

Selection of Flax Genotypes for Pan-Genomic Studies by Sequencing Tagmentation-Based Transcriptome Libraries

 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Preparation of cDNA Libraries Using the Tn5 Transposase
2.2. Mapping the Obtained Illumina Reads to the Flax Genome
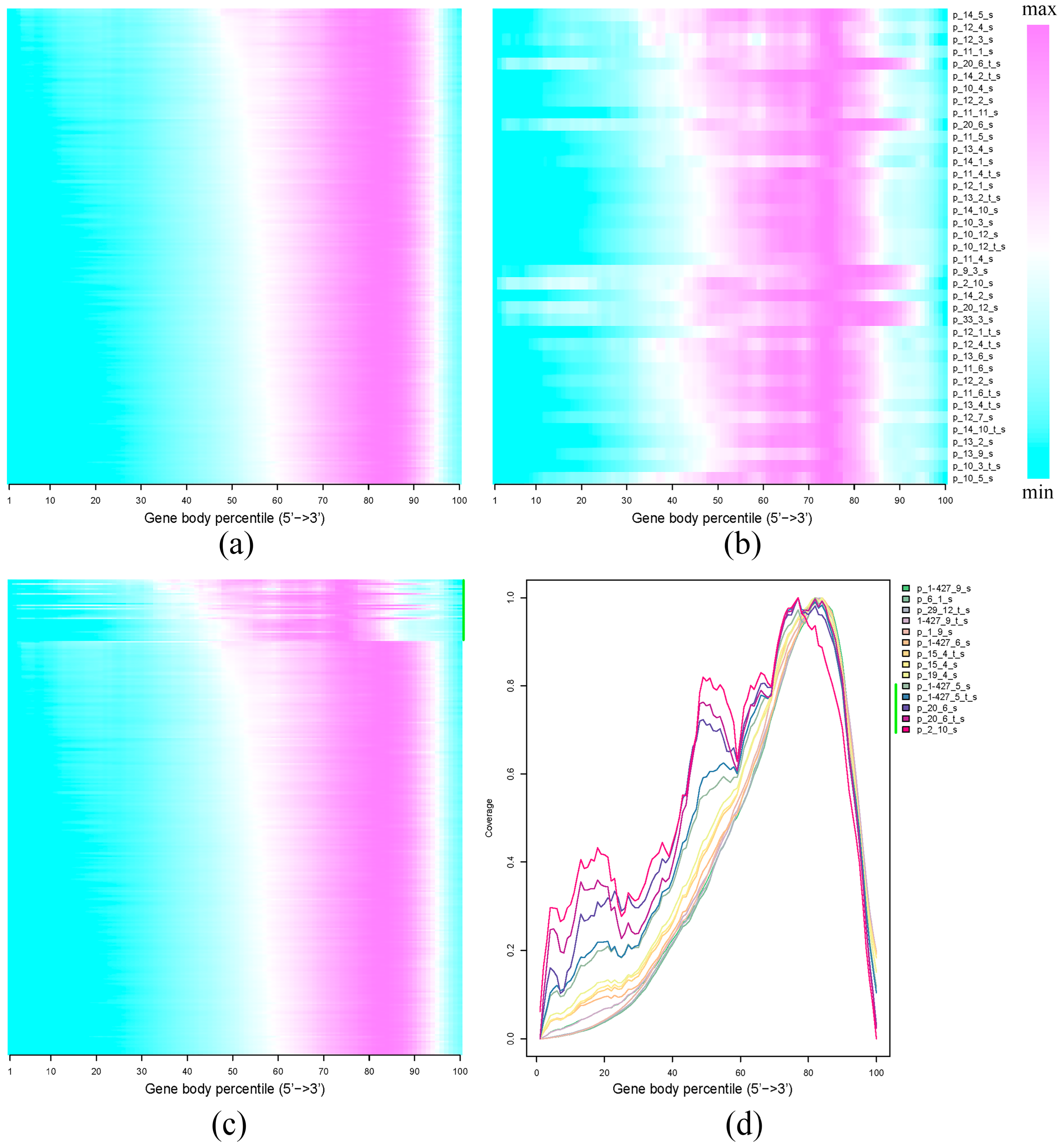
2.3. Gene Coverage Profiles
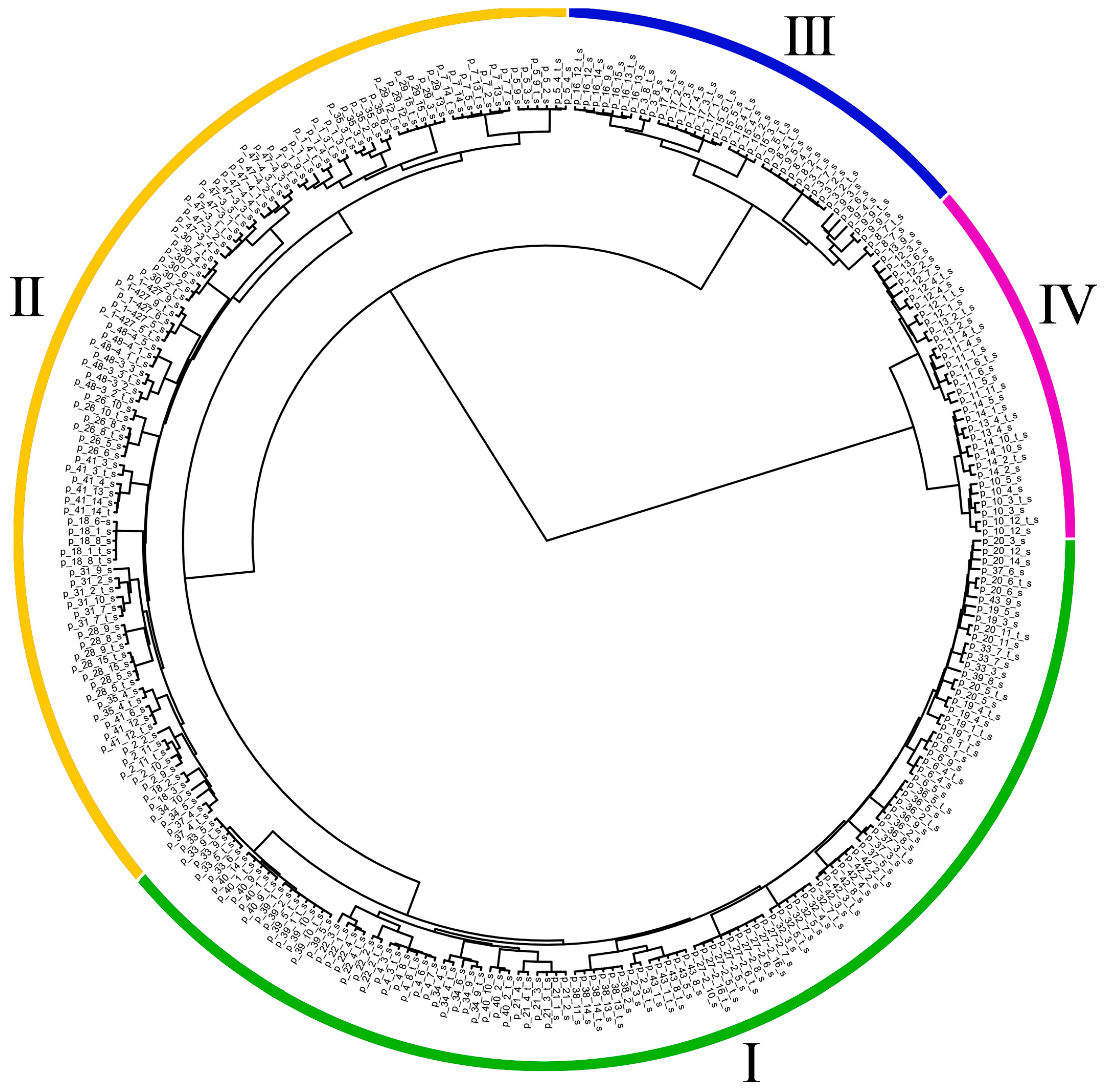
2.4. Identification of DNA Polymorphisms and Clustering of Flax Samples
2.5. Species Identification for Some Accessions
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Preparation and Sequencing of cDNA Libraries
4.3. Bioinformatic Processing of Data
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abdelhamid, A.S.; Martin, N.; Bridges, C.; Brainard, J.S.; Wang, X.; Brown, T.J.; Hanson, S.; Jimoh, O.F.; Ajabnoor, S.M.; Deane, K.H.; et al. Polyunsaturated fatty acids for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2018, 7, CD012345. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.; Severino, P.; Ferreira, C.; Zielinska, A.; Santini, A.; Souto, S.; Souto, E.B. Linseed essential oil—Source of Lipids as Active Ingredients for Pharmaceuticals and Nutraceuticals. Curr. Med. Chem. 2018, 26, 4537–4558. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Sharma, V.; Upadhyay, N.; Gill, S.; Sihag, M. Flax and flaxseed oil: An ancient medicine & modern functional food. J. Food Sci. Technol. 2014, 51, 1633–1653. [Google Scholar] [CrossRef] [PubMed]
- Kezimana, P.; Dmitriev, A.A.; Kudryavtseva, A.V.; Romanova, E.V.; Melnikova, N.V. Secoisolariciresinol Diglucoside of Flaxseed and Its Metabolites: Biosynthesis and Potential for Nutraceuticals. Front. Genet. 2018, 9, 641. [Google Scholar] [CrossRef] [PubMed]
- Parikh, M.; Netticadan, T.; Pierce, G.N. Flaxseed: Its bioactive components and their cardiovascular benefits. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H146–H159. [Google Scholar] [CrossRef]
- Santos, H.O.; Price, J.C.; Bueno, A.A. Beyond Fish Oil Supplementation: The Effects of Alternative Plant Sources of Omega-3 Polyunsaturated Fatty Acids upon Lipid Indexes and Cardiometabolic Biomarkers-An Overview. Nutrients 2020, 12, 3159. [Google Scholar] [CrossRef]
- Muir, A.D.; Westcott, N.D. Flax: The Genus Linum; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Melelli, A.; Jamme, F.; Beaugrand, J.; Bourmaud, A. Evolution of the ultrastructure and polysaccharide composition of flax fibres over time: When history meets science. Carbohydr. Polym. 2022, 291, 119584. [Google Scholar] [CrossRef]
