
Transcriptome Analysis of Wheat–Tilletia indica Interaction Provides Defense and Pathogenesis-Related Genes

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. RNA Sequence Analysis and Mapping with Reference Wheat Genome
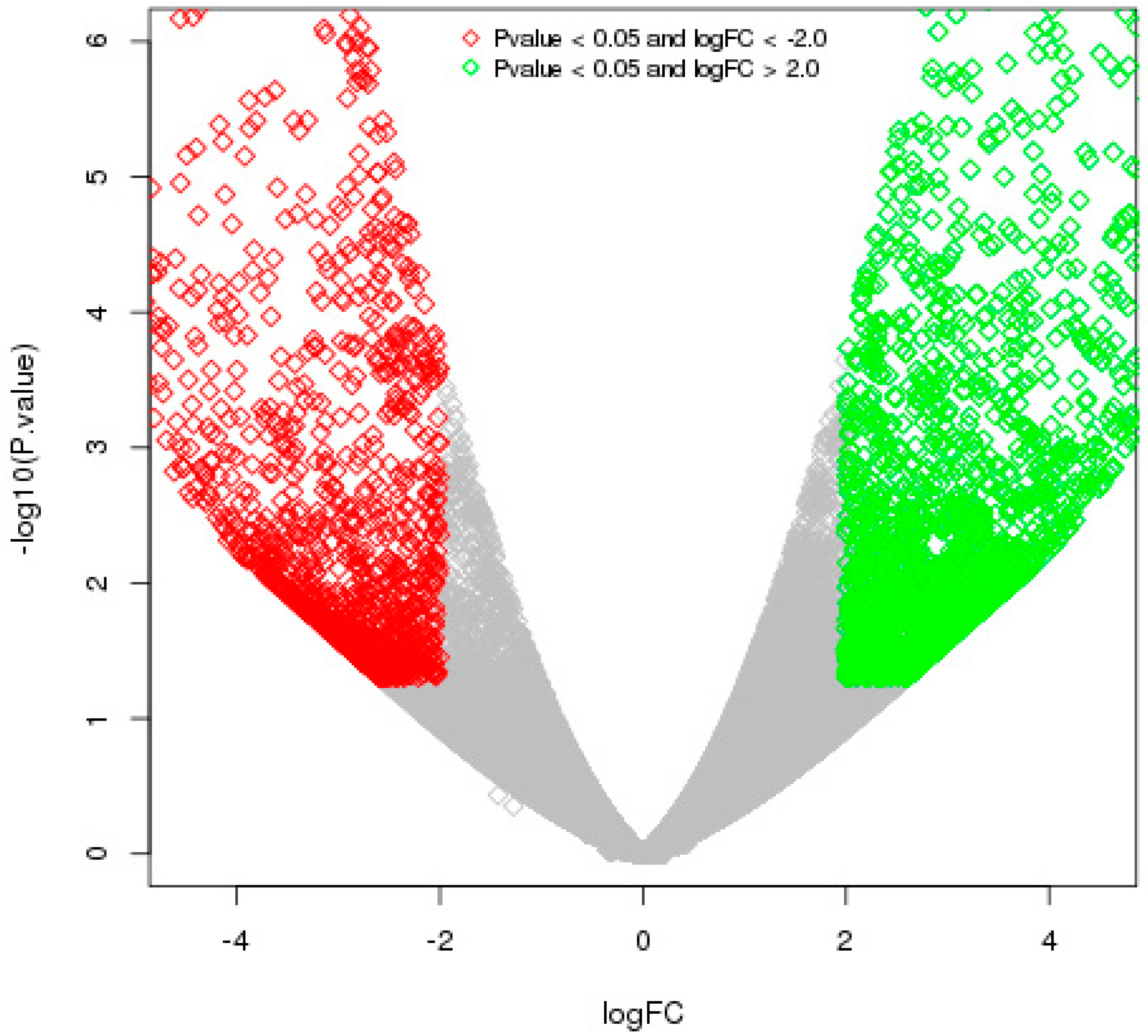
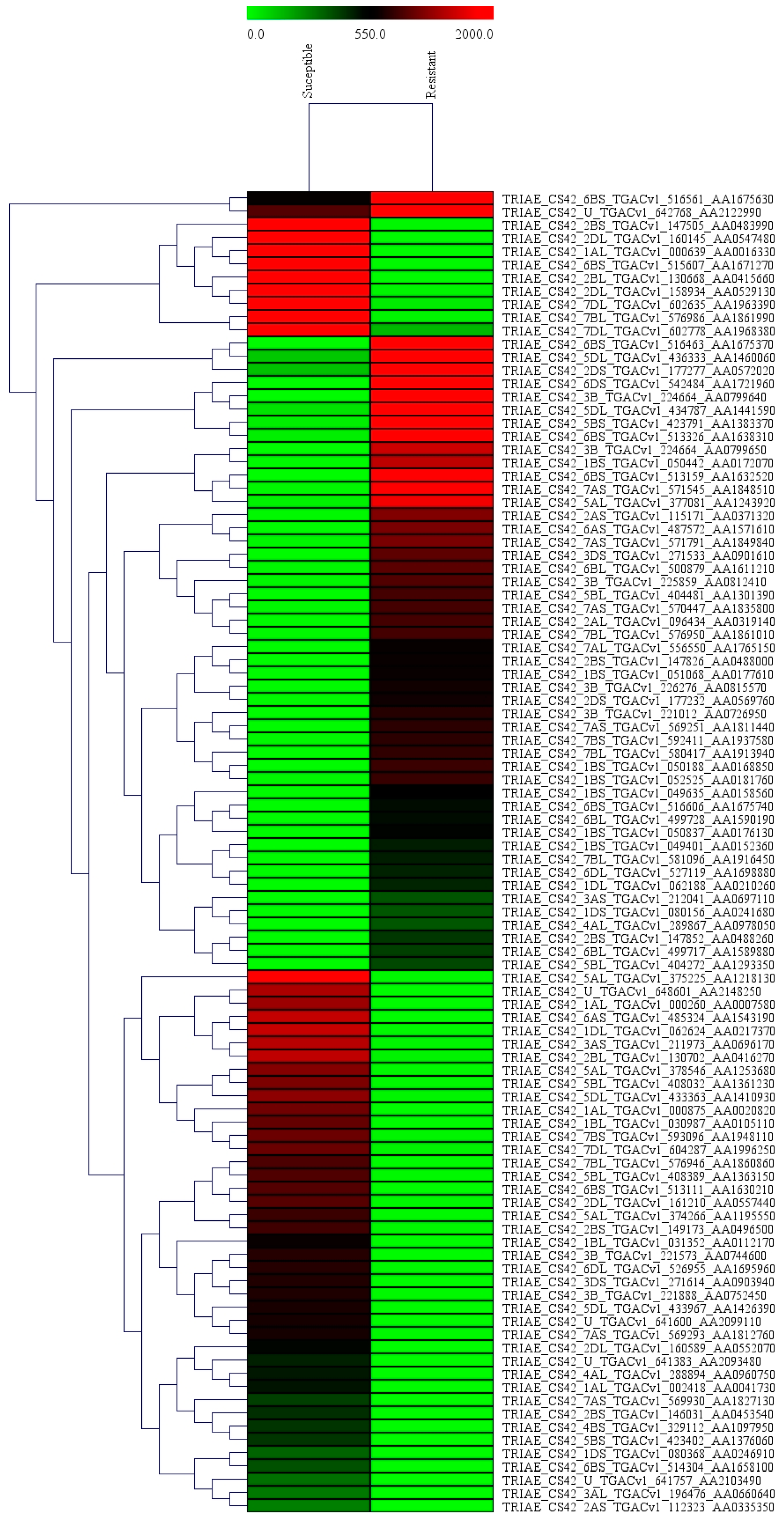
2.2. Differential Gene Expression Patterns
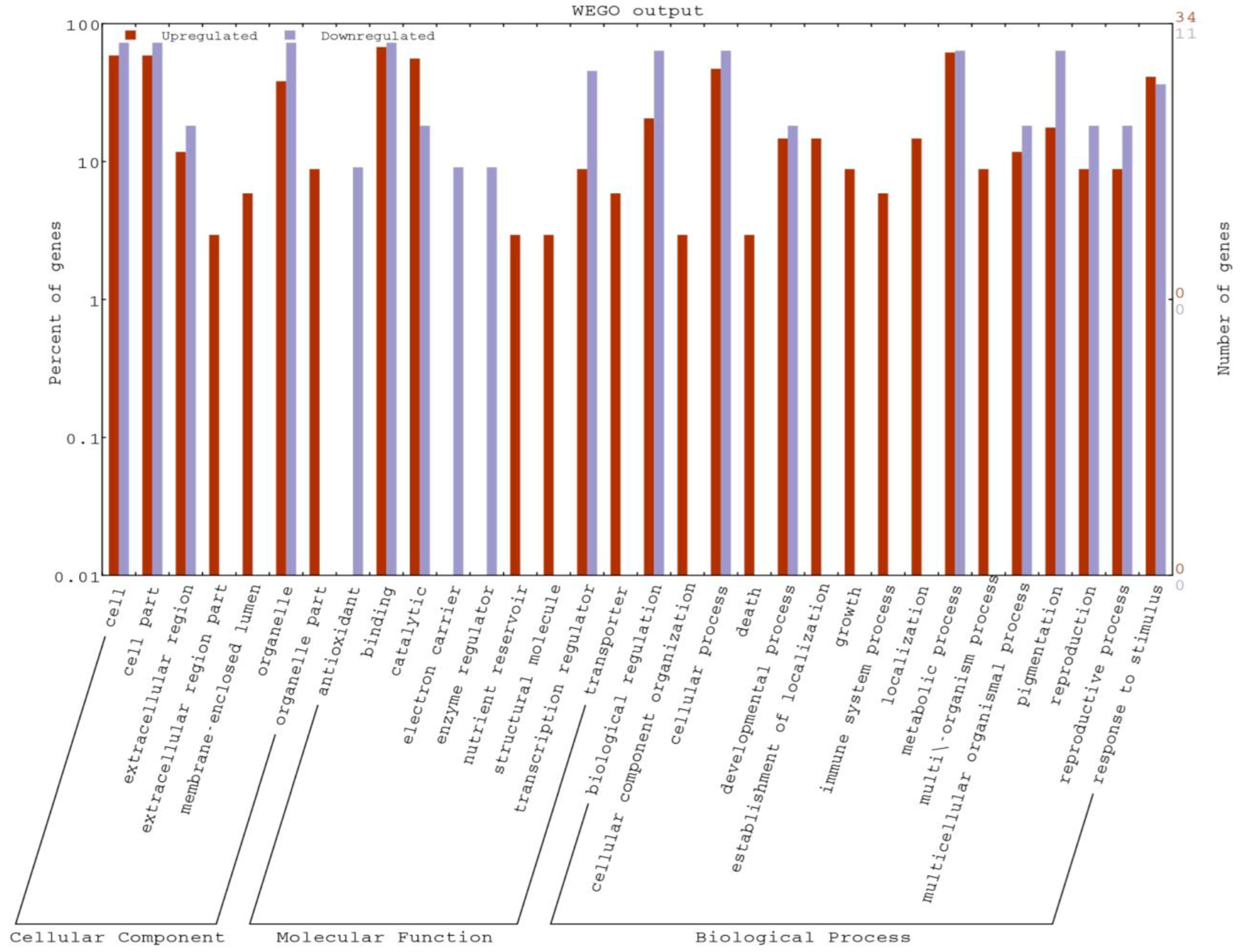
