
Genome-Wide Identification and Development of LTR Retrotransposon-Based Molecular Markers for the Melilotus Genus


Abstract
1. Introduction
2. Results
2.1. Identification and Analysis of LTR Retrotransposons in the Melilotus Genome
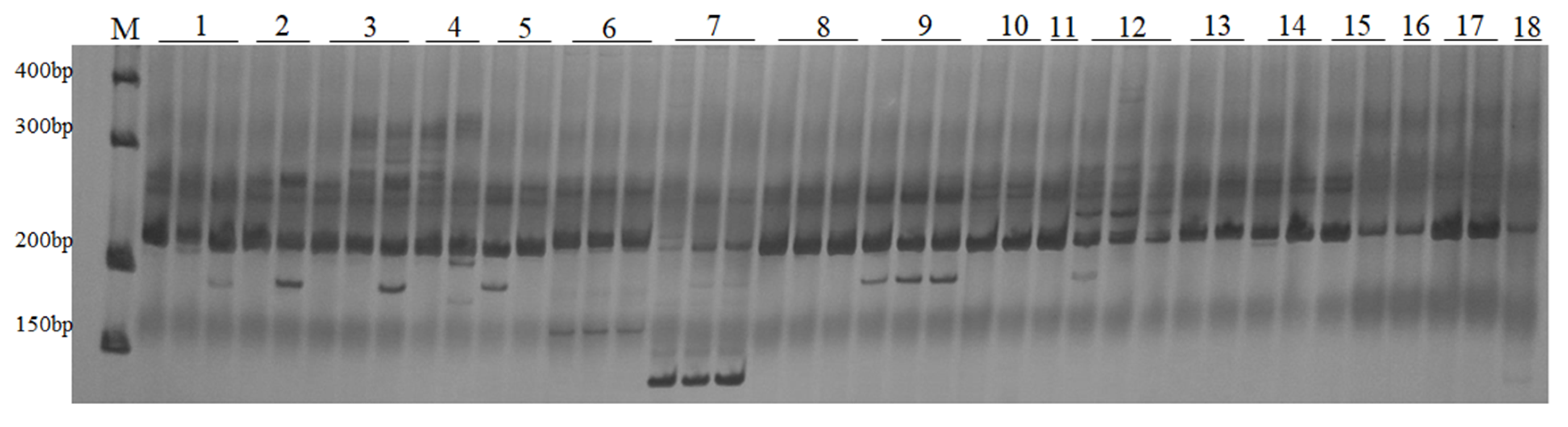
2.2. Amplification with LTR Primers in Melilotus
2.3. Transferability of the Newly Developed LTR Retrotransposon-Based Markers
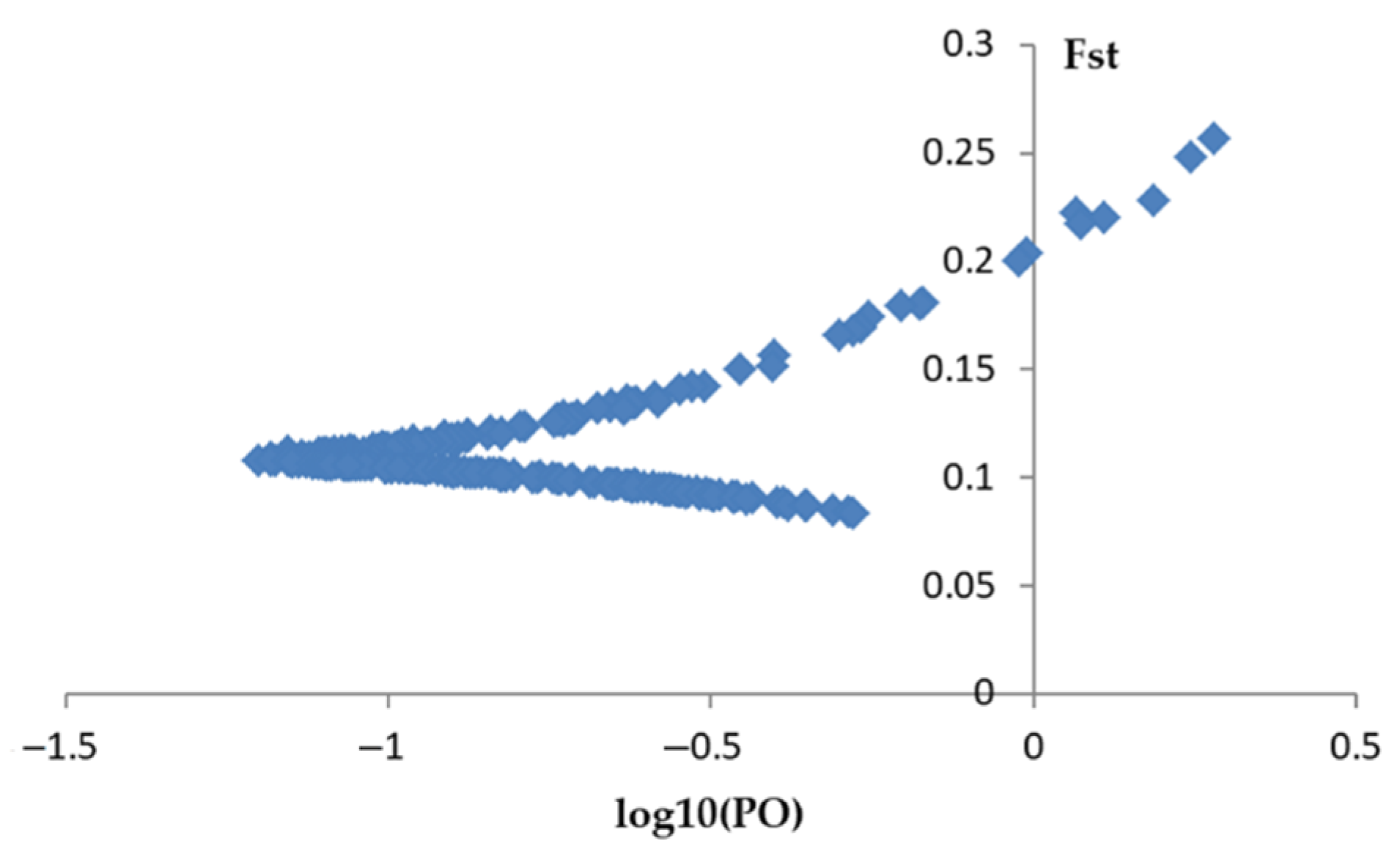
2.4. Outlier Detection
2.5. Genetic Diversity
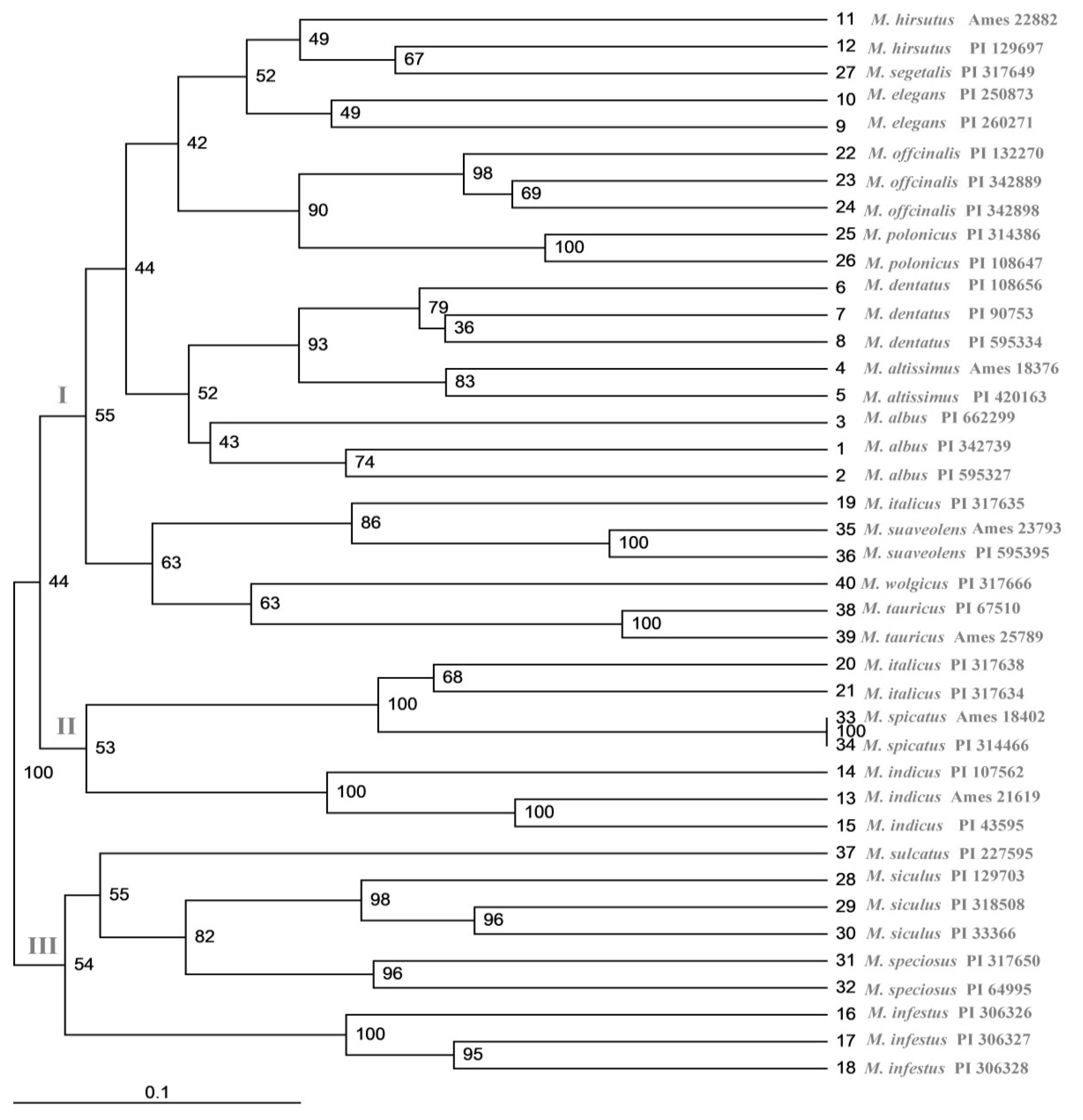
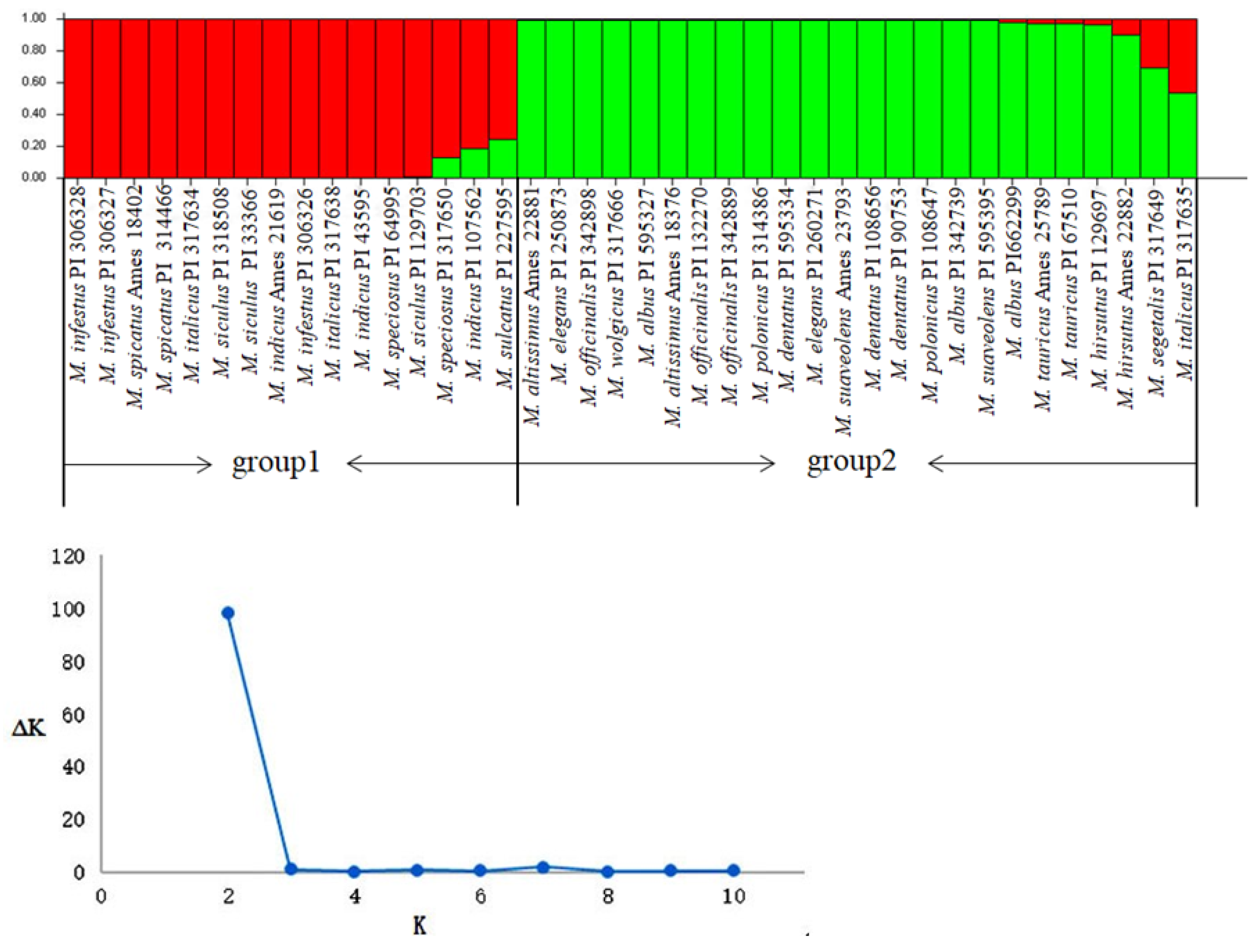
2.6. Cluster and Population Structure Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source of Variation | Degrees of Freedom | Sum of Squares | Variance Components | Percentage of Variation | p-Value |
|---|---|---|---|---|---|
| Among groups | 1 | 167.759 | 5.198 | 12.13 | <0.001 |
| Among species within groups | 16 | 963.857 | 18.976 | 44.29 | <0.001 |
| Within species | 22 | 410.833 | 18.674 | 43.58 | <0.001 |
| Categories | Classification | Subclassification | Species |
