
Genome-Wide Identification and Characterisation of Cytokinin-O-Glucosyltransferase (CGT) Genes of Rice Specific to Potential Pathogens

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
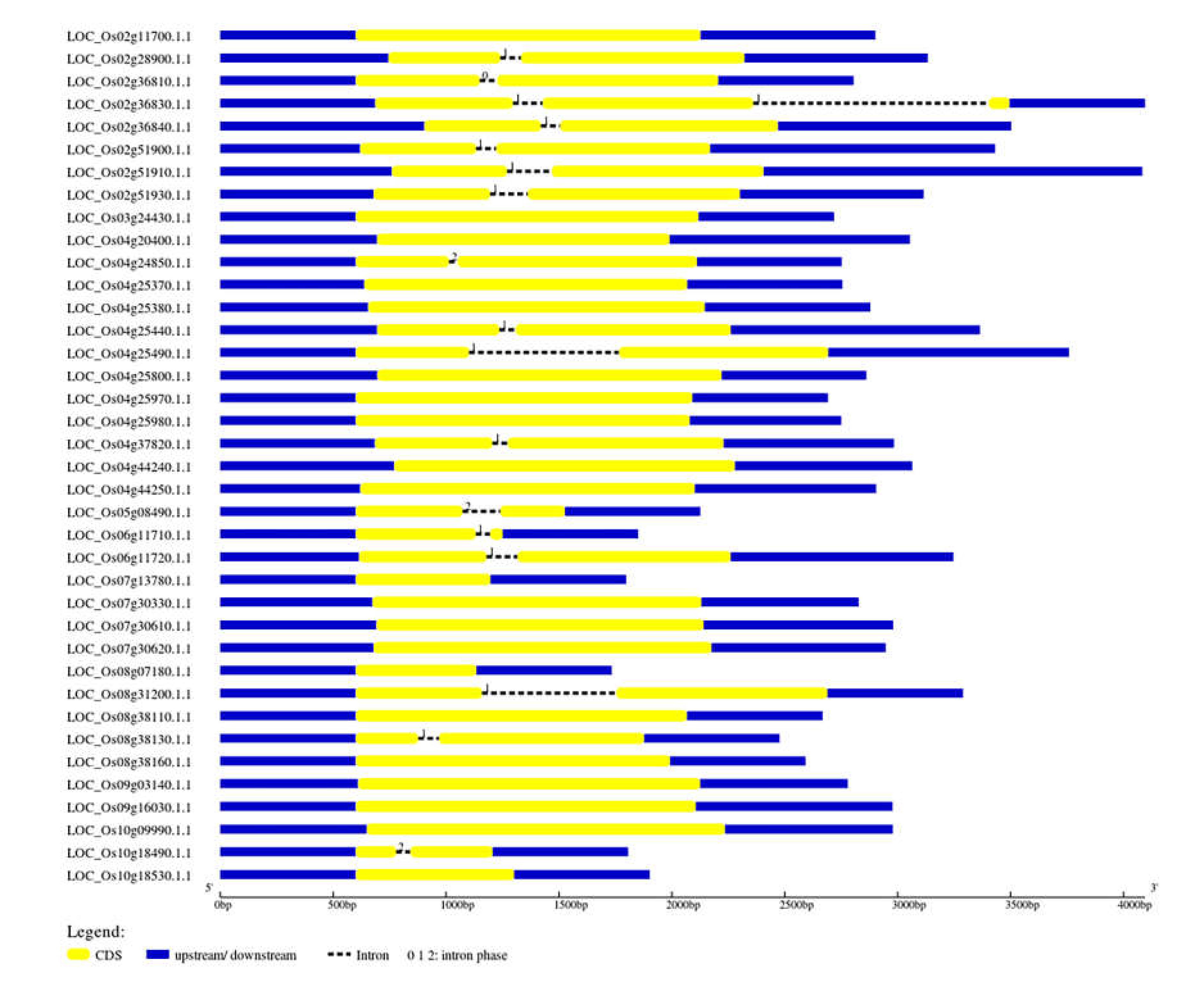
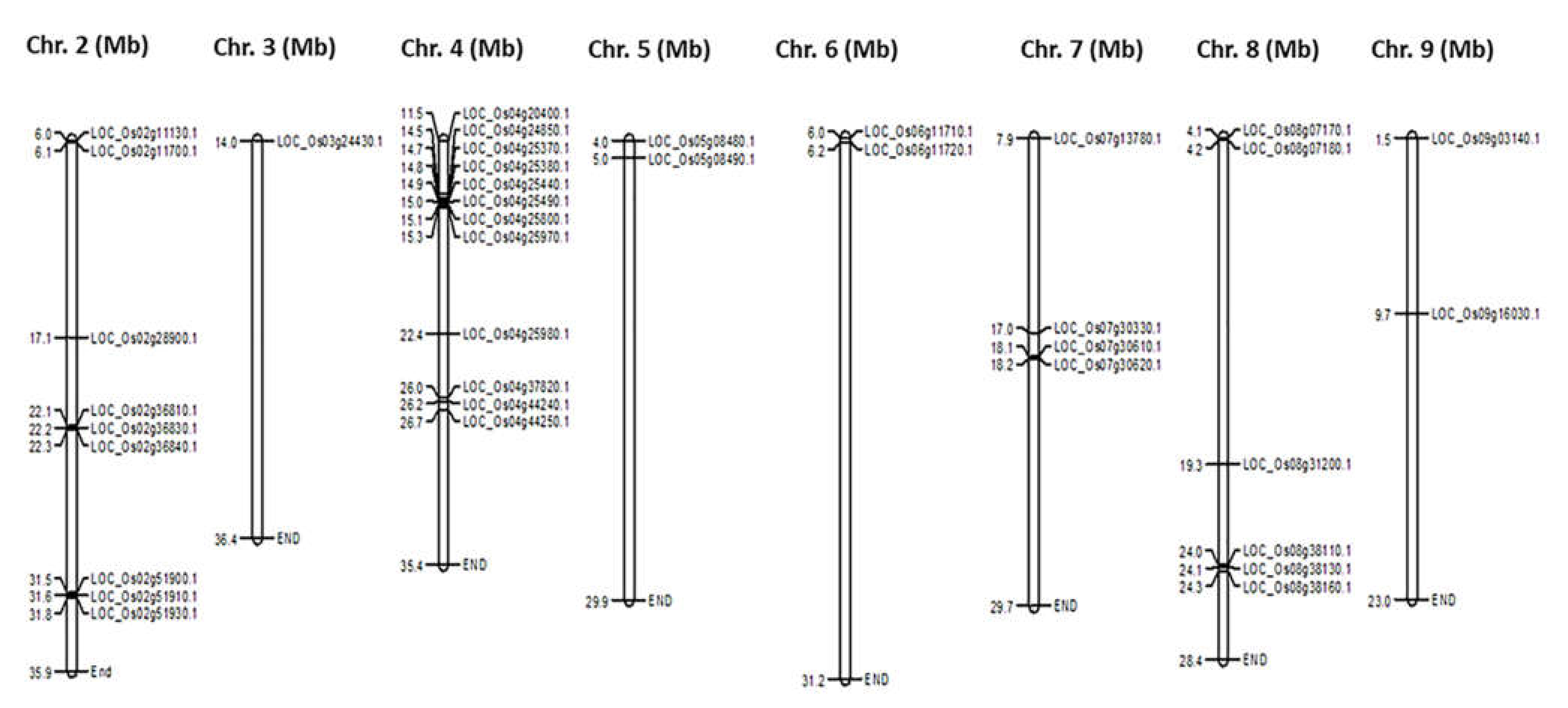
2.1. Genome Identification and Characterisation of CGT Genes
2.2. Physicochemical Characterisation of Rice CGTs
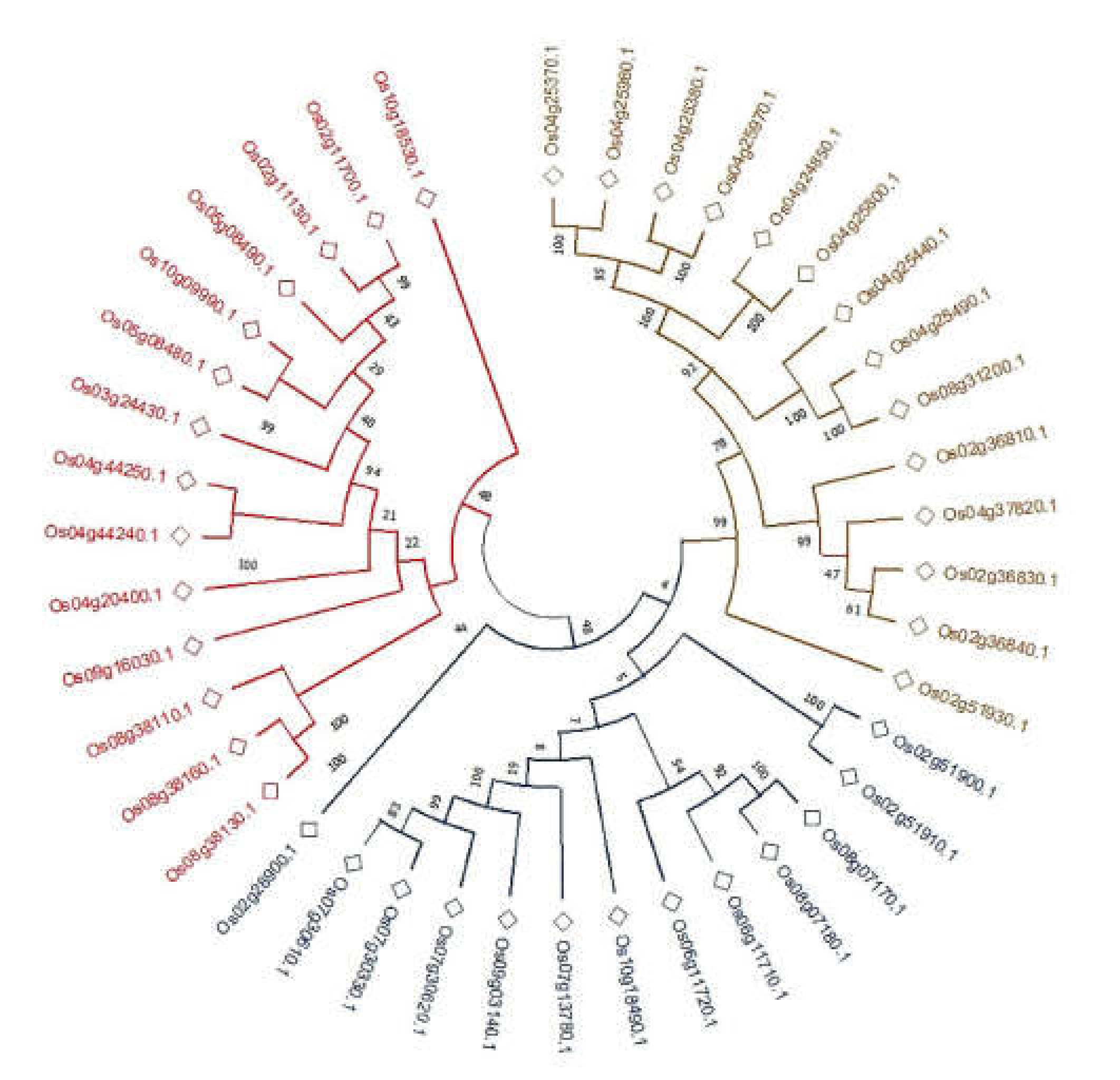
2.3. Phylogenetic Analyses of CGTs
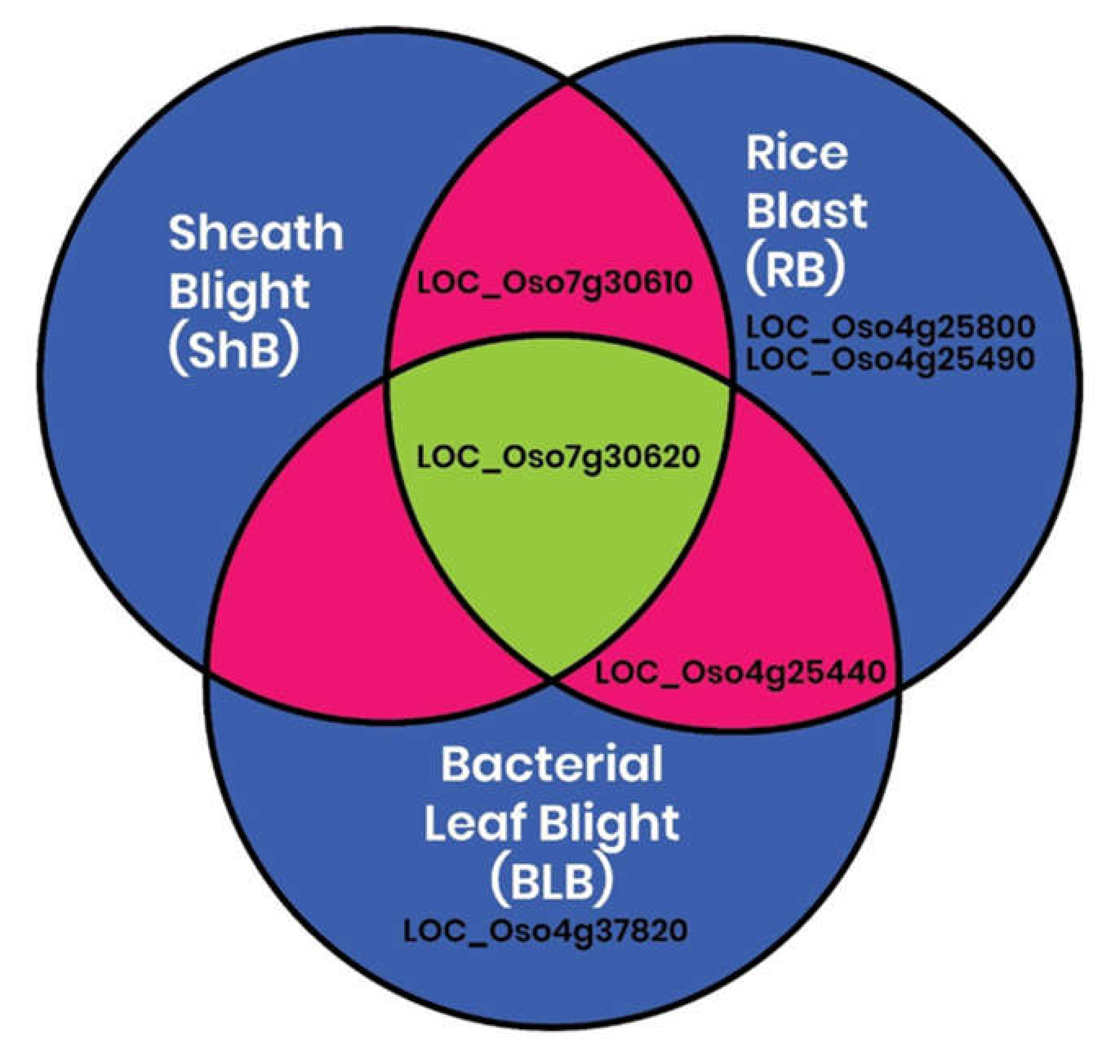
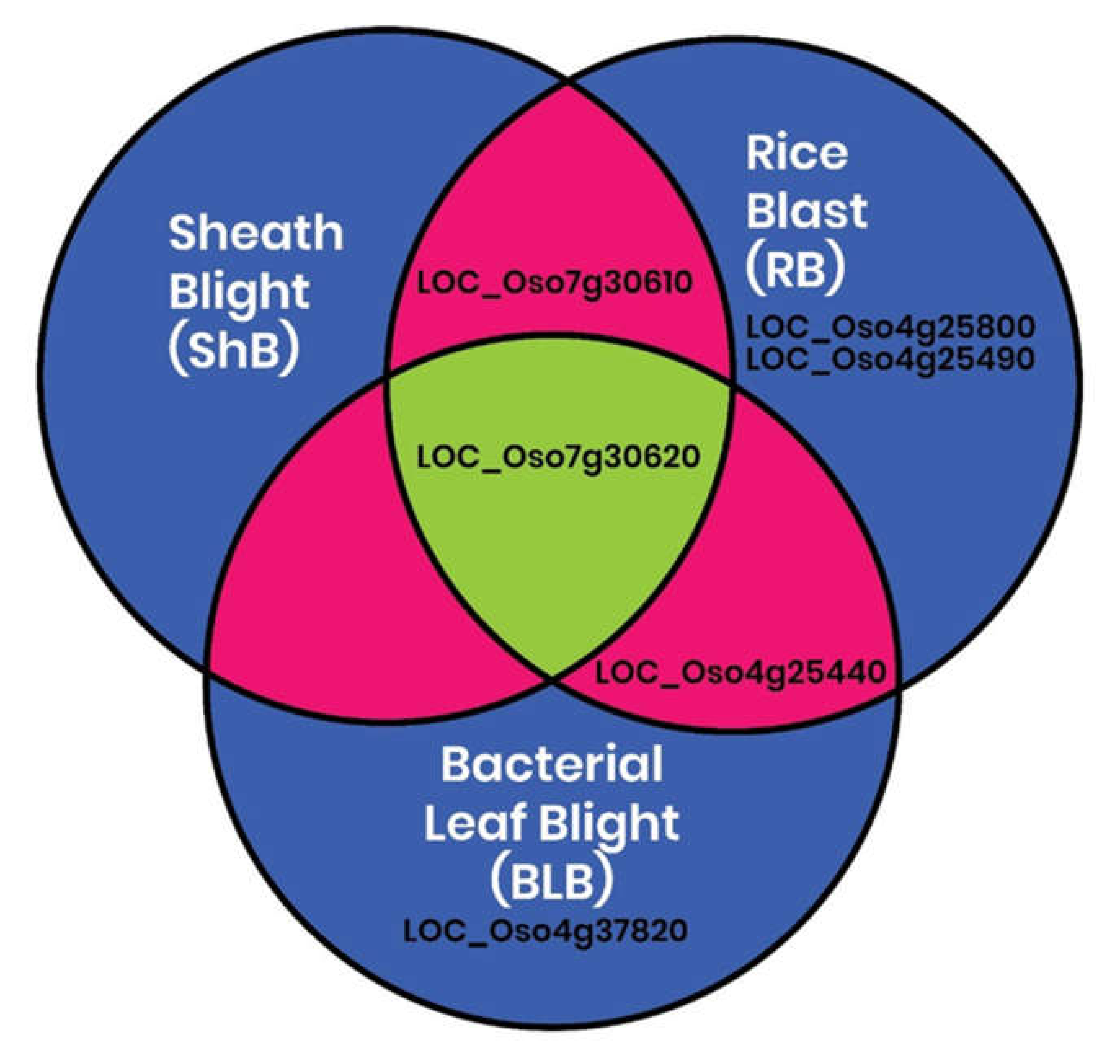
2.4. In Silico Identification of Differential Expression of CGT Genes in Rice during Growth and Pathogen Infection
2.5. Validation of the Expression of the In Silico Regulated CGT Genes by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Genome-Wide Identification and Characterisation of Rice CGT Genes
4.2. Physicochemical Characterisation of CGTs
4.3. Phylogenetic Analysis of CGTs
4.4. In Silico Expression Analysis of CGTs
4.5. Validation of the Expression of the In Silico Regulated CGT Genes
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schneider, P.; Asch, F. Rice production and food security in Asian Mega deltas—A review on characteristics, vulnerabilities and agricultural adaptation options to cope with climate change. J. Agron. Crop Sci. 2020, 206, 491–503. [Google Scholar] [CrossRef]
- Arouna, A.; Fatognon, I.A.; Saito, K.; Futakuchi, K. Moving toward rice self-sufficiency in sub-Saharan Africa by 2030: Lessons learned from 10 years of the coalition for African rice development. World Dev. Perspect. 2021, 21, 100291. [Google Scholar] [CrossRef]
- Narayanan, S.P.; Lung, S.C.; Liao, P.; Lo, C.; Chye, M.L. The overexpression of OsACBP5 protects transgenic rice against necrotrophic, hemibiotrophic and biotrophic pathogens. Sci. Rep. 2020, 10, 14918. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, J.; Genin, S.; Magori, S.; Citovsky, V.; Sriariyanum, M.; Ronald, P.; Dow, M.A.X.; Verdier, V.; Beer, S.V.; Machado, M.A.; et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 614–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jiang, N.; Liu, J.; Liu, W.; Wang, G.L. The role of effectors and host immunity in plant–necrotrophic fungal interactions. Virulence 2014, 5, 722–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djamei, A.; Schipper, K.; Rabe, F.; Ghosh, A.; Vincon, V.; Kahnt, J.; Osorio, S.; Tohge, T.; Fernie, A.R.; Feussner, I.; et al. Metabolic priming by a secreted fungal effector. Nature 2011, 478, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Patkar, R.N.; Benke, P.I.; Qu, Z.; Chen, Y.Y.; Yang, F.; Swarup, S.; Naqvi, N.I. A fungal monooxygenase-derived jasmonate attenuates host innate immunity. Nat. Chem. Biol. 2015, 11, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Uppalapati, S.R.; Ishiga, Y.; Wangdi, T.; Kunkel, B.N.; Anand, A.; Mysore, K.S.; Bender, C.L. The phytotoxin coronatine contributes to pathogen fitness and is required for suppression of salicylic acid accumulation in tomato inoculated with Pseudomonas syringae pv. tomato DC3000. Mol. Plant-Microbe Interact. 