2.3. Gene Ontology Enrichment Study of DEGs and Pathways Analysis
2.4. Mapping and DEGs in T. indica Transcripts
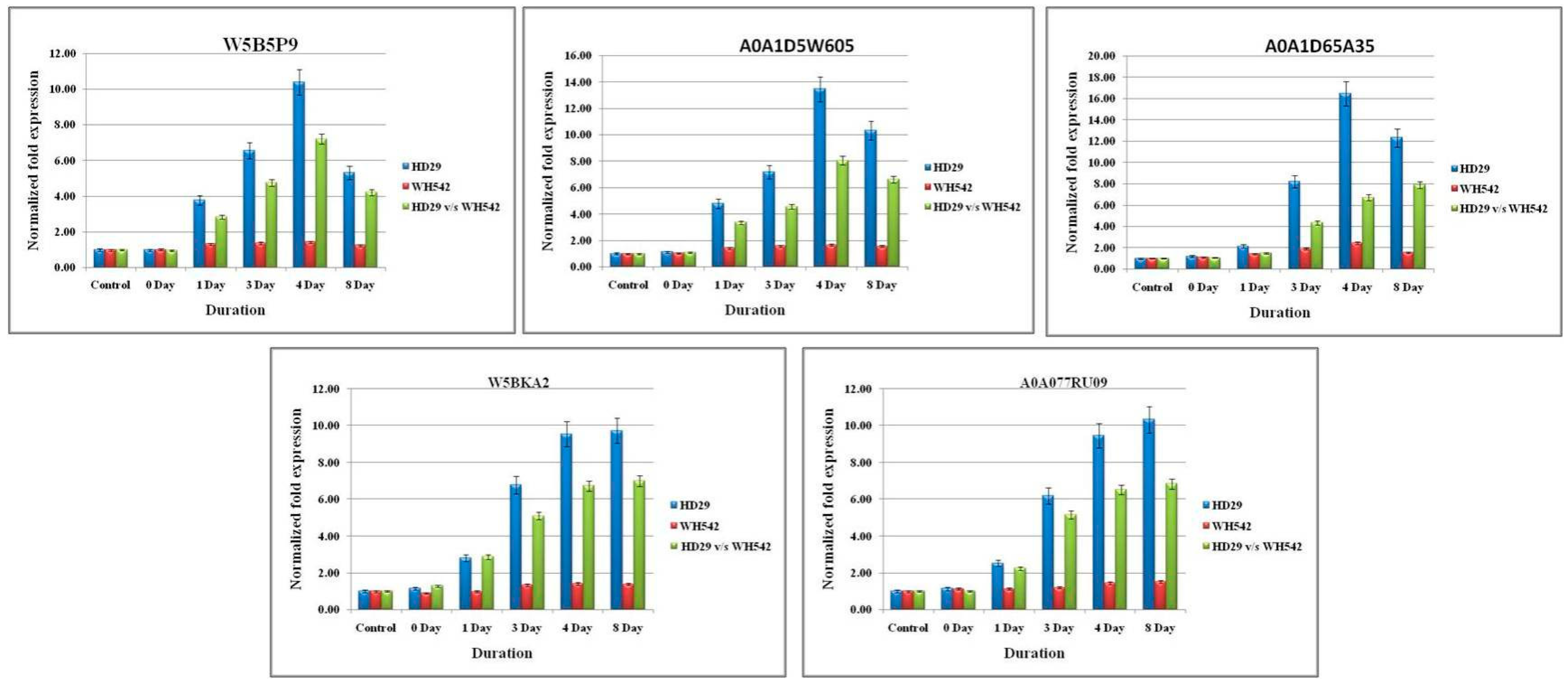
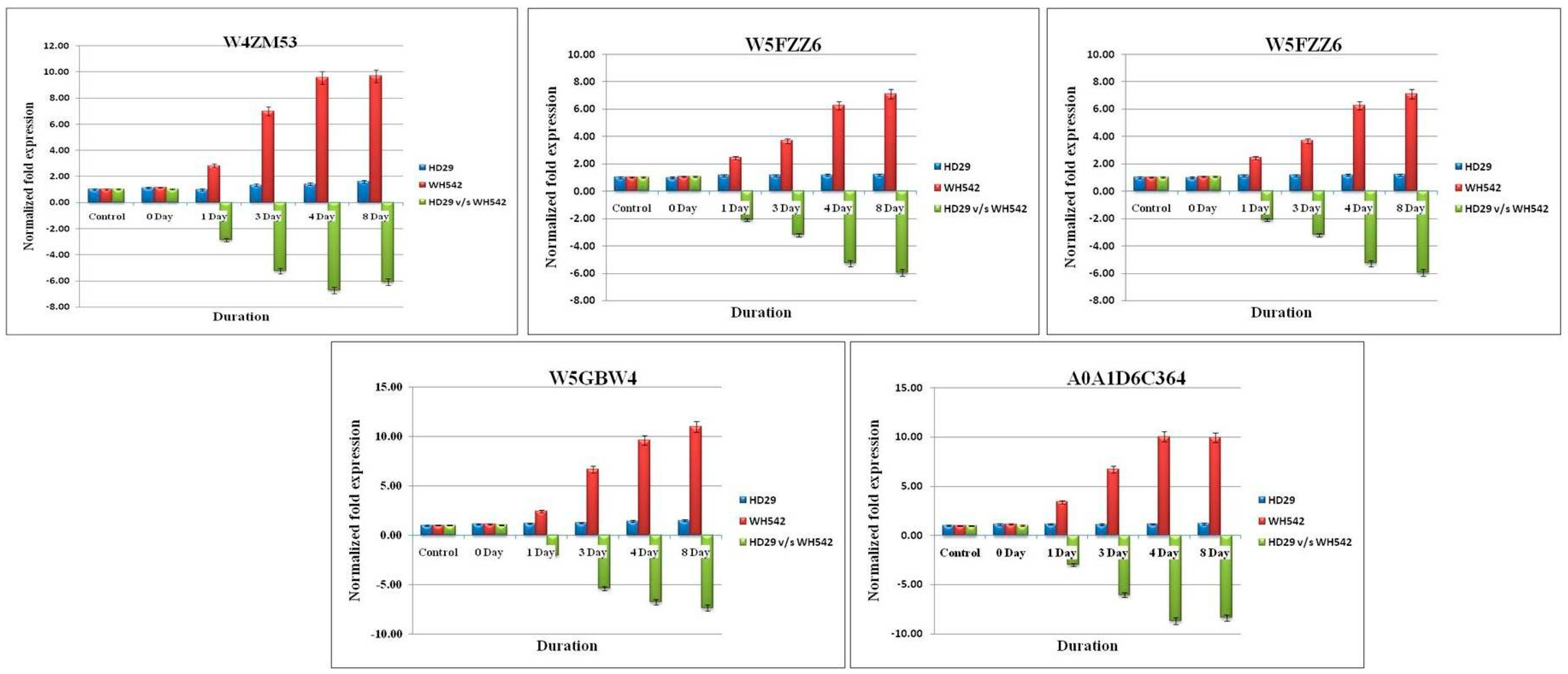
2.5. Validation of Differentially Expressed Genes (DEGs) Using qPCR Analysis
3. Discussion
4. Materials and Methods
4.1. Fungus Material and Inoculum Preparation
4.2. Plant Growth Conditions and Inoculation
4.3. RNA Extraction and Library Construction
4.4. Illumina Sequencing and Reads Mapping
4.5. Analysis of Differentially Expressed Genes (DEGs)
4.6. Functional Annotation and Gene Ontology Enrichment Analysis
4.7. Differential Expression Analysis in T. indica upon Infection
4.8. Validation of Differentially Expressed Genes by Quantitative Real-Time PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anonymous. Annual Report, 2020–2021, Department of Agriculture, Cooperation & Farmers’ Welfare Ministry of Agriculture & Farmers’ Welfare Government of India Krishi Bhawan, New Delhi-110 001; Department of Agriculture, Cooperation & Farmers’ Welfare: Quezon City, Philippines, 2020–2021; p. 6. Available online: www.agricoop.nic.in (accessed on 1 April 2021).