|---|---|---|---|
| Molecular phylogeny [25] | Clade I | M. albus, M. altissimus, M. hirsatus, M.officinalis, M. polonicus, M. suaveolens, M. wolgicus, M. elegans, M. dentatus, M. tauricus | |
| Clade II | Clade 1 | M. spicatus | |
| IIb | M. indicus, M. segetalis | ||
| Clade 2 | M. infestus | ||
| IIa | M. siculus, M. sulcatus, M. speciosus, M. italicus | ||
| SSR makers [26] | A | M. albus, M. altissimus, M. hirsutus, M. officinalis, M. polonicus, M. suaveolens, M. wolgicus, M. elegans, M.infestus, M. spicatus, M. sulcatus, | |
| A1 | M. italicus, M. speciosus | ||
| B | M. dentatus, M. siculus, M. tauricus | ||
| B1 | M. indicus, M. segetalis | ||
| EST-SSR markers [27] | I | M. albus, M. altissimus, M. hirsutus, M. officinalis, M. polonicus, M. suaveolens, M. wolgicus, M. elegans, M. dentatus, M. tauricus | |
| II | M. indicus, M. segetalis, M. italicus, M. spicatus | ||
| III | M. infestus, M. siculus, M. speciosus, M. sulcatus | ||
| LTR-RT markers | GI | M. albus, M. altissimus, M. hirsutus, M. officinalis, M. polonicus, M. suaveolens, M. wolgicus, M. elegans, M. dentatus, M. tauricus, M. segetalis | |
| GIII GII | M. indicus, M. italicus, M. spicatus M. infestus, M. siculus, M. speciosus, M. sulcatus |
3. Discussion
4. Materials and Methods
4.1. Plant Materials, Genomic DNA Isolation, and PCR Primer Design
4.2. Identification of LTRs
4.3. Primer Selection and PCR Conditions
4.4. Sequencing of PCR Amplification Products
4.5. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wicker, T.; Sabot, F.O.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 10, 276. [Google Scholar] [CrossRef]
- Havecker, E.R.; Gao, X.; Voytas, D.F. The diversity of LTR retrotransposons. Genome Biol. 2004, 5, 225. [Google Scholar] [CrossRef][Green Version]
- Özer, G.; Bayraktar, H.; Baloch, F.S. iPBS retrotransposons ‘A Universal Retrotransposons now’ in molecular phylogeny of fungal pathogens. Biochem. Syst. Ecol. 2016. [Google Scholar] [CrossRef]
