
Genome Analysis of Phytophthora nicotianae JM01 Provides Insights into Its Pathogenicity Mechanisms

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Genome Sequencing, Assembly and Validation
2.2. Repeat Elements
2.3. Gene Family Expansion and Contraction
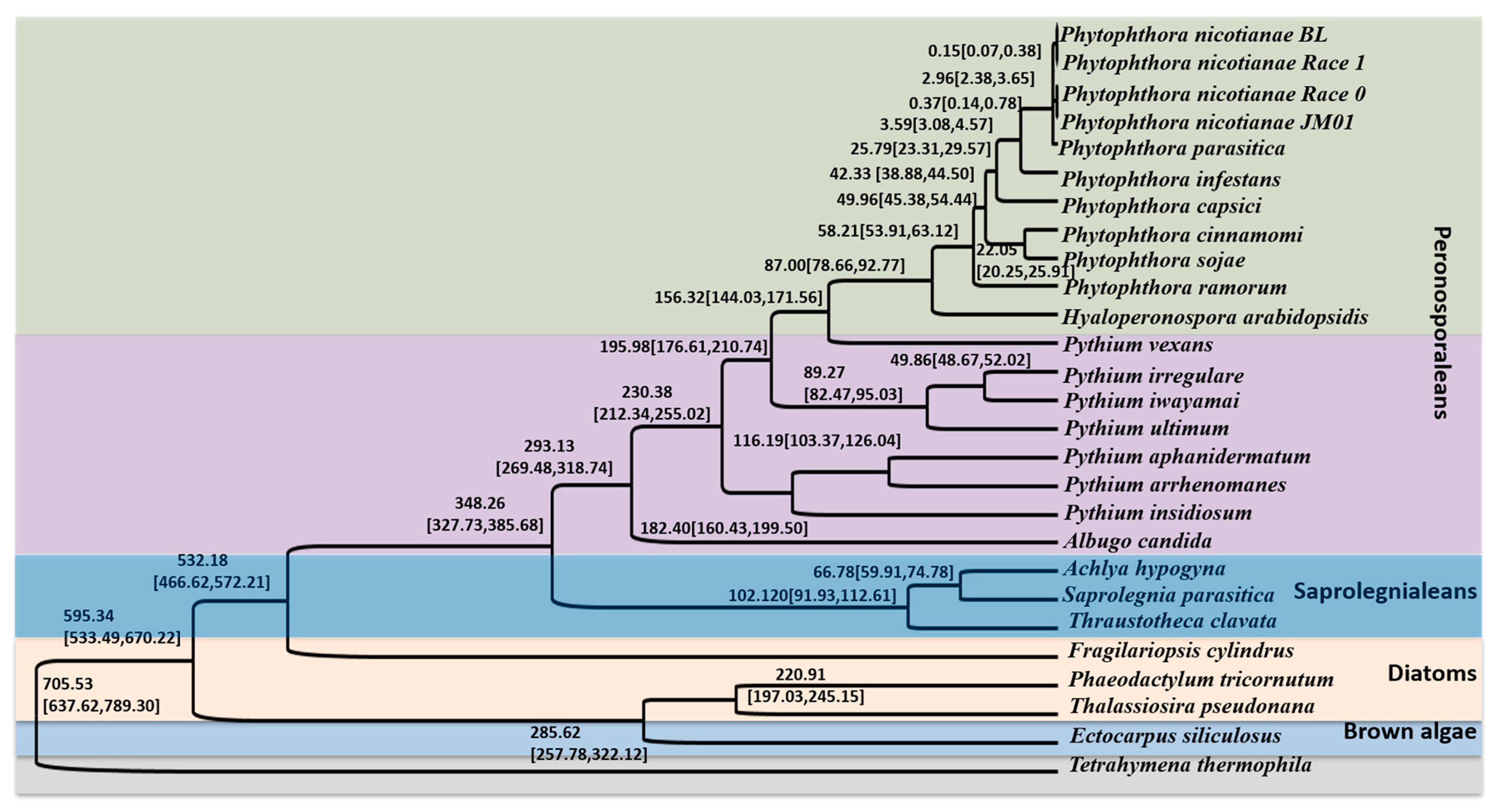
2.4. Phylogenetic Relationship and Divergence Time Analyses
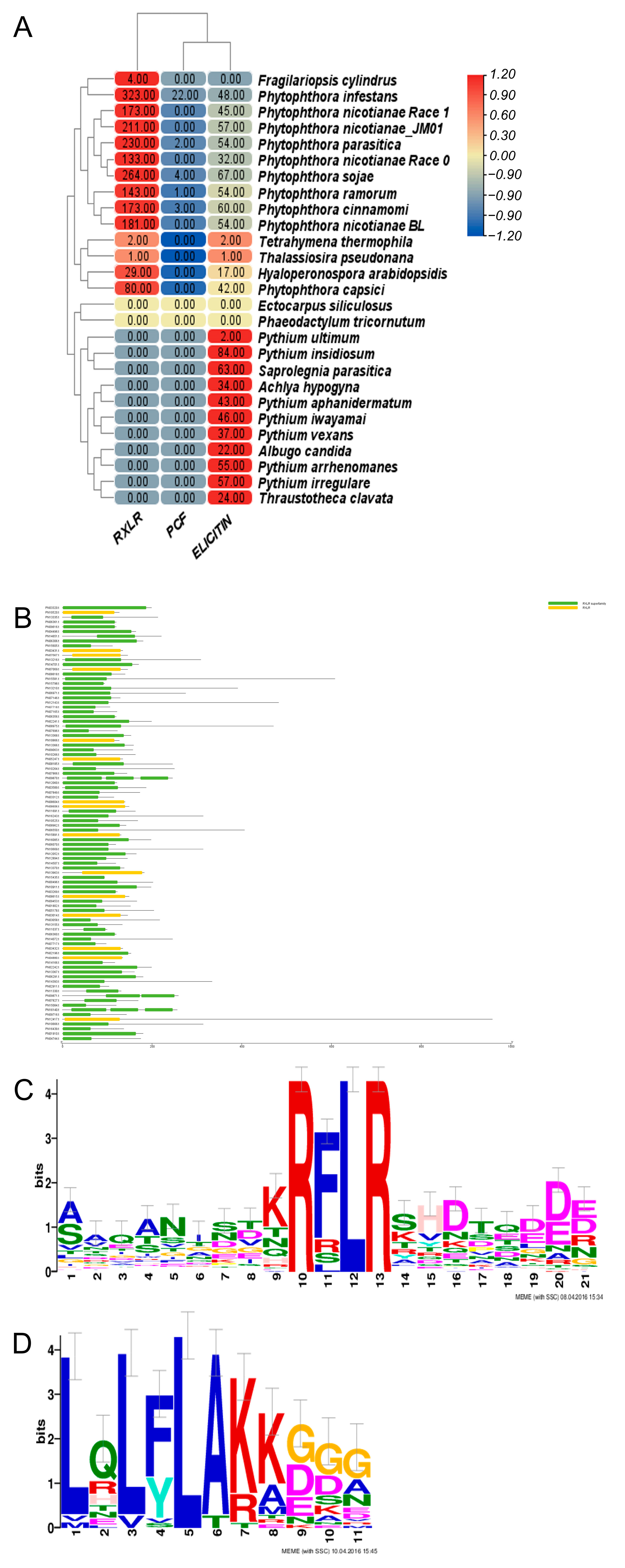
2.5. Analysis of Pathogenicity Mechanisms
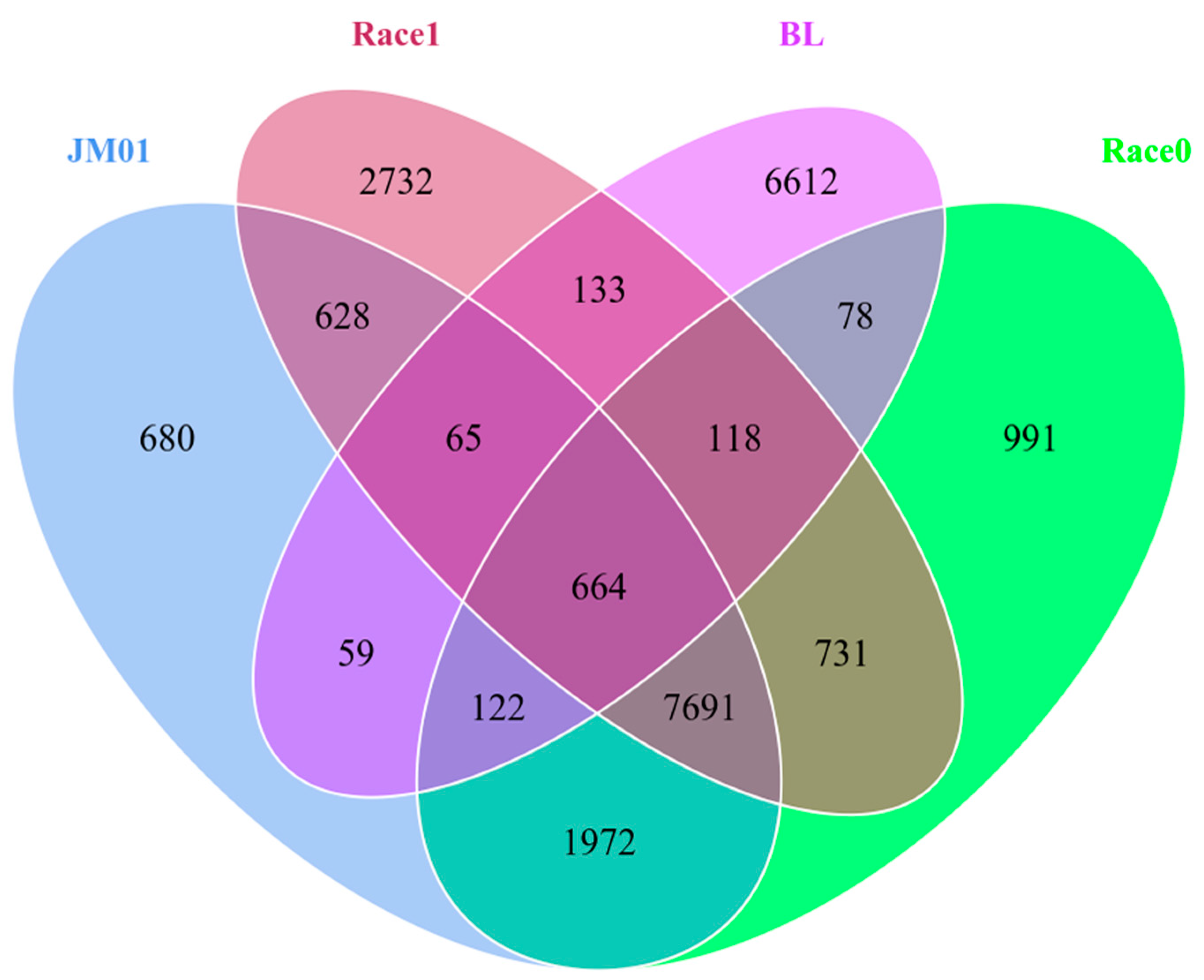
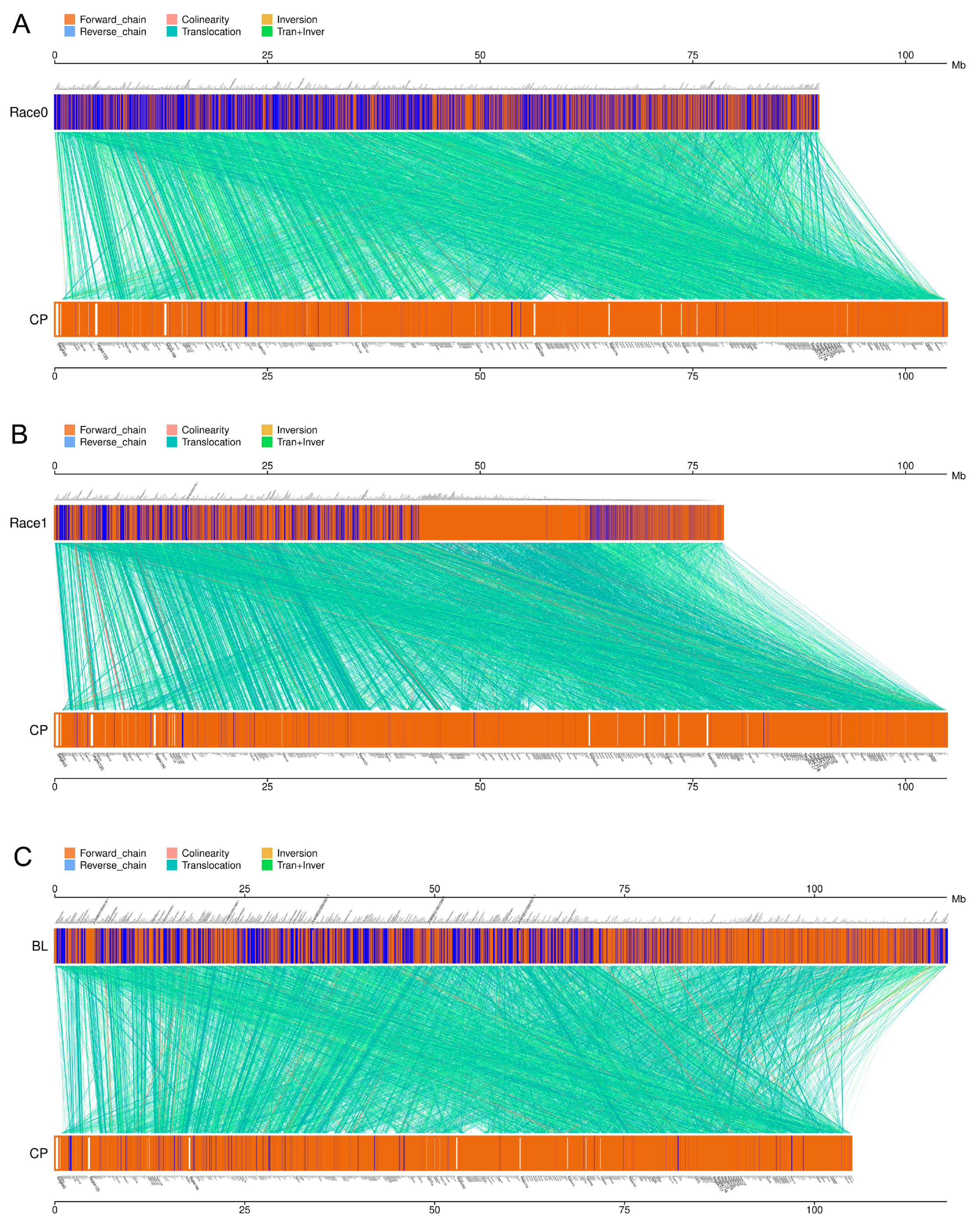
2.6. Comparative Genomes of Four Ph. nicotianae Genomes
3. Discussion
3.1. Genome Features
3.2. Phylogenetic and Divergence Time Analyses
3.3. The Pathogenic Mechanisms of Ph. nicotianae
4. Materials and Methods
4.1. Material Culture
4.2. DNA Extraction and Quantification, Genome Sequencing
4.3. Genome Assembly
4.4. Repeat Element Prediction and Masking
4.5. Gene Prediction
4.6. Gene Annotation
4.7. Gene Expansion and Contraction Analysis
4.8. Phylogenetic and Divergence Time Analyses
4.9. Identification of the Pathogenic Effectors of Ph. nicotianae JM01
4.10. Analysis of Basic Region-Leucine Zipper (bZIP) and V-Myb Avian Myeloblastosis Viral Oncogene Homolog (Myb) Sequences
4.11. Validation of Ph. nicotianae JM01 Effectors by Constructing Recombinant Agrobacterium Tumefaciens Binary PVX Vectors
4.12. Genome-Wide Multicollinearity Analysis and Gene Contents Analysis among Four Ph. nicotianae Strains
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jiang, R.H.; Tyler, B.M. Mechanisms and evolution of virulence in oomycetes. Annu. Rev. Phytopathol. 2012, 50, 295–318. [Google Scholar] [CrossRef]
- Leesutthiphonchai, W.; Vu, A.L.; Ah-Fong, A.M.V.; Judelson, H.S. How does phytophthora infestans evade control efforts? Modern insight into the late blight disease. Phytopathology 2018, 108, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Gascuel, Q.; Martinez, Y.; Boniface, M.C.; Vear, F.; Pichon, M.; Godiard, L. The sunflower downy mildew pathogen plasmopara halstedii. Mol. Plant Pathol. 2015, 16, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Judelson, H.S.; Roberts, S. Novel protein kinase induced during sporangial cleavage in the oomycete phytophthora infestans. Eukaryot. Cell 2002, 1, 687–695. [Google Scholar] [CrossRef]