- Mitra, M. A new bunt of wheat in India. Ann. Appl. Biol. 1931, 18, 178–179. [Google Scholar] [CrossRef]
- Bonde, M.R.; Berner, D.K.; Nester, S.E.; Peterson, G.L.; Olsen, M.W.; Cunfer, B.M.; Sim, T. Survival of T. indica teliospores in different soils. Plant Dis. 2004, 88, 316–324. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Warham, E.J. Studies on Karnal Bunt of Wheat. Ph.D. Thesis, University of Wales, Aberystwyth, UK, 1987. [Google Scholar]
- Tan, M.K.; Brennan, J.P.; Wright, D.; Murray, G.M. A review of the methodology to detect and identify Karnal bunt—A serious biosecurity threat. Australas. Plant Pathol. 2013, 42, 95–102. [Google Scholar] [CrossRef]
- Bishnoi, S.K.; He, X.; Phuke, R.M.; Kashyap, P.L.; Alakonya, A.; Chhokar, V.; Singh, R.P.; Singh, P.K. Karnal bunt: A re-emerging old foe of wheat. Front. Plant Sci. 2020, 11, 569057. [Google Scholar] [CrossRef] [PubMed]
- Gurjar, M.S.; Jogawat, A.; Kulshreshtha, D.; Sharma, S.; Gogoi, R.; Aggarwal, R. Intraspecific variation in T. indica isolates a pathogen causing Karnal bunt of Wheat. Indian Phytopathol. 2016, 69, 352–356. [Google Scholar]
- Bains, S.S.; Dhaliwal, H.S. Release of secondary sporidia of Neovossia indica from inoculated wheat spikes. Plant Soil 1989, 115, 83–87. [Google Scholar] [CrossRef]
- Carris, L.M.; Castlebury, L.A.; Goates, B.J. Nonsystemic bunt fungi- Tilletia indica and T. horrida: A review of history, systematics, and biology. Annu. Rev. Phytopathol. 2006, 44, 113–133. [Google Scholar] [CrossRef]
- Dhaliwal, H.S.; Singh, D.V. Up-to-date life cycle of Neovossia indica (Mitra) Mundkur. Curr. Sci. 1988, 57, 675–677. [Google Scholar]
- Mitra, M. Studies on the stinking smut or bunt of wheat in India. Ind. J. Agric. Sci. 1937, 7, 459–477. [Google Scholar]
- Aujla, S.S.; Grewal, A.S.; Gill, K.S.; Sharma, I. Effect of Karnal bunt on chappati making properties of wheat grains. Crop Improv. 1980, 7, 147–149. [Google Scholar]
- Dhaliwal, H.S. Multiplication of secondary sporidia of Tilletia indica on soil and wheat leaves and spikes and incidence of Karnal bunt. Can. J. Bot. 1989, 67, 2387–2390. [Google Scholar] [CrossRef]
- Babadoost, M.; Mathre, D.E.; Johnston, R.H.; Bonde, M.R. Survival of teliospores of Tilletia indica in soil. Plant Dis. 2004, 88, 56–62. [Google Scholar] [CrossRef]
- Goates, B.J. Survival of secondary sporidia of floret infecting Tilletia species: Implications for epidemiology. Phytopathology 2010, 100, 655–662. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aggarwal, R.; Tripathi, A.; Yadav, A. Pathogenic and genetic variability in T. indica monosporidial culture lines using universal rice primer-PCR. Eur. J. Plant Pathol. 2010, 128, 333–342. [Google Scholar] [CrossRef]
- Thirumalaisamy, P.P.; Singh, D.V. Variability of Indian isolates of T. indica assessed by pathogenicity and molecular markers. J. Phytopathol. 2012, 160, 525–531. [Google Scholar] [CrossRef]
- Gurjar, M.S.; Aggarwal, R.; Jain, S.; Sharma, S.; Singh, J.; Gupta, S.; Agarwal, S.; Saharan, M.S. Multilocus sequence typing and single nucleotide polymorphism analysis in Tilletia indica isolates inciting Karnal bunt of wheat. J. Fungi 2021, 7, 103. [Google Scholar] [CrossRef]
- Sharma, P.; Tiwari, R.; Saharan, M.S.; Sharma, I.; Kumar, J.; Mishra, S.; Muthusamy, S.K. Draft genome sequence of two monosporidial lines of the Karnal bunt fungus Tilletia indica Mitra (PSWKBGH-1 and PSWKBGH-2). Genome Announc. 2016, 4, e00928-16. [Google Scholar] [CrossRef]
- Kumar, A.; Pandey, V.; Singh, M.; Pandey, D.; Saharan, M.S.; Marla, S.S. Draft genome sequence of Karnal bunt pathogen (Tilletia indica) of wheat provides insights into the pathogenic mechanisms of quarantined fungus. PLoS ONE 2017, 12, e0171323. [Google Scholar] [CrossRef]
