
Microbial Community Succession Associated with Poplar Wood Discoloration


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Changes in Microbial Diversity with Successional Stages
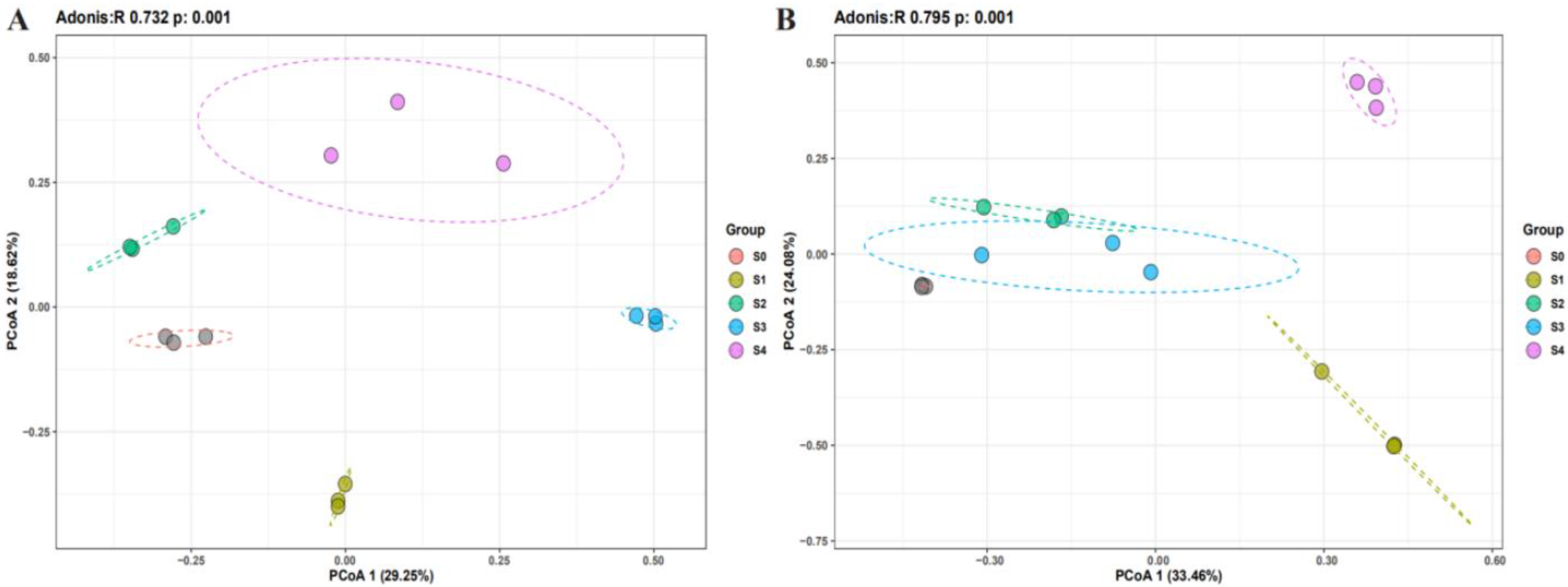
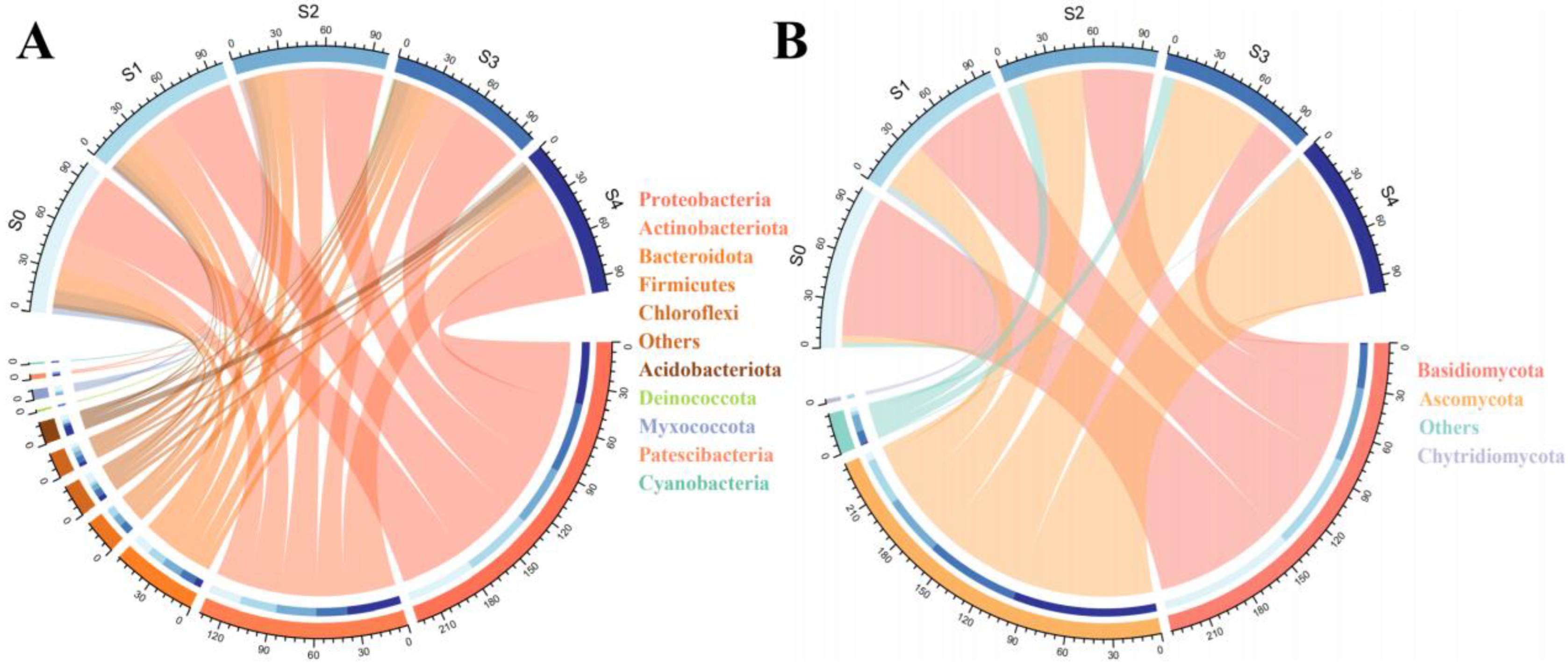
2.2. Changes in Microbial Community Composition with Successional Stages
2.3. Statistically Significant Differences in the Abundance of Bacterial and Fungal Communities between Successional Stages
2.4. The Relative Importance of Deterministic and Stochastic Processes in Shaping the Microbial Assembly with Successional Stages
3. Discussion
3.1. Variation of Microbial Community Diversity across Successional Stages
3.2. Changes in Microbial Community Structure with Succession
3.3. The Different Assembly Processes Underwent by Bacterial and Fungal Communities across Successional Stages
4. Materials and Methods
4.1. Sample Collection and Site Description
4.2. DNA Extraction and High-Throughput Sequencing
4.3. Processing of Sequence Analysis
4.4. Statistical and Bioinformatics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jansson, S.; Douglas, C.J. Populus: A model system for plant biology. Annu. Rev. Plant Biol. 2007, 58, 435–458. [Google Scholar] [CrossRef] [PubMed]
- Strobel, K.; Nyrud, A.Q.; Bysheim, K. Interior wood use: Linking user perceptions to physical properties. Scand. J. For. Res. 2017, 32, 798–806. [Google Scholar] [CrossRef]
- Hu, J.; Liu, Y.; Wu, Z. Structural color for wood coloring: A Review. BioResources 2020, 15, 9917. [Google Scholar] [CrossRef]
- Garbelotto, M.; Slaughter, G.; Popenuck, T.; Cobb, F.W.; Bruns, T.D. Secondary spread of Heterobasidion annosum in white fir root-disease centers. Can. J. For. Res. 1997, 27, 766–773. [Google Scholar] [CrossRef]
- Vasaitis, R.; Bakys, R.; Vasiliauskas, A. Discoloration and associated fungi in stems of silver birch (Betula pendula Roth.) following logging damage. For. Pathol. 2012, 42, 387–392. [Google Scholar] [CrossRef]
- Barua, P.; You, M.P.; Bayliss, K.; Lanoiselet, V.; Barbetti, M.J. A rapid and miniaturized system using Alamar blue to assess fungal spore viability: Implications for biosecurity. Eur. J. Plant Pathol. 2017, 148, 139–150. [Google Scholar] [CrossRef]
- Wingfield, M.J.; Seifert, K.A.; Webber, J.F. Ceratocystis and Ophiostoma: Taxonomy, Ecology, and Pathogenicity; American Phytopathological Society: Saint Paul, MN, USA, 1993. [Google Scholar]
- Savoie, J.-M.; Mata, G.; Mamoun, M. Variability in brown line formation and extracellular laccase production during interaction between white-rot basidiomycetes and Trichoderma harzianum biotype Th2. Mycologia 2001, 93, 243–248. [Google Scholar] [CrossRef]