- Judelson, H.S. Metabolic diversity and novelties in the oomycetes. Annu. Rev. Microbiol. 2017, 71, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.; Handsaker, R.E.; Cano, L.M.; Grabherr, M.; Kodira, C.D.; Raffaele, S.; Torto-Alalibo, T.; et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 2009, 461, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Leonelli, L.; Pelton, J.; Schoeffler, A.; Dahlbeck, D.; Berger, J.; Wemmer, D.E.; Staskawicz, B. Structural elucidation and functional characterization of the Hyaloperonospora arabidopsidis effector protein ATR13. PLoS Pathog. 2011, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, C.A.; Brouwer, H.; Cano, L.; Hamilton, J.P.; Holt, C.; Huitema, E.; Raffaele, S.; Robideau, G.P.; Thines, M.; Win, J.; et al. Genome sequence of the necrotrophic plant pathogen pythium ultimum reveals original pathogenicity mechanisms and effector repertoire. Genome. Biol. 2010, 11, 2010–2011. [Google Scholar] [CrossRef]
- Sudhagar, A.; Kumar, G.; El-Matbouli, M. Transcriptome Analysis based on RNA-Seq in understanding pathogenic mechanisms of diseases and the immune system of fish: A comprehensive review. Int. J. Mol. Sci. 2018, 19, 245. [Google Scholar] [CrossRef]
- Chen, X.R.; Xing, Y.P.; Li, Y.P.; Tong, Y.H.; Xu, J.Y. RNA-Seq reveals infection-related gene expression changes in phytophthora capsici. PLoS ONE 2013, 8, e74588. [Google Scholar] [CrossRef]
- Sun, J.; Gao, Z.; Zhang, X.; Zou, X.; Cao, L.; Wang, J. Transcriptome analysis of Phytophthora litchii reveals pathogenicity arsenals and confirms taxonomic status. PLoS ONE 2017, 12, e0178245. [Google Scholar] [CrossRef]
- Gaulin, E.; Pel, M.J.C.; Camborde, L.; San-Clemente, H.; Courbier, S.; Dupouy, M.A.; Lengellé, J.; Veyssiere, M.; Le Ru, A.; Grandjean, F.; et al. Genomics analysis of aphanomyces spp. identifies a new class of oomycete effector associated with host adaptation. BMC Biol. 2018, 16, 43. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Schlitzer, A.; Placek, K.; Joosten, L.A.B.; Schultze, J.L. Innate and adaptive immune memory: An evolutionary continuum in the host's response to pathogens. Cell Host Microbe 2019, 25, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Rujirawat, T.; Patumcharoenpol, P.; Lohnoo, T.; Yingyong, W.; Lerksuthirat, T.; Tangphatsornruang, S.; Suriyaphol, P.; Grenville-Briggs, L.J.; Garg, G.; Kittichotirat, W.; et al. Draft genome sequence of the pathogenic oomycete pythium insidiosum strain Pi-S, isolated from a patient with pythiosis. Genome Announc. 2015, 3, e00574-15. [Google Scholar] [CrossRef] [PubMed]
- Krajaejun, T.; Kittichotirat, W.; Patumcharoenpol, P.; Rujirawat, T.; Lohnoo, T.; Yingyong, W. Draft genome sequence of the oomycete pythium destruens strain ATCC 64221 from a horse with pythiosis in Australia. BMC Res. Notes 2020, 13, 329. [Google Scholar] [CrossRef]
- Tyler, B.M.; Tripathy, S.; Zhang, X.; Dehal, P.; Jiang, R.H.; Aerts, A.; Arredondo, F.D.; Baxter, L.; Bensasson, D.; Beynon, J.L.; et al. Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science 2006, 313, 1261–1266. [Google Scholar] [CrossRef]
- Lamour, K.H.; Mudge, J.; Gobena, D.; Hurtado-Gonzales, O.P.; Schmutz, J.; Kuo, A.; Miller, N.A.; Rice, B.J.; Raffaele, S.; Cano, L.M.; et al. Genome sequencing and mapping reveal loss of heterozygosity as a mechanism for rapid adaptation in the vegetable pathogen Phytophthora capsici. Mol. Plant-Microbe Interact. 2012, 25, 1350–1360. [Google Scholar] [CrossRef]
- Shi, J.; Ye, W.; Ma, D.; Yin, J.; Zhang, Z.; Wang, Y.; Qiao, Y. Improved whole genome sequence of phytophthora capsici generated by long-read sequencing. Mol. Plant-Microbe Interact. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.S.; Shao, J.; Lary, D.J.; Kronmiller, B.; Shen, D.; Strem, M.D.; Amoako-Attah, I.; Akrofi, A.Y.; Begoude, B.A.; Ten Hoopen, G.M.; et al. Phytophthora megakarya and P. palmivora, closely related causal agents of cacao black pod rot, underwent increases in genome sizes and gene numbers by different mechanisms. Genome Biol. Evol. 2017, 9, 536–557. [Google Scholar] [CrossRef] [PubMed]