2007, 20, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Plihalova, L.; Vylicilova, H.; Dolezal, K.; Zahajska, L.; Zatloukal, M.; Strnad, M. Synthesis of aromatic cytokinins for plant biotechnology. New Biotechnol. 2016, 33, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, M.A.; Nelson, D.C.; Weijers, D. Evolution of plant hormone response pathways. Annu. Rev. Plant Biol. 2020, 71, 327–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhao, J.; Song, J.; Jameson, P.E. Cytokinin glucosyltransferases, key regulators of cytokinin homeostasis, have potential value for wheat improvement. Plant Biotechnol. J. 2021, 19, 878–896. [Google Scholar] [CrossRef]
- Albrecht, T.; Argueso, C.T. Should I fight or should I grow now? The role of cytokinins in plant growth and immunity and in the growth-defence trade-off. Ann. Bot. 2017, 119, 725–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, E.N.; Emery, R.N.; Saville, B.J. Phytohormone involvement in the Ustilago maydis–Zea mays pathosystem: Relationships between abscisic acid and cytokinin levels and strain virulence in infected cob tissue. PLoS ONE 2015, 24, e0130945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinsch, J.; Vrabka, J.; Oeser, B.; Novák, O.; Galuszka, P.; Tudzynski, P. De novo biosynthesis of cytokinins in the biotrophic fungus Claviceps purpurea. Environ. Microbiol. 2015, 17, 2935–2951. [Google Scholar] [CrossRef]
- Devos, S.; Laukens, K.; Deckers, P.; Van Der Straeten, D.; Beeckman, T.; Inzé, D.; Van Onckelen, H.; Witters, E.; Prinsen, E. A hormone and proteome approach to picturing the initial metabolic events during Plasmodiophora brassicae infection on Arabidopsis. Mol. Plant-Microbe Interact. 2006, 19, 1431–1443. [Google Scholar] [CrossRef] [Green Version]
- Pertry, I.; Václavíková, K.; Depuydt, S.; Galuszka, P.; Spíchal, L.; Temmerman, W.; Stes, E.; Schmülling, T.; Kakimoto, T.; Van Montagu, M.C.; et al. Identification of Rhodococcus fascians cytokinins and their modus operandi to reshape the plant. Proc. Natl. Acad. Sci. USA 2009, 106, 929–934. [Google Scholar] [CrossRef] [Green Version]
- Chanclud, E.; Kisiala, A.; Emery, N.R.J.; Chalvon, V.; Ducasse, A.; Romiti-Michel, C.; Gravot, A.; Kroj, T.; Morel, J.B. Cytokinin production by the rice blast fungus is a pivotal requirement for full virulence. PLoS Pathog. 2016, 12, e1005457. [Google Scholar] [CrossRef] [Green Version]
- Morrison, E.N.; Emery, R.J.N.; Saville, B.J. Fungal derived cytokinins are necessary for normal Ustilago maydis infection of maise. Plant Pathol. 2017, 66, 726–742. [Google Scholar] [CrossRef]