- Gurjar, M.S.; Aggarwal, R.; Jogawat, A.; Kulshreshtha, D.; Sharma, S.; Solanke, A.U.; Dubey, H.; Jain, R.K. Genome sequencing and secretome analysis of T. indica inciting Karnal bunt of wheat provides pathogenesis-related genes. 3 Biotech 2019, 9, 219. [Google Scholar] [CrossRef]
- Chittem, K.; Yajima, W.R.; Goswami, R.S.; del Río Mendoza, L.E. Transcriptome analysis of the plant pathogen Sclerotinia sclerotiorum interaction with resistant and susceptible canola (Brassica napus) lines. PLoS ONE 2020, 15, e0229844. [Google Scholar] [CrossRef]
- Singh, J.; Aggarwal, R.; Gurjar, M.S.; Sharma, S.; Jain, S.; Saharan, M.S. Identification and expression analysis of pathogenicity-related genes in Tilletia indica inciting Karnal bunt of wheat. Australas. Plant Pathol. 2020, 49, 393–402. [Google Scholar] [CrossRef]
- Dobon, A.; Bunting, D.C.E.; Cabrera-Quio, L.E.; Uauy, C.; Saunders, D.G.O. The host-pathogen interaction between wheat and yellow rust induces temporally coordinated waves of gene expression. BMC Genom. 2016, 17, 380. [Google Scholar] [CrossRef] [PubMed]
- Desmond, O.J.; Manners, J.M.; Schenk, P.M.; Maclean, D.J.; Kazan, K. Gene expression analysis of the wheat response to infection by Fusarium pseudograminearum. Physiol. Mol. Plant Pathol. 2008, 73, 40–47. [Google Scholar] [CrossRef]
- He, T.; Ren, Z.; Muhae-Ud-Din, G.; Guo, Q.; Liu, T.; Chen, W.; Gao, L. Transcriptomics Analysis of Wheat Tassel Response to Tilletia laevis Kühn, Which Causes Common Bunt of Wheat. Front. Plant Sci. 2022, 13, 823907. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Liu, J.; Din, G.M.U.; Zhang, H.; Du, Z.; Chen, W.; Liu, T.; Zhang, J.; Zhao, S.; Gao, L. Transcriptome analysis of wheat spikes in response to Tilletia controversa Kühn which cause wheat dwarf bunt. Sci. Rep. 2020, 10, 21567. [Google Scholar] [CrossRef] [PubMed]
- Munjal, R.L.; Chatrath, M.S. Studies on the mode of infection of Neovossia indica incitant of Karnal bunt of wheat. J. Nucl. Agric. Biol. 1976, 5, 40–41. [Google Scholar]
- Aggarwal, R.; Singh, D.V.; Srivastava, K.D. Host-pathogen interaction in Karnal bunt of wheat. Indian Phytopathol. 1994, 47, 381–385. [Google Scholar]
- Tripathi, A.; Aggarwal, R.; Yadav, A. Differential expression analysis of defense related genes responsive to T. indica infection in wheat. Turk. J. Biol. 2013, 37, 606–613. [Google Scholar] [CrossRef]
- Muhae-Ud-Din, G.; Chen, D.; Liu, T.; Chen, W.; Gao, L. Characterization of the wheat cultivars against Tilletia controversa Kühn, causal agent of wheat dwarf bunt. Sci. Rep. 2020, 10, 9029. [Google Scholar] [CrossRef]
- Tan, G.; Liu, K.; Kang, J.; Xu, K.; Zhang, Y.; Hu, L.; Zhang, J.; Li, C. Transcriptome analysis of the compatible interaction of tomato with Verticillium dahliae using RNA-sequencing. Front. Plant Sci. 2015, 6, 428. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.F.; Ding, Z.G.; Ma, Q.; Zhang, G.R.; Zhang, S.L.; Li, Z.K.; Wu, L.Q.; Ma, Z.Y. Transcriptome profiling of Gossypium barbadense inoculated with Verticillium dahliae provides a resource for cotton improvement. BMC Genom. 2013, 14, 637. [Google Scholar] [CrossRef] [PubMed]
- Lengeler, K.B.; Davidson, R.C.; D’souza, C.; Harashima, T.; Shen, W.C.; Wang, P.; Pan, X.; Waugh, M.; Heitman, J. Signal transduction cascades regulating fungal development and virulence. Microbiol. Mol. Biol. Rev. 2000, 64, 74685. [Google Scholar] [CrossRef]
- Yadeta, K.A.; Thomma, B.P.J. The xylem as battleground for plant hosts and vascular wilt pathogens. Front. Plant Sci. 2013, 4, 97. [Google Scholar] [CrossRef]