- Vetukuri, R.R.; Tripathy, S.; Malar, C.M.; Panda, A.; Kushwaha, S.K.; Chawade, A.; Andreasson, E.; Grenville-Briggs, L.J.; Whisson, S.C. Draft genome sequence for the tree pathogen phytophthora plurivora. Genome Biol. Evol. 2018, 10, 2432–2442. [Google Scholar] [CrossRef]
- Yang, C.; Lin, L.; Bao, J.; Wang, Z.; Li, Z.; Guo, H.; Lv, L.; Yu, D.; Chen, Q. Genome sequence resource of phytophthora vignae, the causal agent of stem and root rot of cowpea. Mol. Plant-Microbe Interact. 2021. [Google Scholar] [CrossRef]
- Adhikari, B.N.; Hamilton, J.P.; Zerillo, M.M.; Tisserat, N.; Lévesque, C.A.; Buell, C.R. Comparative genomics reveals insight into virulence strategies of plant pathogenic oomycetes. PLoS ONE 2013, 8, e75072. [Google Scholar] [CrossRef]
- Wu, J.; Handique, U.; Graham, J.; Johnson, E. Phytophthora nicotianae infection of citrus leaves and host defense activation compared to root infection. Phytopathology 2020, 110, 1437–1448. [Google Scholar] [CrossRef]
- Yang, J.K.; Tong, Z.J.; Fang, D.H.; Chen, X.J.; Zhang, K.Q.; Xiao, B.G. Transcriptomic profile of tobacco in response to phytophthora nicotianae infection. Sci. Rep. 2017, 7, 401. [Google Scholar] [CrossRef] [PubMed]
- Vanegas-Villa, D.M.; Navarro-Alzate, R.A.; Afanador-Kafuri, L.; Gutierrez-Monsalve, J.A.; Morales-Osorio, J.G.; Uribe-Soto, S.I.; Gaviria-Gutierrez, B.M. Effect of interaction between phytophthora nicotianae and meloidogyne spp. on the productivity and quality of tobacco plants (nicotiana tabacum). Nematology 2020, 22, 1179–1192. [Google Scholar] [CrossRef]
- Liu, H.; Ma, X.; Yu, H.; Fang, D.; Li, Y.; Wang, X.; Wang, W.; Dong, Y.; Xiao, B. Genomes and virulence difference between two physiological races of phytophthora nicotianae. GigaScience 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Drake, K.; Lewis, R.S. An introgressed Nicotiana rustica genomic region confers resistance to Phytophthora nicotianae in cultivated tobacco. Crop. Sci. 2013, 53, 1366–1374. [Google Scholar] [CrossRef]
- Apple, J. Occurrence of race 1 of phytophthora parasitica var. nicotianae in north carolina and its implications in breeding for disease resistance. Tob. Sci. 1967, 11, 79–83. [Google Scholar]