- Wingler, A.; von Schaewen, A.; Leegood, R.C.; Lea, P.J.; Paul Quick, W. Regulation of leaf senescence by cytokinin, sugars, and light: Effects on NADH-dependent hydroxypyruvate reductase. Plant Physiol. 1998, 116, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Hann, D.R.; Domínguez-Ferreras, A.; Motyka, V.; Dobrev, P.I.; Schornack, S.; Jehle, A.; Felix, G.; Chinchilla, D.; Rathjen, J.P.; Boller, T. The Pseudomonas type III effector HopQ1 activates cytokinin signaling and interferes with plant innate immunity. New Phytol. 2014, 201, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Ciaglia, E.; Abate, M.; Laezza, C.; Pisanti, S.; Vitale, M.; Seneca, V.; Torelli, G.; Franceschelli, S.; Catapano, G.; Gazzerro, P.; et al. Antiglioma effects of N6-isopentenyladenosine, an endogenous isoprenoid end product, through the downregulation of epidermal growth factor receptor. Int. J. Cancer 2017, 140, 959–972. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Lei, K.; Li, Y.; He, X.; Wang, S.; Liu, R.; Ji, L.; Hou, B. Identification and characterisation of the first cytokinin glycosyltransferase from rice. Rice 2019, 12, 19. [Google Scholar] [CrossRef]
- Šmehilová, M.; Dobrůšková, J.; Novák, O.; Takáč, T.; Galuszka, P. Cytokinin-specific glycosyltransferases possess different roles in cytokinin homeostasis maintenance. Front. Plant Sci. 2016, 7, 1264. [Google Scholar] [CrossRef] [Green Version]
- Teze, D.; Coines, J.; Fredslund, F.; Dubey, K.D.; Bidart, G.N.; Adams, P.D.; Dueber, J.E.; Svensson, B.; Rovira, C.; Welner, D.H. O-/N-/S-Specificity in glycosyltransferase catalysis: From mechanistic understanding to engineering. ACS Catal. 2021, 11, 1810–1815. [Google Scholar] [CrossRef]
- Caputi, L.; Malnoy, M.; Goremykin, V.; Nikiforova, S.; Martens, S. A genome-wide phylogenetic reconstruction of family 1 UDP-glycosyltransferases revealed the expansion of the family during the adaptation of plants to life on land. Plant J. 2012, 69, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Gachon, C.M.; Langlois-Meurinne, M.; Saindrenan, P. Plant secondary metabolism glycosyltransferases: The emerging functional analysis. Trends Plant Sci. 2005, 10, 542–549. [Google Scholar] [CrossRef]
- Sawada, S.Y.; Suzuki, H.; Ichimaida, F.; Yamaguchi, M.A.; Iwashita, T.; Fukui, Y.; Hemmi, H.; Nishino, T.; Nakayama, T. UDP-glucuronic acid: Anthocyanin glucuronosyltransferase from red daisy (Bellis perennis) flowers: Enzymology and phylogenetics of a novel glucuronosyltransferase involved in flower pigment biosynthesis. J. Biol. Chem. 2005, 280, 899–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terasaka, K.; Mizutani, Y.; Nagatsu, A.; Mizukami, H. In situ UDP-glucose regeneration unravels diverse functions of plant secondary product glycosyltransferases. FEBS Lett. 2012, 586, 4344–4350. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, X.M.; Kojima, M.; Sakakibara, H.; Hou, B.K. N-glucosyltransferase UGT76C2 is involved in cytokinin homeostasis and cytokinin response in Arabidopsis thaliana. Plant Cell Physiol. 2011, 52, 2200–2213. [Google Scholar] [CrossRef] [Green Version]