- Kaur, V.; Singh, M.; Wankhede, D.P.; Gupta, K.; Langyan, S.; Aravind, J.; Thangavel, B.; Yadav, S.K.; Kalia, S.; Singh, K.; et al. Diversity of Linum genetic resources in global genebanks: From agro-morphological characterisation to novel genomic technologies—A review. Front. Nutr. 2023, 10, 1165580. [Google Scholar] [CrossRef]
- Wang, Z.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R.; et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. Cell Mol. Biol. 2012, 72, 461–473. [Google Scholar] [CrossRef]
- You, F.M.; Xiao, J.; Li, P.; Yao, Z.; Jia, G.; He, L.; Kumar, S.; Soto-Cerda, B.; Duguid, S.D.; Booker, H.M.; et al. Genome-Wide Association Study and Selection Signatures Detect Genomic Regions Associated with Seed Yield and Oil Quality in Flax. Int. J. Mol. Sci. 2018, 19, 2303. [Google Scholar] [CrossRef]
- Dmitriev, A.A.; Pushkova, E.N.; Novakovskiy, R.O.; Beniaminov, A.D.; Rozhmina, T.A.; Zhuchenko, A.A.; Bolsheva, N.L.; Muravenko, O.V.; Povkhova, L.V.; Dvorianinova, E.M.; et al. Genome Sequencing of Fiber Flax Cultivar Atlant Using Oxford Nanopore and Illumina Platforms. Front. Genet. 2020, 11, 590282. [Google Scholar] [CrossRef] [PubMed]
- Sa, R.; Yi, L.; Siqin, B.; An, M.; Bao, H.; Song, X.; Wang, S.; Li, Z.; Zhang, Z.; Hazaisi, H.; et al. Chromosome-Level Genome Assembly and Annotation of the Fiber Flax (Linum usitatissimum) Genome. Front. Genet. 2021, 12, 735690. [Google Scholar] [CrossRef] [PubMed]
- Dvorianinova, E.M.; Bolsheva, N.L.; Pushkova, E.N.; Rozhmina, T.A.; Zhuchenko, A.A.; Novakovskiy, R.O.; Povkhova, L.V.; Sigova, E.A.; Zhernova, D.A.; Borkhert, E.V.; et al. Isolating Linum usitatissimum L. Nuclear DNA Enabled Assembling High-Quality Genome. Int. J. Mol. Sci. 2022, 23, 13244. [Google Scholar] [CrossRef] [PubMed]
- Duk, M.; Kanapin, A.; Rozhmina, T.; Bankin, M.; Surkova, S.; Samsonova, A.; Samsonova, M. The Genetic Landscape of Fiber Flax. Front. Plant Sci. 2021, 12, 764612. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Jiang, H.; Yan, W.; Yang, L.; Ye, J.; Wang, Y.; Yan, Q.; Chen, J.; Gao, Y.; Duan, L.; et al. Resequencing 200 Flax Cultivated Accessions Identifies Candidate Genes Related to Seed Size and Weight and Reveals Signatures of Artificial Selection. Front. Plant Sci. 2019, 10, 1682. [Google Scholar] [CrossRef]