- Prinsloo, G.; Pauer, G. Die identifikasie van rasse van phytophthora nicotianae B. De Haan var. nicotianae wat in suid-afrika voorkom. Phytophylactica 1974, 6, 4. [Google Scholar]
- McIntyre, J.L.; Taylor, G. Race 3 of phytophthora parasitica var. nicotianae. Phytopathology 1978, 68, 35–38. [Google Scholar] [CrossRef]
- Bowers, J.H.; Locke, J.C. Effect of formulated plant extracts and oils on population density of phytophthora nicotianae in soil and control of phytophthora blight in the greenhouse. Plant. Dis. 2004, 88, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Feng, C.; Zhang, Z.; Zhang, C. Complete mitochondrial genome of phytophthora nicotianae and identification of molecular markers for the oomycetes. Front. Microbiol. 2017, 8, 1484. [Google Scholar] [CrossRef] [PubMed]
- Qu, T.; Shao, Y.; Csinos, A.S.; Ji, P. Sensitivity of phytophthora nicotianae from tobacco to fluopicolide, mandipropamid, and oxathiapiprolin. Plant. Dis. 2016, 100, 2119–2125. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef]
- Baxter, L.; Tripathy, S.; Ishaque, N.; Boot, N.; Cabral, A.; Kemen, E.; Thines, M.; Ah-Fong, A.; Anderson, R.; Badejoko, W.; et al. Signatures of adaptation to obligate biotrophy in the hyaloperonospora arabidopsidis genome. Science 2010, 330, 1549–1551. [Google Scholar] [CrossRef]
- McGowan, J.; Fitzpatrick, D.A. Recent advances in oomycete genomics. Adv. Genet. 2020, 105, 175–228. [Google Scholar] [CrossRef]
- Matari, N.H.; Blair, J.E. A multilocus timescale for oomycete evolution estimated under three distinct molecular clock models. BMC Evol. Biol. 2014, 14, 1471–2148. [Google Scholar] [CrossRef]
- Höfte, H.; Voxeur, A. Plant cell walls. Curr. Biol. 2017, 27, R865–R870. [Google Scholar] [CrossRef]
- Mateos, F.V.; Rickauer, M.; Esquerré-Tugayé, M.T. Cloning and characterization of a cDNA encoding an elicitor of phytophthora parasitica var. nicotianae that shows cellulose-binding and lectin-like activities. Mol. Plant-Microbe Interact. 1997, 10, 1045–1053. [Google Scholar] [CrossRef]
- Links, M.G.; Holub, E.; Jiang, R.H.; Sharpe, A.G.; Hegedus, D.; Beynon, E.; Sillito, D.; Clarke, W.E.; Uzuhashi, S.; Borhan, M.H. De novo sequence assembly of albugo candida reveals a small genome relative to other biotrophic oomycetes. BMC Genom. 2011, 12, 1471–2164. [Google Scholar] [CrossRef]