- Zeier, J. Metabolic regulation of systemic acquired resistance. Curr. Opin. Plant Biol. 2021, 62, 102050. [Google Scholar] [CrossRef]
- Costanzo, S.; Jackson, A.K.; Brooks, S.A. High-resolution mapping of Rsn1, a locus controlling sensitivity of rice to a necrosis-inducing phytotoxin from Rhizoctonia solani AG1-IA. Theor. Appl. Genet. 2011, 123, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Vidhyasekaran, P.; Ponmalar, T.R.; Samiyappan, R.; Velazhahan, R.; Vimala, R.; Ramanathan, A.; Paranidharan, V.; Muthukrishnan, S. Host-specific toxin production by Rhizoctonia solani, the rice sheath blight pathogen. Phytopathology 1997, 87, 1258–1263. [Google Scholar] [CrossRef] [Green Version]
- Ke, Y.; Hui, S.; Yuan, M. Xanthomonas oryzae pv. oryzae inoculation and growth rate on rice by leaf clipping method. Bio-protocol 2017, 7, e2568. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.S.; Kumar, P.; Singh, I. Structural and functional analysis of protein. In Bioinformatics; Academic Press: Cambridge, MA, USA, 2022; pp. 189–206. [Google Scholar]
- Moon, S.; Kim, S.R.; Zhao, G.; Yi, J.; Yoo, Y.; Jin, P.; Lee, S.W.; Jung, K.H.; Zhang, D.; An, G. Rice glycosyltransferase1 encodes a glycosyltransferase essential for pollen wall formation. Plant Physiol. 2013, 161, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Ullah, F.; Zhou, D.X.; Yi, M.; Zhao, Y. Mechanisms of ROS regulation of plant development and stress responses. Front. Plant Sci. 2019, 10, 800. [Google Scholar] [CrossRef]
- Vogt, T.; Jones, P. Glycosyltransferases in plant natural product synthesis: Characterisation of a supergene family. Trends Plant Sci. 2000, 5, 380–386. [Google Scholar] [CrossRef]
- Yonekura-Sakakibara, K.; Hanada, K. An evolutionary view of functional diversity in family 1 glycosyltransferases. Plant J. 2011, 66, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.J.; Bartley, L.E.; Jung, K.H.; Ronald, P.C. Construction of a rice glycosyltransferase phylogenomic database and identification of rice-diverged glycosyltransferases. Mol. Plant 2008, 1, 858–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, P.I.; Owens, I.S.; Burchell, B.; Bock, K.W.; Bairoch, A.; Belanger, A.; Fournel-Gigleux, S.; Green, M.; Hum, D.W.; Iyanagi, T.; et al. The UDP glycosyltransferase gene superfamily: Recommended nomenclature update based on evolutionary divergence. Pharmacogenetics 1997, 7, 255–269. [Google Scholar] [CrossRef]
- He, Y.; Ahmad, D.; Zhang, X.; Zhang, Y.; Wu, L.; Jiang, P.; Ma, H. Genome-wide analysis of family-1 UDP glycosyltransferases (UGT) and identification of UGT genes for FHB resistance in wheat (Triticum aestivum L.). BMC Plant Biol. 2018, 18, 67. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Lu, Q.; Liu, R.; Gong, J.; Gong, W.; Liu, A.; Ge, Q.; Li, J.; Shang, H.; Li, P.; et al. Genome-wide characterisation of the UDP-glycosyltransferase gene family in upland cotton. 3 Biotech 2019, 9, 453. [Google Scholar] [CrossRef] [PubMed]