- Shi, J.; Tian, Z.; Lai, J.; Huang, X. Plant pan-genomics and its applications. Mol. Plant 2023, 16, 168–186. [Google Scholar] [CrossRef]
- Sun, Y.; Shang, L.; Zhu, Q.H.; Fan, L.; Guo, L. Twenty years of plant genome sequencing: Achievements and challenges. Trends Plant Sci. 2022, 27, 391–401. [Google Scholar] [CrossRef]
- Dmitriev, A.A.; Pushkova, E.N.; Melnikova, N.V. Plant Genome Sequencing: Modern Technologies and Novel Opportunities for Breeding. Mol. Biol. 2022, 56, 495–507. [Google Scholar] [CrossRef]
- Lei, L.; Goltsman, E.; Goodstein, D.; Wu, G.A.; Rokhsar, D.S.; Vogel, J.P. Plant Pan-Genomics Comes of Age. Annu. Rev. Plant Biol. 2021, 72, 411–435. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Zhang, H.; Liu, Z.; Wang, Y.; Xing, L.; He, Q.; Du, H. Plant pan-genomics: Recent advances, new challenges, and roads ahead. J. Genet. Genom. 2022, 49, 833–846. [Google Scholar] [CrossRef]
- Sizov, I.A. Len; Sel’hozgiz: Moscow, Russia; Leningrad, Russia, 1955; p. 256. [Google Scholar]
- Gill, K.S.; Yermanos, D.M. Cytogenetic Studies on the Genus Linum II. Hybrids Among Taxa with Nine as the Haploid Chromosome Number. Crop Sci. 1967, 7, 627–631. [Google Scholar] [CrossRef]
- Gill, K.S.; Yermanos, D.M. Cytogenetic Studies on the Genus Linum I. Hybrids Among Taxa with 15 as the Haploid Chromosome Number1. Crop Sci. 1967, 7, 623–627. [Google Scholar] [CrossRef]
- Diederichsen, A.; Hammer, K. Variation of cultivated flax (Linum usitatissimum L. subsp. usitatissimum) and its wild progenitor pale flax (subsp. angustifolium (Huds.) Thell.). Genet. Resour. Crop Evol. 2006, 42, 263–272. [Google Scholar] [CrossRef]
- Fu, Y.-B.; Peterson, G.; Diederichsen, A.; Richards, K. RAPD analysis of genetic relationships of seven flax species in the genus Linum L. Genet. Resour. Crop Evol. 2002, 49, 253–259. [Google Scholar] [CrossRef]
- Bolsheva, N.L.; Melnikova, N.V.; Kirov, I.V.; Speranskaya, A.S.; Krinitsina, A.A.; Dmitriev, A.A.; Belenikin, M.S.; Krasnov, G.S.; Lakunina, V.A.; Snezhkina, A.V.; et al. Evolution of blue-flowered species of genus Linum based on high-throughput sequencing of ribosomal RNA genes. BMC Evol. Biol. 2017, 17, 253. [Google Scholar] [CrossRef]