- Kale, S.D.; Gu, B.; Capelluto, D.G.; Dou, D.; Feldman, E.; Rumore, A.; Arredondo, F.D.; Hanlon, R.; Fudal, I.; Rouxel, T.; et al. External lipid PI3P mediates entry of eukaryotic pathogen effectors into plant and animal host cells. Cell 2010, 142, 284–295. [Google Scholar] [CrossRef]
- Evangelisti, E.; Govetto, B.; Minet-Kebdani, N.; Kuhn, M.L.; Attard, A.; Ponchet, M.; Panabières, F.; Gourgues, M. The phytophthora parasitica RXLR effector penetration-specific effector 1 favours arabidopsis thaliana infection by interfering with auxin physiology. New Phytol. 2013, 199, 476–489. [Google Scholar] [CrossRef]
- Jiang, R.H.; de Bruijn, I.; Haas, B.J.; Belmonte, R.; Löbach, L.; Christie, J.; van den Ackerveken, G.; Bottin, A.; Bulone, V.; Díaz-Moreno, S.M.; et al. Distinctive expansion of potential virulence genes in the genome of the oomycete fish pathogen saprolegnia parasitica. PLoS Genet. 2013, 9, e1003272. [Google Scholar] [CrossRef] [PubMed]
- Tani, S.; Yatzkan, E.; Judelson, H.S. Multiple pathways regulate the induction of genes during zoosporogenesis in phytophthora infestans. Mol. Plant-Microbe Interact. 2004, 17, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.A.; Judelson, H.S. A bZIP transcription factor from phytophthora interacts with a protein kinase and is required for zoospore motility and plant infection. Mol. Microbiol. 2005, 56, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lu, J.; Tao, K.; Ye, W.; Li, A.; Liu, X.; Kong, L.; Dong, S.; Zheng, X.; Wang, Y. A Myb transcription factor of Phytophthora sojae, regulated by MAP kinase PsSAK1, is required for zoospore development. PLoS ONE 2012, 7, e40246. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Wang, Y.; Dong, S.; Tyler, B.M. Phylogenetic and transcriptional analysis of an expanded bZIP transcription factor family in phytophthora sojae. BMC Genom. 2013, 14, 1471–2164. [Google Scholar] [CrossRef] [PubMed]
