak, M.A. Diverse genome organization following 13 independent mesopolyploid events in Brassicaceae contrasts with convergent patterns of gene retention. Plant J. 2017, 91, 3–21. [Google Scholar] [CrossRef]
- Mandáková, T.; Lysak, M.A. Post-polyploid diploidization and diversification through dysploid changes. Curr. Opin. Plant Biol. 2018, 42, 55–65. [Google Scholar] [CrossRef]
- Zou, J.; Hu, D.; Liu, P.; Raman, H.; Liu, Z.; Liu, X.; Parkin, I.A.; Chalhoub, B.; Meng, J. Co-linearity and divergence of the A subgenome of Brassica juncea compared with other Brassica species carrying different A subgenomes. Bmc Genom. 2016, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, N.; Wolf, E.M.; Lysak, M.A.; Koch, M.A. A Time-Calibrated Road Map of Brassicaceae Species Radiation and Evolutionary History. Plant Cell 2015, 27, 2770–2784. [Google Scholar] [CrossRef] [PubMed]
- Walden, N.; German, D.A.; Wolf, E.M.; Kiefer, M.; Rigault, P.; Huang, X.C.; Kiefer, C.; Schmickl, R.; Franzke, A.; Neuffer, B.; et al. Nested whole-genome duplications coincide with diversification and high morphological disparity in Brassicaceae. Nat. Commun. 2020, 11, 3795. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, J.; Schemske, D.W. Neopolyploidy in flowering plants. Annu. Rev. Ecol. Syst. 2002, 33, 589–639. [Google Scholar] [CrossRef]
- Baduel, P.; Bray, S.; Vallejo-Marin, M.; Kolář, F.; Yant, L. The “Polyploid Hop”: Shifting challenges and opportunities over the evolutionary lifespan of genome duplications. Front. Ecol. Evol. 2018, 6, 117. [Google Scholar] [CrossRef]
- Tank, D.C.; Eastman, J.M.; Pennell, M.W.; Soltis, P.S.; Soltis, D.E.; Hinchliff, C.E.; Brown, J.W.; Sessa, E.B.; Harmon, L.J. Nested radiations and the pulse of angiosperm diversification: Increased diversification rates often follow whole genome duplications. New Phytol. 2015, 207, 454–467. [Google Scholar] [CrossRef]
- Schranz, M.E.; Mohammadin, S.; Edger, P.P. Ancient whole genome duplications, novelty and diversification: The WGD Radiation Lag-Time Model. Curr. Opin. Plant Biol. 2012, 15, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Estep, M.C.; McKain, M.R.; Diaz, D.V.; Zhong, J.S.; Hodge, J.G.; Hodkinson, T.R.; Layton, D.J.; Malcomber, S.T.; Pasquet, R.; Kellogg, E.A. Allopolyploidy, diversification, and the Miocene grassland expansion. Proc. Natl. Acad. Sci. USA 2014, 111, 15149–15154. [Google Scholar] [CrossRef]
- Zachos, J.; Pagani, M.; Sloan, L.; Thomas, E.; Billups, K. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 2001, 292, 686–693. [Google Scholar] [CrossRef]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Ratzka, A.; Vogel, H.; Kliebenstein, D.J.; Mitchell-Olds, T.; Kroymann, J. Disarming the mustard oil bomb. Proc. Natl. Acad. Sci. USA 2002, 99, 11223–11228. [Google Scholar] [CrossRef] [PubMed]
- Lambrix, V.; Reichelt, M.; Mitchell-Olds, T.; Kliebenstein, D.J.; Gershenzon, J. The Arabidopsis epithiospecifier protein promotes the hydrolysis of glucosinolates to nitriles and influences Trichoplusia ni herbivory. Plant Cell 2001, 13, 2793–2807. [Google Scholar] [CrossRef] [PubMed]