- Zhang, J.; Qi, Y.; Wang, L.; Wang, L.; Yan, X.; Dang, Z.; Li, W.; Zhao, W.; Pei, X.; Li, X.; et al. Genomic Comparison and Population Diversity Analysis Provide Insights into the Domestication and Improvement of Flax. iScience 2020, 23, 100967. [Google Scholar] [CrossRef]
- Kutuzova, S.N.; Chukhina, I.G. Updating the intraspecific classification of cultivated flax (Linum usitatissimum L.) using methods of classical genetics. Proc. Appl. Bot. Genet. Breed. 2017, 178, 97–109. [Google Scholar] [CrossRef]
- Diederichsen, A.; Richards, K. Cultivated flax and the genus Linum L.: Taxonomy and germplasm conservation. In Flax; CRC Press: Boca Raton, FL, USA, 2003; pp. 22–54. [Google Scholar]
- Zelentsov, S.V.; Zelentsov, V.S.; Moshnenko, E.V.; Ryabenko, L.G. Modern understanding of the phylogeny and taxonomy of genus Linum L. and flax (Linum usitatissimum L.). Oil Crop. 2016, 1, 106–121. [Google Scholar]
- Alexeev, Y.E.; Geltman, D.V.; Grabovskaja (Borodina), A.E.; Grintalj, A.R.; Gusev, Y.D.; Egorova, T.V.; Ikonnikov, S.S.; Krupkina, L.I.; Linczevski, I.A.; Medvedeva, N.A.; et al. Flora of Eastern Europe (Flora Europae Orientalis); Tzvelev, N.N., Ed.; Mir i Semya-95: St. Petersburg, Russia, 1996; Volume 9, p. 456. [Google Scholar]
- Tutin, T.; Heywood, V.; Burges, N.; Moore, D.; Valentine, D.; Walters, S.; Webb, D. Flora Europaea; Cambridge University Press: London, UK, 1968; Volume 2, p. 486. [Google Scholar]
- Shishkin, B.K.; Bobrov, E.G.E. Flora of the USSR: Geraniales, Sapindales, Rhamnales; Israel Program for Scientific Translations: Jerusalem, Israel, 1963; Volume 14. [Google Scholar]
- Sinskaya, E.N. Istoricheskaya Geografiya Kul’turnoj Flory (Na Zare Zemledeliya); Kolos: Leningrad, Russia, 1959; p. 480. [Google Scholar]
- Bolsheva, N.L.; Melnikova, N.V.; Dvorianinova, E.M.; Mironova, L.N.; Yurkevich, O.Y.; Amosova, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Muravenko, O.V. Clarification of the Position of Linum stelleroides Planch. within the Phylogeny of the Genus Linum L. Plants 2022, 11, 652. [Google Scholar] [CrossRef]
- Dodsworth, S.; Pokorny, L.; Johnson, M.G.; Kim, J.T.; Maurin, O.; Wickett, N.J.; Forest, F.; Baker, W.J. Hyb-Seq for Flowering Plant Systematics. Trends Plant Sci. 2019, 24, 887–891. [Google Scholar] [CrossRef]