- Kerkhof, L.; Ward, B.B. Comparison of nucleic acid hybridization and fluorometry for measurement of the relationship between RNA/DNA ratio and growth rate in a marine bacterium. Appl. Environ. Microbiol. 1993, 59, 1303–1309. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- de Sena Brandine, G.; Smith, A.D. Falco: High-speed FastQC emulation for quality control of sequencing data. F1000Research 2019, 8, 1874. [Google Scholar] [CrossRef]
- Chaisson, M.J.; Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinform. 2012, 13, 238. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 1–18. [Google Scholar] [CrossRef]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef]
- English, A.C.; Richards, S.; Han, Y.; Wang, M.; Vee, V.; Qu, J.; Qin, X.; Muzny, D.M.; Reid, J.G.; Worley, K.C.; et al. Mind the gap: Upgrading genomes with pacific biosciences RS long-read sequencing technology. PLoS ONE 2012, 7, e47768. [Google Scholar] [CrossRef]
- Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4.10. 1–4.10. 14. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C. The universal protein resource (UniProt). Nucleic Acids Res. 2008, 36, D190–D195. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Slater, G.S.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 1471–2105. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 1471–2148. [Google Scholar] [CrossRef] [PubMed]
- Torto, T.A.; Li, S.; Styer, A.; Huitema, E.; Testa, A.; Gow, N.A.; van West, P.; Kamoun, S. EST mining and functional expression assays identify extracellular effector proteins from the plant pathogen phytophthora. Genome Res. 2003, 13, 1675–1685. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, 18. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable venn and euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Categories | Scaffold | Contig |
|---|---|---|
| Total Number | 2105 | 3868 |
| Total length (bp) | 95,318,700 | 58,288,754 |
| Average Length (bp) | 29,463.56 | 15,069.48 |
| N50 Length (kb) | 113.15 | 54.23 |
| N90 Length (kb) | 28.94 | 8.54 |
| Maximum Length (bp) | 540,321 | 383,516 |
| Minimum Length (bp) | 1002 | 200 |
| GC content | 49.02% | 49.78% |
| Class | Type | Number of Elements | Length Occupied | Percentage of Sequence |
|---|---|---|---|---|