- Brischke, C.; Welzbacher, C.; Huckfeldt, T. Influence of fungal decay by different basidiomycetes on the structural integrity of Norway spruce wood. Holz Als Roh-Und Werkst. 2008, 66, 433–438. [Google Scholar] [CrossRef]
- Pánek, M.; Reinprecht, L.; Hulla, M. Ten essential oils for beech wood protection-efficacy against wood-destroying fungi and moulds, and effect on wood discoloration. BioResources 2014, 9, 5588–5603. [Google Scholar] [CrossRef]
- Humar, M.; Vek, V.; Bučar, B. Properties of blue-stained wood. Drv. Ind. 2008, 59, 75–79. [Google Scholar]
- Chittenden, C.; Singh, T. Antifungal activity of essential oils against wood degrading fungi and their applications as wood preservatives. Int. Wood Prod. J. 2011, 2, 44–48. [Google Scholar] [CrossRef]
- Singh, T.; Chittenden, C. In-vitro antifungal activity of chilli extracts in combination with Lactobacillus casei against common sapstain fungi. Int. Biodeterior. Biodegrad. 2008, 62, 364–367. [Google Scholar] [CrossRef]
- Lepinay, C.; Tláskal, V.; Vrška, T.; Brabcová, V.; Baldrian, P. Successional development of wood-inhabiting fungi associated with dominant tree species in a natural temperate floodplain forest. Fungal Ecol. 2021, 59, 101116. [Google Scholar] [CrossRef]
- Ottosson, E.; Norden, J.; Dahlberg, A.; Edman, M.; Jönsson, M.; Larsson, K.H.; Olsson, J.; Penttilä, R.; Stenlid, J.; Ovaskainen, O. Species associations during the succession of wood-inhabiting fungal communities. Fungal Ecol. 2014, 11, 17–28. [Google Scholar] [CrossRef]
- Wang, G.; Liu, Y.; Cui, M.; Zhou, Z.; Zhang, Q.; Li, Y.; Ha, W.; Pang, D.; Luo, J.; Zhou, J. Effects of secondary succession on soil fungal and bacterial compositions and diversities in a karst area. Plant Soil 2022, 475, 91–102. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, G.; Xue, S.; Wang, G. Soil bacterial community dynamics reflect changes in plant community and soil properties during the secondary succession of abandoned farmland in the Loess Plateau. Soil Biol. Biochem. 2016, 97, 40–49. [Google Scholar] [CrossRef]
- Hu, Y.; Xiang, D.; Veresoglou, S.D.; Chen, F.; Chen, Y.; Hao, Z.; Zhang, X.; Chen, B. Soil organic carbon and soil structure are driving microbial abundance and community composition across the arid and semi-arid grasslands in northern China. Soil Biol. Biochem. 2014, 77, 51–57. [Google Scholar] [CrossRef]
- Shen, C.; Xiong, J.; Zhang, H.; Feng, Y.; Lin, X.; Li, X.; Liang, W.; Chu, H. Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol. Biochem. 2013, 57, 204–211. [Google Scholar] [CrossRef]
- Cui, Y.; Fang, L.; Guo, X.; Wang, X.; Wang, Y.; Li, P.; Zhang, Y.; Zhang, X. Responses of soil microbial communities to nutrient limitation in the desert-grassland ecological transition zone. Sci. Total Environ. 2018, 642, 45–55. [Google Scholar] [CrossRef]