- Burow, M.; Markert, J.; Gershenzon, J.; Wittstock, U. Comparative biochemical characterization of nitrile-forming proteins from plants and insects that alter myrosinase-catalysed hydrolysis of glucosinolates. FEBS J. 2006, 273, 2432–2446. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.; Batley, J.; Parkin, I.; Kole, C. Genetics, Genomics and Breeding of Oilseed Brassicas; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Hofberger, J.A.; Lyons, E.; Edger, P.P.; Pires, J.C.; Schranz, M.E. Whole Genome and Tandem Duplicate Retention Facilitated Glucosinolate Pathway Diversification in the Mustard Family. Genome Biol. Evol. 2013, 5, 2155–2173. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Calvo, P.; Chini, A.; Fernandez-Barbero, G.; Chico, J.M.; Gimenez-Ibanez, S.; Geerinck, J.; Eeckhout, D.; Schweizer, F.; Godoy, M.; Franco-Zorrilla, J.M.; et al. The Arabidopsis bHLH transcription factors MYC3 and MYC4 are targets of JAZ repressors and act additively with MYC2 in the activation of jasmonate responses. Plant Cell 2011, 23, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.G.; Kliebenstein, D.J.; Halkier, B.A. Identification of a flavin-monooxygenase as the S-oxygenating enzyme in aliphatic glucosinolate biosynthesis in Arabidopsis. Plant J. 2007, 50, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Pfalz, M.; Mukhaimar, M.; Perreau, F.; Kirk, J.; Hansen, C.I.C.; Olsen, C.E.; Agerbirk, N.; Kroymann, J. Methyl Transfer in Glucosinolate Biosynthesis Mediated by Indole Glucosinolate O-Methyltransferase 5. Plant Physiol. 2016, 172, 2190–2203. [Google Scholar] [CrossRef] [PubMed]
- Nour-Eldin, H.H.; Andersen, T.G.; Burow, M.; Madsen, S.R.; Jorgensen, M.E.; Olsen, C.E.; Dreyer, I.; Hedrich, R.; Geiger, D.; Halkier, B.A. NRT/PTR transporters are essential for translocation of glucosinolate defence compounds to seeds. Nature 2012, 488, 531–534. [Google Scholar] [CrossRef]
- Jorgensen, M.E.; Xu, D.; Crocoll, C.; Ramirez, D.; Motawia, M.S.; Olsen, C.E.; Nour-Eldin, H.H.; Halkier, B.A. Origin and evolution of transporter substrate specificity within the NPF family. eLife 2017, 6, e19466. [Google Scholar] [CrossRef]
- de Kraker, J.W.; Gershenzon, J. From amino acid to glucosinolate biosynthesis: Protein sequence changes in the evolution of methylthioalkylmalate synthase in Arabidopsis. Plant Cell 2011, 23, 38–53. [Google Scholar] [CrossRef]
- Barco, B.; Clay, N.K. Evolution of Glucosinolate Diversity via Whole-Genome Duplications, Gene Rearrangements, and Substrate Promiscuity. Annu. Rev. Plant Biol. 2019, 70, 585–604. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.C.; Ooi, M.K.; Guja, L.K. Polyploidy but not range size is associated with seed and seedling traits that affect performance of Pomaderris species. Front. Plant Sci. 2022, 12, 779651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xue, H.; Lu, X.J.; Zhang, B.; Wang, F.; Ma, Y.; Zhang, Z.H. Autotetraploidization enhances drought stress tolerance in two apple cultivars. Trees-Struct. Funct. 2015, 29, 1773–1780. [Google Scholar] [CrossRef]
- Ha, M.; Kim, E.D.; Chen, Z.J. Duplicate genes increase expression diversity in closely related species and allopolyploids. Proc. Natl. Acad. Sci. USA 2009, 106, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Salman-Minkov, A.; Sabath, N.; Mayrose, I. Whole-genome duplication as a key factor in crop domestication. Nat. Plants 2016, 2, 16115. [Google Scholar] [CrossRef] [PubMed]