- Bjornson, M.; Kajala, K.; Zipfel, C.; Ding, P. Low-cost and High-throughput RNA-seq Library Preparation for Illumina Sequencing from Plant Tissue. Bio-Protocol 2020, 10, e3799. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S. Full-Length Single-Cell RNA Sequencing with Smart-seq2. Methods Mol. Biol. 2019, 1979, 25–44. [Google Scholar] [CrossRef]
- Di, L.; Fu, Y.; Sun, Y.; Li, J.; Liu, L.; Yao, J.; Wang, G.; Wu, Y.; Lao, K.; Lee, R.W.; et al. RNA sequencing by direct tagmentation of RNA/DNA hybrids. Proc. Natl. Acad. Sci. USA 2020, 117, 2886–2893. [Google Scholar] [CrossRef] [PubMed]
- Sveinsson, S.; McDill, J.; Wong, G.K.; Li, J.; Li, X.; Deyholos, M.K.; Cronk, Q.C. Phylogenetic pinpointing of a paleopolyploidy event within the flax genus (Linum) using transcriptomics. Ann. Bot. 2014, 113, 753–761. [Google Scholar] [CrossRef]
- Picelli, S.; Bjorklund, A.K.; Reinius, B.; Sagasser, S.; Winberg, G.; Sandberg, R. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 2014, 24, 2033–2040. [Google Scholar] [CrossRef]
- Hennig, B.P.; Velten, L.; Racke, I.; Tu, C.S.; Thoms, M.; Rybin, V.; Besir, H.; Remans, K.; Steinmetz, L.M. Large-Scale Low-Cost NGS Library Preparation Using a Robust Tn5 Purification and Tagmentation Protocol. G3 Genes Genomes Genet. 2018, 8, 79–89. [Google Scholar] [CrossRef]
- Li, N.; Jin, K.; Bai, Y.; Fu, H.; Liu, L.; Liu, B. Tn5 Transposase Applied in Genomics Research. Int. J. Mol. Sci. 2020, 21, 8329. [Google Scholar] [CrossRef]
- Penkov, D.; Zubkova, E.; Parfyonova, Y. Tn5 DNA Transposase in Multi-Omics Research. Methods Protoc. 2023, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Feng, S.; Li, S.; Chen, G.; Wang, J.; Xu, L.; Fu, X.; Yu, J.; Xu, S. A novel strand-specific RNA-sequencing protocol using dU-adaptor-assembled Tn5. J. Exp. Bot. 2023, 74, 1806–1820. [Google Scholar] [CrossRef]
- Brouilette, S.; Kuersten, S.; Mein, C.; Bozek, M.; Terry, A.; Dias, K.R.; Bhaw-Rosun, L.; Shintani, Y.; Coppen, S.; Ikebe, C.; et al. A simple and novel method for RNA-seq library preparation of single cell cDNA analysis by hyperactive Tn5 transposase. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2012, 241, 1584–1590. [Google Scholar] [CrossRef]
- Lu, B.; Dong, L.; Yi, D.; Zhang, M.; Zhu, C.; Li, X.; Yi, C. Transposase-assisted tagmentation of RNA/DNA hybrid duplexes. eLife 2020, 9, e54919. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Yi, C. TRACE-seq: Rapid, Low-Input, One-Tube RNA-seq Library Construction Based on Tagmentation of RNA/DNA Hybrids. Curr. Protoc. 2023, 3, e735. [Google Scholar] [CrossRef] [PubMed]