- Ren, C.; Liu, W.; Zhao, F.; Zhong, Z.; Deng, J.; Han, X.; Yang, G.; Feng, Y.; Ren, G. Soil bacterial and fungal diversity and compositions respond differently to forest development. Catena 2019, 181, 104071. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, G.; Hai, X.; Li, J.; Shangguan, Z.; Peng, C.; Deng, L. Long-term forest succession improves plant diversity and soil quality but not significantly increase soil microbial diversity: Evidence from the Loess Plateau. Ecol. Eng. 2020, 142, 105631. [Google Scholar] [CrossRef]
- Cai, Z.-Q.; Zhang, Y.-H.; Yang, C.; Wang, S. Land-use type strongly shapes community composition, but not always diversity of soil microbes in tropical China. Catena 2018, 165, 369–380. [Google Scholar] [CrossRef]
- Guo-Mei, J.; Zhang, P.-D.; Gang, W.; Jing, C.; Jing-Cheng, H.; Huang, Y.-P. Relationship between microbial community and soil properties during natural succession of abandoned agricultural land. Pedosphere 2010, 20, 352–360. [Google Scholar]
- Zhou, Z.; Wang, C.; Jiang, L.; Luo, Y. Trends in soil microbial communities during secondary succession. Soil Biol. Biochem. 2017, 115, 92–99. [Google Scholar] [CrossRef]
- Zhong, Z.; Zhang, X.; Wang, X.; Fu, S.; Wu, S.; Lu, X.; Ren, C.; Han, X.; Yang, G. Soil bacteria and fungi respond differently to plant diversity and plant family composition during the secondary succession of abandoned farmland on the Loess Plateau, China. Plant Soil 2020, 448, 183–200. [Google Scholar] [CrossRef]
- Zechmeister-Boltenstern, S.; Keiblinger, K.M.; Mooshammer, M.; Peñuelas, J.; Richter, A.; Sardans, J.; Wanek, W. The application of ecological stoichiometry to plant–microbial–soil organic matter transformations. Ecol. Monogr. 2015, 85, 133–155. [Google Scholar] [CrossRef]
- Mikluscak, M.; Dawson-Andoh, B.E. Microbial colonizers of freshly sawn yellow-poplar (Liriodendron tulipifera L.) lumber in two seasons: Part 2. Bacteria 2004, 58, 182–188. [Google Scholar]
- Kolton, M.; Harel, Y.M.; Pasternak, Z.; Graber, E.R.; Elad, Y.; Cytryn, E. Impact of biochar application to soil on the root-associated bacterial community structure of fully developed greenhouse pepper plants. Appl. Environ. Microbiol. 2011, 77, 4924–4930. [Google Scholar] [CrossRef]
- Kim, T.G.; Jeong, S.-Y.; Cho, K.-S. Functional rigidity of a methane biofilter during the temporal microbial succession. Appl. Microbiol. Biotechnol. 2014, 98, 3275–3286. [Google Scholar] [CrossRef]
- Al Farraj, D.A.; Alkufeidy, R.M.; Alkubaisi, N.A.; Alshammari, M.K. Polynuclear aromatic anthracene biodegradation by psychrophilic Sphingomonas sp., cultivated with tween-80. Chemosphere 2021, 263, 128115. [Google Scholar] [CrossRef]