- Yim, W.C.; Swain, M.L.; Ma, D.; An, H.; Bird, K.A.; Curdie, D.D.; Wang, S.; Ham, H.D.; Luzuriaga-Neira, A.; Kirkwood, J.S.; et al. The final piece of the Triangle of U: Evolution of the tetraploid Brassica carinata genome. Plant Cell 2022, 34, 4143–4172. [Google Scholar] [CrossRef] [PubMed]
- Bonnema, G.; Del Carpio, D.P.; Zhao, J. Diversity analysis and molecular taxonomy of Brassica vegetable crops. Genet. Genom. Breed. Veg. Brassicas 2011, 1, 81–124. [Google Scholar]
- Wang, T.; van Dijk, A.D.J.; Bucher, J.; Liang, J.; Wu, J.; Bonnema, G.; Wang, X. Interploidy Introgression Shaped Adaptation during the Origin and Domestication History of Brassica napus. Mol. Biol. Evol. 2023, 40, msad199. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Gao, J.; Li, W.; Liu, Y.; Fang, P.; Peng, Z. Colchicine-induced tetraploidy influences morphological and cytological characteristics and enhances accumulation of anthocyanins in a red-fleshed radish (Raphanus sativus L.). Hortic. Environ. Biotechnol. 2021, 62, 937–948. [Google Scholar] [CrossRef]
- Chen, Z.J.; Ni, Z.F. Mechanisms of genomic rearrangements and gene expression changes in plant polyploids. Bioessays 2006, 28, 240–252. [Google Scholar] [CrossRef]
- Cui, X.B.; Hu, M.; Yao, S.L.; Zhang, Y.Y.; Tang, M.Q.; Liu, L.J.; Cheng, X.H.; Tong, C.B.; Liu, S.Y. BnaOmics: A comprehensive platform combining pan-genome and multi-omics data from. Plant Commun. 2023, 4, 100609. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Liu, Q.Q.; He, Z.S.; Raman, R.; Wang, H.; Long, X.X.; Qin, H.; Raman, H.; Parkin, I.A.P.; Bancroft, I.; et al. A Brassica carinata pan-genome platform for Brassica crop improvement. Plant Commun. 2024, 5, 100725. [Google Scholar] [CrossRef] [PubMed]
- Claros, M.G.; Bautista, R.; Guerrero-Fernández, D.; Benzerki, H.; Seoane, P.; Fernández-Pozo, N. Why Assembling Plant Genome Sequences Is So Challenging. Biology 2012, 1, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Song, Q.; Ye, W.; Chen, Z.J. Concerted genomic and epigenomic changes accompany stabilization of Arabidopsis allopolyploids. Nat. Ecol. Evol. 2021, 5, 1382–1393. [Google Scholar] [CrossRef] [PubMed]
- Song, J.M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ying, H.; Yang, X.; Gao, Y.; Li, T.; Wu, B.; Ren, M.; Zhang, Z.; Ding, J.; Gao, J.; et al. The Cardamine enshiensis genome reveals whole genome duplication and insight into selenium hyperaccumulation and tolerance. Cell Discov. 2021, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Hori, T.; Yamamoto, S.; Toyoda, A.; Yano, K.; Yamane, K.; Itoh, T. Haplotype-resolved chromosomal-level assembly of wasabi (Eutrema japonicum) genome. Sci. Data 2023, 10, 441. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Ma, Y.; Mandáková, T.; Shi, S.; Chen, C.; Sun, P.; Zhang, L.; Feng, L.; Zheng, Y.; Feng, X.; et al. Genome evolution of the psammophyte Pugionium for desert adaptation and further speciation. Proc. Natl. Acad. Sci. USA 2021, 118, e2025711118. [Google Scholar] [CrossRef]