- Soto-Cerda, B.J.; Diederichsen, A.; Ragupathy, R.; Cloutier, S. Genetic characterization of a core collection of flax (Linum usitatissimum L.) suitable for association mapping studies and evidence of divergent selection between fiber and linseed types. BMC Plant Biol. 2013, 13, 78. [Google Scholar] [CrossRef] [PubMed]
- Povkhova, L.V.; Melnikova, N.V.; Rozhmina, T.A.; Novakovskiy, R.O.; Pushkova, E.N.; Dvorianinova, E.M.; Zhuchenko, A.A.; Kamionskaya, A.M.; Krasnov, G.S.; Dmitriev, A.A. Genes associated with the flax plant type (oil or fiber) identified based on genome and transcriptome sequencing data. Plants 2021, 10, 2616. [Google Scholar] [CrossRef]
- Guo, C.; Luo, Y.; Gao, L.M.; Yi, T.S.; Li, H.T.; Yang, J.B.; Li, D.Z. Phylogenomics and the flowering plant tree of life. J. Integr. Plant Biol. 2023, 65, 299–323. [Google Scholar] [CrossRef]
- McDill, J.; Repplinger, M.; Simpson, B.; Kadereit, J. The Phylogeny of Linum and Linaceae Subfamily Linoideae, with Implications for Their Systematics, Biogeography, and Evolution of Heterostyly. Syst. Bot. 2009, 34, 386–405. [Google Scholar] [CrossRef]
- Good, J.M. Reduced representation methods for subgenomic enrichment and next-generation sequencing. Methods Mol. Biol. 2011, 772, 85–103. [Google Scholar] [CrossRef]
- Kumar, S.; You, F.M.; Cloutier, S. Genome wide SNP discovery in flax through next generation sequencing of reduced representation libraries. BMC Genom. 2012, 13, 684. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, Q.; Wu, G.; Zhang, S.; Jiang, T. Development of Novel SSR Markers for Flax (Linum usitatissimum L.) Using Reduced-Representation Genome Sequencing. Front. Plant Sci. 2016, 7, 2018. [Google Scholar] [CrossRef]
- Johnson, M.G.; Pokorny, L.; Dodsworth, S.; Botigue, L.R.; Cowan, R.S.; Devault, A.; Eiserhardt, W.L.; Epitawalage, N.; Forest, F.; Kim, J.T.; et al. A Universal Probe Set for Targeted Sequencing of 353 Nuclear Genes from Any Flowering Plant Designed Using k-Medoids Clustering. Syst. Biol. 2019, 68, 594–606. [Google Scholar] [CrossRef]