- Gurjar, M.S.; Jogawat, A.; Saharan, M.S.; Aggarwal, R. Response of putative pathogenicity-related genes in T. indica inciting Karnal Bunt of wheat. Cereal Res. Commun. 2018, 46, 89–103. [Google Scholar] [CrossRef]
- Gurjar, M.S.; Jogawat, A.; Sharma, S.; Aggarwal, R. Functional expression of MAP kinase TiHOG1 gene in T. indica inciting Karnal bunt of wheat. Indian Phytopathol. 2018, 71, 325–335. [Google Scholar] [CrossRef]
- Maeda, H.; Dudareva, N. The shikimate pathway and aromatic amino acid biosynthesis in plants. Annu. Rev. Plant Biol. 2012, 63, 73–105. [Google Scholar] [CrossRef]
- Trovato, M.; Funck, D.; Forlani, G.; Okumoto, S.; Amir, R. Editorial: Amino acids in plants: Regulation and functions in development and stress defense. Front. Plant Sci. 2021, 12, 772810. [Google Scholar] [CrossRef]
- Gayoso, C.; Pomar, F.; Novo Uzal, E.; Merino, F.; de Ilárduya, O.M. The Ve-mediated resistance response of the tomato to Verticillium dahliae involves H2O2, peroxidase and lignins and drives PAL gene expression. BMC Plant Biol. 2010, 10, 232–250. [Google Scholar] [CrossRef]
- De Cremer, K.; Mathys, J.; Vos, C.; Froenicke, L.; Michelmore, R.W.; Cammue, B.P.A.; De Coninck, B. RNAseq-based transcriptome analysis of Lactuca sativa infected by the fungal necrotroph Botrytis cinerea. Plant Cell Environ. 2013, 36, 1992–2007. [Google Scholar]
- Zadoks, J.C.; Chang, T.T.; Konzak, C.F. A decimal code for the growth stages of cereals. Weed Res. 1974, 14, 415–421. [Google Scholar] [CrossRef]
- Aujla, S.S.; Grewal, A.S.; Gill, K.S.; Sharma, I. Artificial creation of Karnal bunt disease of wheat. Cereal Res. Commun. 1982, 10, 171–175. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Kharbikar, L.L.; Majumdar, B.A. Low-cost disruption of rhizospheric microorganisms for the extraction of total RNA using modified RNeasy mini kit protocols. Ann. Microbiol. 2015, 65, 1797–1801. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Resistant Genotype (HD29) | Susceptible Genotype (WH542) |
|---|---|---|
| Total Reads | 29,321,161 (100%) | 29,062,661 (100%) |
| Number of bases | 8,611,180,199 | 8,531,647,691 |
| Total data in Gb | 8.6 Gb | 8.5 Gb |
| Mapped reads | 93.67% | 92.11% |
| Uniquely mapped reads | 92.94% | 90.47% |
| Multiple mapped reads | 0.72% | 1.63% |
| Unmapped reads | 6.32% | 7.88% |
| % GC content | 51.38% | 49.48% |
| GO Terms | Ontology | Descriptions | Total Input List |
|---|---|---|---|
| GO:0050896 | P | response to stimulus | 13 |
| GO:0006950 | P | response to stress | 11 |
| GO:0005975 | P | carbohydrate metabolic process | 9 |
| GO:0008152 | P | metabolic process | 21 |
| GO:0042221 | P | response to chemical stimulus | 7 |
| GO:0009628 | P | response to abiotic stimulus | 5 |
| GO:0044238 | P | primary metabolic process | 17 |
| GO:0032502 | P | developmental process | 5 |
| GO:0009056 | P | catabolic process | 6 |
| GO:0065007 | P | biological regulation | 7 |
| GO:0004553 | F | hydrolase activity, hydrolyzing O-glycosyl compounds | 7 |
| GO:0016798 | F | hydrolase activity, acting on glycosyl bonds | 7 |
| GO:0005488 | F | Binding | 23 |
| GO:0003824 | F | catalytic activity | 19 |
| GO:0016787 | F | hydrolase activity | 9 |
| Particulars | Number of Mapped Reads | % Mapping |
|---|---|---|
| Resistant genotype (HD29) | 4,146,257 | 7.07 |
| Susceptible genotype (WH542) | 4,434,294 | 7.63 |
| Descriptions | Number of Genes |
|---|---|
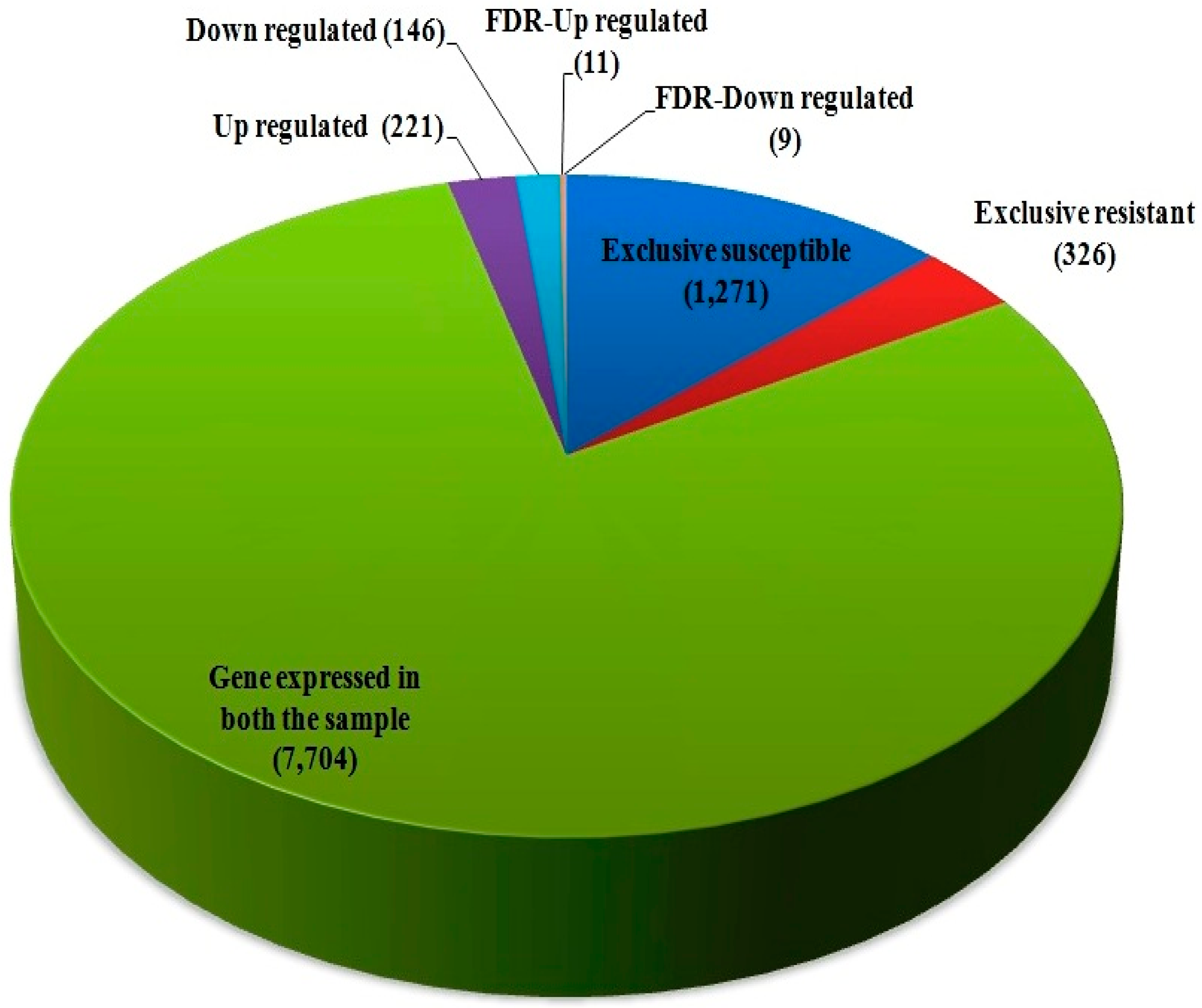
| Exclusive susceptible (HD29) genotype | 1271 |
| Exclusive resistant (WH542) genotype | 326 |