- Wu, H.; Sun, B.; Li, J. Influence of polycyclic aromatic hydrocarbon pollution on the diversity and function of bacterial communities in urban wetlands. Environ. Sci. Pollut. Res. 2021, 28, 56281–56293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, M.-Y.; Khan, N.; Tan, L.-L.; Yang, S. Potentials, utilization, and bioengineering of plant growth-Promoting Methylobacterium for sustainable agriculture. Sustainability 2021, 13, 3941. [Google Scholar] [CrossRef]
- Liu, J.; Jia, X.; Yan, W.; Zhong, Y.; Shangguan, Z. Changes in soil microbial community structure during long-term secondary succession. Land Degrad. Dev. 2020, 31, 1151–1166. [Google Scholar] [CrossRef]
- Brunner, I.; Plötze, M.; Rieder, S.; Zumsteg, A.; Furrer, G.; Frey, B. Pioneering fungi from the Damma glacier forefield in the Swiss Alps can promote granite weathering. Geobiology 2011, 9, 266–279. [Google Scholar] [CrossRef]
- Singh, T.; Vesentini, D.; Singh, A.P.; Daniel, G. Effect of chitosan on physiological, morphological, and ultrastructural characteristics of wood-degrading fungi. Int. Biodeterior. Biodegrad. 2008, 62, 116–124. [Google Scholar] [CrossRef]
- Xenopoulos, S.; Tsopelas, P. Sphaeropsis canker, a new disease of cypress in Greece. For. Pathol. 2000, 30, 121–126. [Google Scholar] [CrossRef]
- Nemergut, D.R.; Schmidt, S.K.; Fukami, T.; O’Neill, S.P.; Bilinski, T.M.; Stanish, L.F.; Knelman, J.E.; Darcy, J.L.; Lynch, R.C.; Wickey, P. Patterns and processes of microbial community assembly. Microbiol. Mol. Biol. Rev. 2013, 77, 342–356. [Google Scholar] [CrossRef]
- Chesson, P. Mechanisms of maintenance of species diversity. Annu. Rev. Ecol. Syst. 2000, 31, 343–366. [Google Scholar] [CrossRef]
- Jackson, C.R.; Churchill, P.F.; Roden, E.E. Successional changes in bacterial assemblage structure during epilithic biofilm development. Ecology 2001, 82, 555–566. [Google Scholar] [CrossRef]
- Sigler, W.; Zeyer, J. Microbial diversity and activity along the forefields of two receding glaciers. Microb. Ecol. 2002, 43, 397–407. [Google Scholar] [CrossRef]
- Chase, J.M.; Myers, J.A. Disentangling the importance of ecological niches from stochastic processes across scales. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 2351–2363. [Google Scholar] [CrossRef] [PubMed]
- Xun, W.; Li, W.; Xiong, W.; Ren, Y.; Liu, Y.; Miao, Y.; Xu, Z.; Zhang, N.; Shen, Q.; Zhang, R. Diversity-triggered deterministic bacterial assembly constrains community functions. Nat. Commun. 2019, 10, 3833. [Google Scholar] [CrossRef]
- Glassman, S.I.; Peay, K.G.; Talbot, J.M.; Smith, D.P.; Chung, J.A.; Taylor, J.W.; Vilgalys, R.; Bruns, T.D. A continental view of pine-associated ectomycorrhizal fungal spore banks: A quiescent functional guild with a strong biogeographic pattern. New Phytol. 2015, 205, 1619–1631. [Google Scholar] [CrossRef] [PubMed]