- Grabski, D.F.; Broseus, L.; Kumari, B.; Rekosh, D.; Hammarskjold, M.L.; Ritchie, W. Intron retention and its impact on gene expression and protein diversity: A review and a practical guide. Wiley Interdiscip. Rev. RNA 2021, 12, e1631. [Google Scholar] [CrossRef]
- Kumar, V.; Jain, P.; Venkadesan, S.; Karkute, S.G.; Bhati, J.; Abdin, M.Z.; Sevanthi, A.M.; Mishra, D.C.; Chaturvedi, K.K.; Rai, A.; et al. Understanding rice-Magnaporthe oryzae interaction in resistant and susceptible cultivars of rice under panicle blast infection using a time-course transcriptome analysis. Genes 2021, 12, 301. [Google Scholar] [CrossRef] [PubMed]
- Nedvěd, D.; Hošek, P.; Klíma, P.; Hoyerová, K. Differential subcellular distribution of cytokinins: How does membrane transport fit into the big picture? Int. J. Mol. Sci. 2021, 22, 3428. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Juez, E.; Pyke, K.A. Plastids unleashed: Their development and their integration in plant development. Int. J. Dev. Biol. 2004, 49, 557–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Sheng, Z.; Cai, Y.; Li, Q.; Wei, X.; Xie, L.; Jiao, G.; Shao, G.; Tang, S.; Wang, J.; et al. Rice morphogenesis and chlorophyll accumulation is regulated by the protein encoded by NRL3 and its interaction with NAL9. Front. Plant Sci. 2019, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Liang, G.; Yang, S.; Yu, D. Arabidopsis WRKY57 functions as a node of convergence for jasmonic acid- and auxin-mediated signaling in jasmonic acid-induced leaf senescence. Plant Cell. 2014, 26, 230–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babiychuk, E.; Vandepoele, K.; Wissing, J.; Garcia-Diaz, M.; De Rycke, R.; Akbari, H.; Joubès, J.; Beeckman, T.; Jänsch, L.; Frentzen, M.; et al. Plastid gene expression and plant development require a plastidic protein of the mitochondrial transcription termination factor family. Proc. Natl. Acad. Sci. USA 2011, 108, 6674–6679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, W.G.; Romanov, G.A.; Köllmer, I.; Bürkle, L.; Schmülling, T. Immediate-early and delayed cytokinin response genes of Arabidopsis thaliana identified by genome-wide expression profiling reveal novel cytokinin-sensitive processes and suggest cytokinin action through transcriptional cascades. Plant J. 2005, 44, 314–333. [Google Scholar] [CrossRef] [PubMed]
- Polanská, L.; Vičánková, A.; Nováková, M.; Malbeck, J.; Dobrev, P.I.; Brzobohatý, B.; Vaňková, R.; Macháčková, I. Altered cytokinin metabolism affects cytokinin, auxin, and abscisic acid contents in leaves and chloroplasts, and chloroplast ultrastructure in transgenic tobacco. J. Exp. Bot. 2007, 58, 637–649. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.E.; Tian, L. Phylogenomic analysis of UDP-dependent glycosyltransferases provides insights into the evolutionary landscape of glycosylation in plant metabolism. Plant J. 2019, 100, 1273–1288. [Google Scholar] [CrossRef]