- Lyu, M.J.; Gowik, U.; Kelly, S.; Covshoff, S.; Mallmann, J.; Westhoff, P.; Hibberd, J.M.; Stata, M.; Sage, R.F.; Lu, H.; et al. RNA-Seq based phylogeny recapitulates previous phylogeny of the genus Flaveria (Asteraceae) with some modifications. BMC Evol. Biol. 2015, 15, 116. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.C.; Okada, S.; Taylor, M.C.; Menon, A.; Mathew, A.; Cullerne, D.; Stephen, S.J.; Allen, R.S.; Zhou, X.R.; Liu, Q.; et al. Seed-specific RNAi in safflower generates a superhigh oleic oil with extended oxidative stability. Plant Biotechnol. J. 2018, 16, 1788–1796. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Jung, J.; Kim, M.S.; Lee, J.M.; Choi, D.; Yeam, I. Molecular marker development and genetic diversity exploration by RNA-seq in Platycodon grandiflorum. Genome 2015, 58, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Scintu, A.; Posadinu, C.M.; Xu, Y.; Nguyen, C.V.; Sun, H.; Bitocchi, E.; Bellucci, E.; Papa, R.; Fei, Z.; et al. GWAS Based on RNA-Seq SNPs and High-Throughput Phenotyping Combined with Climatic Data Highlights the Reservoir of Valuable Genetic Diversity in Regional Tomato Landraces. Genes 2020, 11, 1387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zeng, C.; Li, L.; Zhu, W.; Xu, L.; Wang, Y.; Zeng, J.; Fan, X.; Sha, L.; Wu, D.; et al. RNA-seq analysis revealed considerable genetic diversity and enabled the development of specific KASP markers for Psathyrostachys huashanica. Front. Plant Sci. 2023, 14, 1166710. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar] [CrossRef]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Accession Name | Species/Form | Origin |
|---|---|---|---|
| p_1 | K-1230 | L. crepitans | Portugal |
| p_2 | K-1531 | L. crepitans | Spain |
| p_3 | K-4731 | L. bienne | - |
| p_4 | Caesar | fiber | Russia |
| p_5 | VNIIMK 620 | intermediate | Russia |
| p_6 | K-495 | intermediate | Ukraine |
| p_7 | K-1295 | intermediate | Russia |
| p_8 | K-5696 | L. angustifolium | Iran |
| p_9 | K-6036 | L. angustifolium | Spain |
| p_10 | K-6060 | L. angustifolium | Italy |
| p_11 | K-6065 | L. angustifolium | France |
| p_12 | K-6066 | L. angustifolium | Canary Islands |
| p_13 | K-6077 | L. angustifolium | France |
| p_14 | K-6085 | L. angustifolium | Italy |
| p_15 | K-6099 | L. angustifolium | Tunisia |
| p_16 | K-6375 | L. angustifolium | Netherlands |
| p_17 | K-6097 | L. angustifolium | Portugal |
| p_18 | Severny | intermediate | Russia |
| p_19 | K-2864 | fiber | Ukraine |
| p_20 | K-470 | fiber | Russia |
| p_21 | Atlant | fiber | Russia |
| p_22 | Alizee | fiber | Netherlands |
| p_26 | K-1570 | crown | China |
| p_27-2 | K-2787 | crown | Georgia |