- Robinson, D.O.; Coate, J.E.; Singh, A.; Hong, L.L.; Bush, M.; Doyle, J.J.; Roeder, A.H.K. Ploidy and Size at Multiple Scales in the Arabidopsis Sepal. Plant Cell 2018, 30, 2308–2329. [Google Scholar] [CrossRef] [PubMed]
- Otto, S.P. The evolutionary consequences of polyploidy. Cell 2007, 131, 452–462. [Google Scholar] [CrossRef]
- Yuan, S.X.; Liu, Y.M.; Fang, Z.Y.; Yang, L.M.; Zhuang, M.; Zhang, Y.Y.; Sun, P.T. Study on the Relationship Between the Ploidy Level of Microspore-Derived Plants and the Number of Chloroplast in Stomatal Guard Cells in Brassica oleracea. Agric. Sci. China 2009, 8, 939–946. [Google Scholar] [CrossRef]
- Roddy, A.B.; Théroux-Rancourt, G.; Abbo, T.; Benedetti, J.W.; Brodersen, C.R.; Castro, M.; Castro, S.; Gilbride, A.B.; Jensen, B.; Jiang, G.F.; et al. The Scaling of Genome Size and Cell Size Limits Maximum Rates of Photosynthesis with Implications for Ecological Strategies. Int. J. Plant Sci. 2020, 181, 75–87. [Google Scholar] [CrossRef]
- Williams, J.H.; Oliveira, P.E. For things to stay the same, things must change: Polyploidy and pollen tube growth rates. Ann. Bot. 2020, 125, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, Y.; Azumatani, M.; Suyama, C.; Adamec, L. Determination of Ploidy Level and Nuclear DNA Content in the Droseraceae by Flow Cytometry. Cytologia 2017, 82, 321–327. [Google Scholar] [CrossRef]
- Li, W.; Liu, L.; Wang, Y.; Fan, G.; Zhang, S.; Wang, Y.; Liao, K. Determination of genome size and chromosome ploidy of selected taxa from Prunus armeniaca by flow cytometry. Sci. Hortic. 2020, 261, 108987. [Google Scholar] [CrossRef]
- Song, K.M.; Lu, P.; Tang, K.L.; Osborn, T.C. Rapid Genome Change in Synthetic Polyploids of Brassica and Its Implications for Polyploid Evolution. Proc. Natl. Acad. Sci. USA 1995, 92, 7719–7723. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Yi, S.V.; Zhang, M.; Zhou, X. Development of novel EST-SSR markers for ploidy identification based on de novo transcriptome assembly for Misgurnus anguillicaudatus. PLoS ONE 2018, 13, e0195829. [Google Scholar] [CrossRef]
- Shafey, S.; El-Maaty, S.A.; El Habbasha, S.; Elarabi, N.I. Evaluation of the growth, yield traits and the genetic diversity of some Brassica napus genotypes under Egyptian conditions. Beni-Suef Univ. J. Basic Appl. Sci. 2023, 12, 48. [Google Scholar] [CrossRef]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef]
- Niedringhaus, T.P.; Milanova, D.; Kerby, M.B.; Snyder, M.P.; Barron, A.E. Landscape of Next-Generation Sequencing Technologies. Anal. Chem. 2011, 83, 4327–4341. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.S.; Ulahannan, N.; Pendleton, M.; Dai, X.G.; Ly, L.; Behr, J.M.; Schwenk, S.; Liao, W.; Augello, M.A.; Tyer, C.; et al. Identifying synergistic high-order 3D chromatin conformations from genome-scale nanopore concatemer sequencing. Nat. Biotechnol. 2022, 40, 1488–1499. [Google Scholar] [CrossRef]
- Jia, K.-H.; Liu, H.; Zhang, R.-G.; Xu, J.; Zhou, S.-S.; Jiao, S.-Q.; Yan, X.-M.; Tian, X.-C.; Shi, T.-L.; Luo, H. Chromosome-scale assembly and evolution of the tetraploid Salvia splendens (Lamiaceae) genome. Hortic. Res. 2021, 8, 177. [Google Scholar] [CrossRef]