- Passardi, F.; Longet, D.; Penel, C.; Dunand, C. The class III peroxidase multigenic family in rice and its evolution in land plants. Phytochemistry 2004, 65, 1879–1893. [Google Scholar] [CrossRef] [PubMed]
- Kurakawa, T.; Ueda, N.; Maekawa, M.; Kobayashi, K.; Kojima, M.; Nagato, Y.; Sakakibara, H.; Kyozuka, J. Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature 2007, 445, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.Y.; Chen, H.W.; Ng, C.Y.; Lin, C.Y.; Tseng, T.H.; Li, W.H.; Ku, M.S.B. Down-regulation of cytokinin oxidase 2 expression increases tiller number and improves rice yield. Rice 2015, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Harrop, T.W.; Ud Din, I.; Gregis, V.; Osnato, M.; Jouannic, S.; Adam, H.; Kater, M.M. Gene expression profiling of reproductive meristem types in early rice inflorescences by laser microdissection. Plant J. 2016, 86, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Pizarro, L.; Leibman-Markus, M.; Marash, I.; Bar, M. Cytokinin response induces immunity and fungal pathogen resistance, and modulates trafficking of the PRR LeEIX2 in tomato. Mol. Plant Pathol. 2020, 21, 1287–1306. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Choi, D.; Lee, S.; Ryu, C.-M.; Hwang, I. Cytokinins and plant immunity: Old foes or new friends? Trends Plant Sci. 2011, 16, 388–394. [Google Scholar]
- Robert-Seilaniantz, A.; Navarro, L.; Bari, R.; Jones, J.D. Pathological hormone imbalances. Curr. Opin. Plant Biol. 2007, 10, 372–379. [Google Scholar] [CrossRef]
- Kong, W.; Ding, L.; Xia, X. Identification and characterisation of genes frequently responsive to Xanthomonas oryzae pv. oryzae and Magnaporthe oryzae infections in rice. BMC Genom. 2020, 21, 21. [Google Scholar] [CrossRef]
- Brooks, S.A. Sensitivity to a phytotoxin from Rhizoctonia solani correlates with sheath blight susceptibility in rice. Phytopathology 2007, 97, 1207–1212. [Google Scholar] [CrossRef] [Green Version]
- Vleeshouwers, V.G.; Oliver, R.P. Effectors as tools in disease resistance breeding against biotrophic, hemibiotrophic, and necrotrophic plant pathogens. Mol. Plant-Microbe Interact. 2014, 27, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, B.; Lim, E.K.; Higgins, G.S.; Bowles, D.J. N-glucosylation of cytokinins by glycosyltransferases of Arabidopsis thaliana. J. Biol. Chem. 2004, 279, 47822–47832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, F.J.; Wang, X.L.; Xu, W.L.; Li, X.B. Characterization and expression analysis of two cotton genes encoding putative UDP-Glycosyltransferases. Mol. Biol. 2008, 42, 44–51. [Google Scholar] [CrossRef]
- Ishii, K.; Kubo, H.; Yamasaki, R. Synthesis of α-lactosyl-(1→3)-l-glycero-α-d-manno-heptopyranoside, a partial oligosaccharide structure expressed within the lipooligosaccharide produced by Neisseria gonorrhoeae strain 15253. Carbohydr. Res. 2002, 337, 11–20. [Google Scholar] [CrossRef]