| Interspersed repeats | DNA elements | |||
| Crypton | 535 | 320,797 | 0.34% | |
| Maverick | 110 | 143,341 | 0.15% | |
| MuLE-MuDR | 1243 | 587,998 | 0.62% | |
| MuLE-NOF | 535 | 60,260 | 0.06% | |
| PIF-Harbinger | 294 | 231,089 | 0.24% | |
| PiggyBac | 450 | 257,994 | 0.27% | |
| Sola | 275 | 91,598 | 0.10% | |
| TcMar-ISRm11 | 791 | 398,902 | 0.42% | |
| Merlin | 198 | 88,846 | 0.09% | |
| TcMar-Pogo | 201 | 54,855 | 0.06% | |
| TcMar-Ant1 | 272 | 138,354 | 0.15% | |
| TcMar-Tc1 | 807 | 449,528 | 0.47% | |
| TcMar-Tc2 | 469 | 215,152 | 0.23% | |
| TcMar-Stowawa | 197 | 113,580 | 0.12% | |
| hAT-Ac | 324 | 136,041 | 0.14% | |
| hAT-Tag1 | 176 | 68,793 | 0.07% | |
| DNA | 543 | 303,342 | 0.32% | |
| LTR # | ||||
| LTR | 283 | 238,869 | 0.25% | |
| Copia | 1179 | 787,427 | 0.83% | |
| Gypsy | 7785 | 6,079,720 | 6.38% | |
| Ngaro | 214 | 148,614 | 0.16% | |
| RC $ | ||||
| Helitron | 654 | 444,976 | 0.47% | |
| Unknown | 20279 | 6,888,937 | 7.23% | |
| Tandem repeats | Microsatellite | 717 | 52,963 | 0.06% |
| Minisatellite | 7437 | 598,915 | 0.63% | |
| Satellite | 576 | 255,356 | 0.27% | |
| Total | 46544 | 19,156,247 | 20.10% | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, X.-L.; Zhang, C.-S.; Kong, F.-Y.; Zhang, Z.-F.; Wang, F.-L. Genome Analysis of Phytophthora nicotianae JM01 Provides Insights into Its Pathogenicity Mechanisms. Plants 2021, 10, 1620. https://doi.org/10.3390/plants10081620
Yuan X-L, Zhang C-S, Kong F-Y, Zhang Z-F, Wang F-L. Genome Analysis of Phytophthora nicotianae JM01 Provides Insights into Its Pathogenicity Mechanisms. Plants. 2021; 10(8):1620. https://doi.org/10.3390/plants10081620
Chicago/Turabian StyleYuan, Xiao-Long, Cheng-Sheng Zhang, Fan-Yu Kong, Zhong-Feng Zhang, and Feng-Long Wang. 2021. "Genome Analysis of Phytophthora nicotianae JM01 Provides Insights into Its Pathogenicity Mechanisms" Plants 10, no. 8: 1620. https://doi.org/10.3390/plants10081620
APA StyleYuan, X.-L., Zhang, C.-S., Kong, F.-Y., Zhang, Z.-F., & Wang, F.-L. (2021). Genome Analysis of Phytophthora nicotianae JM01 Provides Insights into Its Pathogenicity Mechanisms. Plants, 10(8), 1620. https://doi.org/10.3390/plants10081620