- Mitros, T.; Session, A.M.; James, B.T.; Wu, G.A.; Belaffif, M.B.; Clark, L.V.; Shu, S.Q.; Dong, H.X.; Barling, A.; Holmes, J.R.; et al. Genome biology of the paleotetraploid perennial biomass crop. Nat. Commun. 2020, 11, 5442. [Google Scholar] [CrossRef]
- Campomayor, N.B.; Waminal, N.E.; Kang, B.Y.; Nguyen, T.H.; Lee, S.S.; Huh, J.H.; Kim, H.H. Subgenome Discrimination in Brassica and Raphanus Allopolyploids Using Microsatellites. Cells 2021, 10, 2358. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.X.; Bennetzen, J.L. Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 12404–12410. [Google Scholar] [CrossRef]
- Jia, K.H.; Wang, Z.X.; Wang, L.; Li, G.Y.; Zhang, W.; Wang, X.L.; Xu, F.J.; Jiao, S.Q.; Zhou, S.S.; Liu, H.; et al. SubPhaser: A robust allopolyploid subgenome phasing method based on subgenome-specific k-mers. New Phytol. 2022, 235, 801–809. [Google Scholar] [CrossRef]
- Slotte, T.; Ceplitis, A.; Neuffer, B.; Hurka, H.; Lascoux, M. Intrageneric phylogeny of Capsella (Brassicaceae) and the origin of the tetraploid C. bursa-pastoris based on chloroplast and nuclear DNA sequences. Am. J. Bot. 2006, 93, 1714–1724. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, C.; Zhang, M.; Zhou, H.; Zuo, Z.; Ding, X.; Zhang, R.; Li, F.; Gao, Y. Chromosome-level genome assembly of the Colorado potato beetle, Leptinotarsa decemlineata. Sci. Data 2023, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Lu, R.; Li, C.; Yang, D.; Zeng, Z.; Lin, W.; Cheng, J.; Yang, Z.; Wang, L.; Gao, Y.; et al. Haplotype-resolved and chromosome-level genome assembly of Colorado potato beetle. J. Genet. Genom. 2023, 50, 532–535. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Estimated Genome Size | Ploidy Level | Long-Read Sequencing Platform | Chromosome Capture Sequencing Platform | Ref. |
|---|---|---|---|---|---|
| Arabidopsis suecica | ~272 Mb | Allotetraploid | PacBio Sequel | Hi-C | [55] |
| Armoracia rusticana | ~636 Mbp | Allotetraploid | ONT, PacBio HiFi | Hi-C | [12] |
| Brassica carinata | ~1.31 Gbp | Allotetraploid | PacBio | Hi-C | [47] |
| Brassica juncea | ~922 Mb | Allotetraploid | PacBio RSII | Hi-C | [10] |
| Brassica napus | ~1132 Mb | Allotetraploid | PacBio SMRT | Hi-C | [56] |
| Cardamine enshiensis | ~443 Mb | Allotetraploid | PacBio Sequel | Hi-C | [57] |
| Eutrema japonicum | ~1512.1 Mb | Allotetraploid | PacBio CLR | Hi-C | [58] |
| Pugionium cornutum | ~570 Mb | Allotetraploid | GridION, PacBio RS II | ||
| Pugionium dolabratum | ~606 Mb | Allotetraploid | GridION, PacBio RS II | [59] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, D.; Kim, C. Polyploids of Brassicaceae: Genomic Insights and Assembly Strategies. Plants 2024, 13, 2087. https://doi.org/10.3390/plants13152087
Jeon D, Kim C. Polyploids of Brassicaceae: Genomic Insights and Assembly Strategies. Plants. 2024; 13(15):2087. https://doi.org/10.3390/plants13152087
Chicago/Turabian StyleJeon, Donghyun, and Changsoo Kim. 2024. "Polyploids of Brassicaceae: Genomic Insights and Assembly Strategies" Plants 13, no. 15: 2087. https://doi.org/10.3390/plants13152087
APA StyleJeon, D., & Kim, C. (2024). Polyploids of Brassicaceae: Genomic Insights and Assembly Strategies. Plants, 13(15), 2087. https://doi.org/10.3390/plants13152087