- Barvkar, V.T.; Pardeshi, V.C.; Kale, S.M.; Kadoo, N.Y.; Gupta, V.S. Phylogenomic analysis of UDP glycosyltransferase 1 multigene family in Linum usitatissimum identified genes with varied expression patterns. BMC Genom. 2012, 13, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2006, 93, 77–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Zhang, Z.; Mei, Q.; Chen, M. PSI: A comprehensive and integrative approach for accurate plant subcellular localisation prediction. PLoS ONE 2013, 8, e75826. [Google Scholar] [CrossRef] [PubMed]
- Sureshkumar, V.; Dutta, B.; Kumar, V.; Prakash, G.; Mishra, D.C.; Chaturvedi, K.K.; Rai, A.; Sevanthi, A.M.; Solanke, A.U. RiceMetaSysB: A database of blast and bacterial blight responsive genes in rice and its utilisation in identifying key blast-resistant WRKY genes. Database 2019, 2019, baz015. [Google Scholar] [CrossRef]
- Hruz, T.; Laule, O.; Szabo, G.; Wessendorp, F.; Bleuler, S.; Oertle, L.; Widmayer, P.; Gruissem, W.; Zimmermann, P. Genevestigator v3: A reference expression database for the meta-analysis of transcriptomes. Adv. Bioinform. 2008, 2008, 420747. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Stable ID/ Locus Name | MW (kDa) | PL | PI | II | AI | GRAVY | SL |
|---|---|---|---|---|---|---|---|
| LOC_Os02g11130.1 | 54.60 | 501 | 5.1286 | 35.06 | 90.58 | 0.035 | Plasma membrane |
| LOC_Os02g11700.1 | 55.10 | 508 | 5.1228 | 39.92 | 84.97 | −0.043 | Plasma membrane |
| LOC_Os02g28900.1 | 53.30 | 494 | 4.7694 | 36.37 | 86.54 | 0.034 | Plastid |
| LOC_Os02g36810.1 | 54.20 | 508 | 5.7206 | 40.99 | 84.13 | 0.006 | Plasma membrane |
| LOC_Os02g36830.1 | 59.70 | 544 | 5.7414 | 37.71 | 82.27 | −0.081 | Plastid |
| LOC_Os02g36840.1 | 54.30 | 493 | 5.9562 | 40.3 | 80.52 | −0.141 | Mitochondria |
| LOC_Os02g51900.1 | 53.50 | 486 | 5.1939 | 43.07 | 82.82 | −0.089 | Plasma membrane |
| LOC_Os02g51910.1 | 52.80 | 482 | 5.6463 | 41.46 | 81.46 | −0.091 | Plastid |
| LOC_Os02g51930.1 | 53.50 | 485 | 5.8247 | 40.92 | 83.95 | −0.18 | Plasma membrane |
| LOC_Os03g24430.1 | 54.00 | 505 | 6.511 | 40.82 | 88.44 | 0.06 | Plastid |
| LOC_Os04g20400.1 | 47.30 | 431 | 6.6482 | 58.07 | 84.79 | −0.192 | Plastid |
| LOC_Os04g24850.1 | 53.00 | 490 | 5.7159 | 37.36 | 89.84 | −0.05 | Plastid |
| LOC_Os04g25370.1 | 52.10 | 476 | 5.9424 | 38.31 | 86.56 | −0.052 | Plasma membrane |
| LOC_Os04g25380.1 | 53.90 | 496 | 6.8259 | 42.23 | 88.62 | −0.033 | Plastid |
| LOC_Os04g25440.1 | 54.00 | 497 | 6.1201 | 45.32 | 83.44 | −0.043 | Mitochondria |
| LOC_Os04g25490.1 | 51.30 | 475 | 4.8767 | 43.16 | 86.26 | −0.029 | Plasma membrane |
| LOC_Os04g25800.1 | 54.70 | 507 | 5.5869 | 37.86 | 89.73 | −0.009 | Plastid |
| LOC_Os04g25970.1 | 53.80 | 496 | 6.6935 | 41.24 | 88.24 | −0.03 | Plastid |
| LOC_Os04g25980.1 | 53.90 | 492 | 6.3781 | 40.63 | 86.93 | −0.057 | Mitochondria |
| LOC_Os04g37820.1 | 54.10 | 491 | 5.5712 | 44.92 | 78.24 | −0.136 | Plasma membrane |
| LOC_Os04g44240.1 | 53.90 | 502 | 6.1602 | 39.13 | 87.54 | 0.086 | Cytosol |