| p_28 | TR-42713 | intermediate | Turkey |
| p_29 | K-1119 | crown | Armenia |
| p_30 | K-432 | crown | Kazakhstan |
| p_31 | K-3774 | crown | Uzbekistan |
| p_32 | K-115 | crown | Tajikistan |
| p_33 | K-5543 | large-seeded | India |
| p_34 | K-568 | crown | India |
| p_35 | K-1024 | large-seeded | Morocco |
| p_36 | K-1146 | crown | Ethiopia |
| p_37 | K-519 | large-seeded | Egypt |
| p_38 | K-1743 | crown | Egypt |
| p_39 | Norlin | intermediate | Canada |
| p_40 | K-550 | crown | Argentina |
| p_41 | K-1034 | large-seeded | Argentina |
| p_42 | K-1404 | intermediate | Ukraine |
| p_43 | K-3018 | winter | Yugoslavia |
| p_47-3 | 47-3 | crown | Russia |
| p_47-4 | 47-4 | crown | Russia |
| p_48-3 and p_48-4 | 48-981 | crown | Russia |
| p_1-427 | 1-427/10 | crown | Russia |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pushkova, E.N.; Borkhert, E.V.; Novakovskiy, R.O.; Dvorianinova, E.M.; Rozhmina, T.A.; Zhuchenko, A.A.; Zhernova, D.A.; Turba, A.A.; Yablokov, A.G.; Sigova, E.A.; et al. Selection of Flax Genotypes for Pan-Genomic Studies by Sequencing Tagmentation-Based Transcriptome Libraries. Plants 2023, 12, 3725. https://doi.org/10.3390/plants12213725
Pushkova EN, Borkhert EV, Novakovskiy RO, Dvorianinova EM, Rozhmina TA, Zhuchenko AA, Zhernova DA, Turba AA, Yablokov AG, Sigova EA, et al. Selection of Flax Genotypes for Pan-Genomic Studies by Sequencing Tagmentation-Based Transcriptome Libraries. Plants. 2023; 12(21):3725. https://doi.org/10.3390/plants12213725
Chicago/Turabian StylePushkova, Elena N., Elena V. Borkhert, Roman O. Novakovskiy, Ekaterina M. Dvorianinova, Tatiana A. Rozhmina, Alexander A. Zhuchenko, Daiana A. Zhernova, Anastasia A. Turba, Arthur G. Yablokov, Elizaveta A. Sigova, and et al. 2023. "Selection of Flax Genotypes for Pan-Genomic Studies by Sequencing Tagmentation-Based Transcriptome Libraries" Plants 12, no. 21: 3725. https://doi.org/10.3390/plants12213725
APA StylePushkova, E. N., Borkhert, E. V., Novakovskiy, R. O., Dvorianinova, E. M., Rozhmina, T. A., Zhuchenko, A. A., Zhernova, D. A., Turba, A. A., Yablokov, A. G., Sigova, E. A., Krasnov, G. S., Bolsheva, N. L., Melnikova, N. V., & Dmitriev, A. A. (2023). Selection of Flax Genotypes for Pan-Genomic Studies by Sequencing Tagmentation-Based Transcriptome Libraries. Plants, 12(21), 3725. https://doi.org/10.3390/plants12213725