| Gene expressed both resistant and susceptible hst (HD29 & WH542) | 7704 |
| Upregulated | 221 |
| Downregulated | 146 |
| FDR-Upregulated | 11 |
| FDR-Downregulated | 09 |
| Gene IDS | Log2 Fold Change | UniProtKB/TrEMBL ID | GO Terms | Forward Primer (5′-3′) | Reverse Primer (5′-3′) | Protein Name | Functions |
|---|---|---|---|---|---|---|---|
| Five Up Regulated Genes in Wheat-T. indica Interaction | |||||||
| TRIAE_CS42_2AS_TGACv1_115171_AA0371320 | 6.590434 | W5B5P9 | Integral component of membrane | GTGATGTGTCCATTGCATGTG | AAACGCAGTCCATATAGCCAG | Uncharacterized protein | - |
| TRIAE_CS42_3B_TGACv1_224664_AA0799640 | 9.915167 | A0A1D5W605 | - | CGCAGCTTATCACGACAATG | GCTAGTAAAAGGGACAGGGAC | Uncharacterized protein | - |
| TRIAE_CS42_6BS_TGACv1_513159_AA1632520 | 8.262696 | A0A1D65A35 | 3′-5′ exonuclease activity, nucleic acid binding | GAATCCACCTCTCACCATCC | ACGGAACAGACCAGCATAAG | Uncharacterized protein | - |
| TRIAE_CS42_2BS_TGACv1_147826_AA0488000 | 6.692584 | W5BKA2 | integral component of membrane | ACGGCCTCAGAGCTTATGTG | GGTTTGCACTTGACCACAACA | Uncharacterized protein | - |
| TRIAE_CS42_3B_TGACv1_221012_AA0726950 | 6.968122 | A0A077RU09 | DNA binding, protein heterodimerization activity | TCACCTCCTCGTAGAGTGCT | AGGCAGGCACCGTTACTATT | Histone H2B | Core component of nucleosome. DNA repair, DNA replication and chromosomal stability. |
| Five Down Regulated Genes in Wheat-T. indica Interaction | |||||||
| TRIAE_CS42_1AL_TGACv1_000260_AA0007580 | −7.39315 | W4ZM53 | transferase activity, transferring acyl groups other than amino-acyl groups | GCGGCTTTCAATCTTTTCAGG | TGCTCTGTTTGGTGATGGTG | Uncharacterized protein | - |
| TRIAE_CS42_1BL_TGACv1_030856_AA0102410 | −5.23889 | W5A6W5 | serine-type, carboxypeptidase activity | AACTTGCTGTTCCTGGACTC | TCGCTAACTGTGGAATGTAGTG | Carboxypeptidase | cleave peptides from the C-terminus at a speed of one residue each time |
| TRIAE_CS42_5DL_TGACv1_435993_AA1457050 | −5.12949 | W5FZZ6 | coenzyme binding | TCTTCCACATTGCATCACCTG | GTTTCGTCCCTAATCCTCACTG | Uncharacterized protein | - |
| TRIAE_CS42_6BS_TGACv1_515607_AA1671270 | −7.7181 | W5GBW4 | metal ion binding, ATP binding, methionine, adenosyl transferase activity | CACAATGACAATGGTGCTATGG | ATGGGTTGAGATGGAAGATGG | S-adenosylmethionine synthase (EC 2.5.1.6) | Catalyzes the formation of S-adenosylmethionine from methionine and ATP. |
| TRIAE_CS42_7BL_TGACv1_576986_AA1861990 | −9.32268 | A0A1D6C364 | - | CTTGTCTTATTCCCTCACCCAC | ACCTGACCCCTAATTGCTTG | Uncharacterized protein | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurjar, M.S.; Jain, S.; Aggarwal, R.; Saharan, M.S.; Kumar, T.P.J.; Kharbikar, L. Transcriptome Analysis of Wheat–Tilletia indica Interaction Provides Defense and Pathogenesis-Related Genes. Plants 2022, 11, 3061. https://doi.org/10.3390/plants11223061
Gurjar MS, Jain S, Aggarwal R, Saharan MS, Kumar TPJ, Kharbikar L. Transcriptome Analysis of Wheat–Tilletia indica Interaction Provides Defense and Pathogenesis-Related Genes. Plants. 2022; 11(22):3061. https://doi.org/10.3390/plants11223061
Chicago/Turabian StyleGurjar, Malkhan Singh, Shekhar Jain, Rashmi Aggarwal, Mahender Singh Saharan, Tej Pratap Jitendra Kumar, and Lalit Kharbikar. 2022. "Transcriptome Analysis of Wheat–Tilletia indica Interaction Provides Defense and Pathogenesis-Related Genes" Plants 11, no. 22: 3061. https://doi.org/10.3390/plants11223061
APA StyleGurjar, M. S., Jain, S., Aggarwal, R., Saharan, M. S., Kumar, T. P. J., & Kharbikar, L. (2022). Transcriptome Analysis of Wheat–Tilletia indica Interaction Provides Defense and Pathogenesis-Related Genes. Plants, 11(22), 3061. https://doi.org/10.3390/plants11223061