- Dini-Andreote, F.; Stegen, J.C.; van Elsas, J.D.; Salles, J.F. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc. Natl. Acad. Sci. USA 2015, 112, E1326–E1332. [Google Scholar] [CrossRef] [PubMed]
- Vellend, M. Conceptual synthesis in community ecology. Q. Rev. Biol. 2010, 85, 183–206. [Google Scholar] [CrossRef]
- Jackson, C.R. Changes in community properties during microbial succession. Oikos 2003, 101, 444–448. [Google Scholar] [CrossRef]
- Shang, R.; Li, S.; Huang, X.; Liu, W.; Lang, X.; Su, J. Effects of soil properties and plant diversity on soil microbial community composition and diversity during secondary succession. Forests 2021, 12, 805. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Highlander and E. Sodergren. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Yang, S.; Liebner, S.; Alawi, M.; Ebenhöh, O.; Wagner, D. Taxonomic database and cut-off value for processing mcrA gene 454 pyrosequencing data by MOTHUR. J. Microbiol. Methods 2014, 103, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Vegan: Community Ecology Package. R package Version 1. 15-4. 2009. Available online: https://CRAN.R-project.org/src/contrib/Archive/MVPARTwrap (accessed on 29 July 2022).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Stegen, J.C.; Lin, X.; Konopka, A.E.; Fredrickson, J.K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Chen, X.; Kennedy, D.W.; Murray, C.J.; Rockhold, M.L.; Konopka, A. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013, 7, 2069–2079. [Google Scholar] [CrossRef]
- Zhou, J.; Ning, D. Stochastic community assembly: Does it matter in microbial ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002-17. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Liu, H.; Han, H.; Zhang, B.; Zhang, C.; He, J.; Li, S.; Cao, H. Microbial Community Succession Associated with Poplar Wood Discoloration. Plants 2022, 11, 2420. https://doi.org/10.3390/plants11182420
Zhang X, Liu H, Han H, Zhang B, Zhang C, He J, Li S, Cao H. Microbial Community Succession Associated with Poplar Wood Discoloration. Plants. 2022; 11(18):2420. https://doi.org/10.3390/plants11182420
Chicago/Turabian StyleZhang, Xiaohua, Hao Liu, Heming Han, Bo Zhang, Cunzhi Zhang, Jian He, Shunpeng Li, and Hui Cao. 2022. "Microbial Community Succession Associated with Poplar Wood Discoloration" Plants 11, no. 18: 2420. https://doi.org/10.3390/plants11182420
APA StyleZhang, X., Liu, H., Han, H., Zhang, B., Zhang, C., He, J., Li, S., & Cao, H. (2022). Microbial Community Succession Associated with Poplar Wood Discoloration. Plants, 11(18), 2420. https://doi.org/10.3390/plants11182420