| LOC_Os04g44250.1 | 53.10 | 493 | 6.857 | 43.54 | 89.88 | 0.096 | Extracellular |
| LOC_Os05g08480.1 | 56.70 | 544 | 6.6946 | 44.16 | 92.14 | 0.189 | Plasma membrane |
| LOC_Os05g08490.1 | 28.60 | 252 | 5.841 | 39.17 | 89.8 | −0.033 | Plastid |
| LOC_Os06g11710.1 | 20.80 | 195 | 9.5658 | 52.36 | 83.89 | −0.088 | Cytosol |
| LOC_Os06g11720.1 | 54.30 | 502 | 5.683 | 52.75 | 85.23 | −0.108 | Mitochondria |
| LOC_Os07g13780.1 | 20.80 | 198 | 9.0927 | 52.87 | 104.43 | 0.266 | Plastid |
| LOC_Os07g30330.1 | 52.10 | 485 | 5.7028 | 40.05 | 94.41 | 0.194 | Mitochondria |
| LOC_Os07g30610.1 | 51.60 | 482 | 5.2655 | 50.19 | 89.79 | 0.071 | Plastid |
| LOC_Os07g30620.1 | 52.90 | 498 | 6.1141 | 46.23 | 89.9 | 0.134 | Plastid |
| LOC_Os08g07170.1 | 23.20 | 217 | 7.7053 | 27.25 | 87.5 | −0.065 | Plasma membrane |
| LOC_Os08g07180.1 | 19.00 | 177 | 7.6701 | 26.17 | 92.22 | −0.049 | Cytosol |
| LOC_Os08g31200.1 | 54.10 | 497 | 5.2116 | 42.04 | 85.18 | −0.038 | Cytosol |
| LOC_Os08g38110.1 | 50.90 | 488 | 7.2976 | 49.36 | 88.78 | 0.191 | Plasma membrane |
| LOC_Os08g38130.1 | 41.90 | 394 | 6.0123 | 41.66 | 89.32 | 0.059 | Extracellular |
| LOC_Os08g38160.1 | 49.00 | 463 | 6.1379 | 39.73 | 95.82 | 0.238 | Plastid |
| LOC_Os09g03140.1 | 54.90 | 504 | 4.8158 | 48.76 | 96.19 | 0.023 | Plastid |
| LOC_Os09g16030.1 | 54.10 | 501 | 5.3122 | 34.3 | 91.19 | 0.1 | Plastid |
| LOC_Os10g09990.1 | 56.30 | 528 | 4.768 | 32.3 | 92.86 | 0.085 | Plasma membrane |
| LOC_Os10g18490.1 | 19.50 | 180 | 6.7946 | 45.69 | 90.6 | −0.014 | Plasma membrane |
| LOC_Os10g18530.1 | 25.80 | 233 | 6.1427 | 87.16 | 87.16 | 0.134 | Extracellular |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dauda, W.P.; Shanmugam, V.; Tyagi, A.; Solanke, A.U.; Kumar, V.; Krishnan, S.G.; Bashyal, B.M.; Aggarwal, R. Genome-Wide Identification and Characterisation of Cytokinin-O-Glucosyltransferase (CGT) Genes of Rice Specific to Potential Pathogens. Plants 2022, 11, 917. https://doi.org/10.3390/plants11070917
Dauda WP, Shanmugam V, Tyagi A, Solanke AU, Kumar V, Krishnan SG, Bashyal BM, Aggarwal R. Genome-Wide Identification and Characterisation of Cytokinin-O-Glucosyltransferase (CGT) Genes of Rice Specific to Potential Pathogens. Plants. 2022; 11(7):917. https://doi.org/10.3390/plants11070917
Chicago/Turabian StyleDauda, Wadzani Palnam, Veerubommu Shanmugam, Aditya Tyagi, Amolkumar U. Solanke, Vishesh Kumar, Subbaiyan Gopala Krishnan, Bishnu Maya Bashyal, and Rashmi Aggarwal. 2022. "Genome-Wide Identification and Characterisation of Cytokinin-O-Glucosyltransferase (CGT) Genes of Rice Specific to Potential Pathogens" Plants 11, no. 7: 917. https://doi.org/10.3390/plants11070917
APA StyleDauda, W. P., Shanmugam, V., Tyagi, A., Solanke, A. U., Kumar, V., Krishnan, S. G., Bashyal, B. M., & Aggarwal, R. (2022). Genome-Wide Identification and Characterisation of Cytokinin-O-Glucosyltransferase (CGT) Genes of Rice Specific to Potential Pathogens. Plants, 11(7), 917. https://doi.org/10.3390/plants11070917