- Wu, J. Construction and application of identification and analysis process of full-length LTR-retrotransposons (D). Anhui Agric. Univ. 2017, 56. [Google Scholar]
- Mccarthy, E.M.; Liu, J.; Gao, L.; Mcdonald, J.F. Long terminal repeat retrotransposons of Oryza sativa. Genome Biol. 2002, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nie, Q.; Qiao, G.; Peng, L.; Wen, X. Transcriptional activation of long terminal repeat retrotransposon sequences in the genome of pitaya under abiotic stress. Plant Physiol. Bioch. 2018. [Google Scholar] [CrossRef]
- Arystanbekkyzy, M.; Nadeem, M.A.; Aktaş, H.; Yeken, M.Z.; Zencirci, N. Phylogenetic and taxonomic relationship of Turkish wild and cultivated Emmer (Triticum turgidum ssp. dicoccoides) revealed by iPBS-Retrotransposons markers. Int. J. Agric. Biol. 2018, 1814–9596. [Google Scholar] [CrossRef]
- Zhao, M.; Zhi, H.; Zhang, X.; Jia, G.; Diao, X. Retrotransposon-mediated DELLA transcriptional reprograming underlies semi-dominant dwarfism in foxtail millet. Crop J. 2019, 7, 458–468. [Google Scholar] [CrossRef]
- Flavia, M.; Tommaso, G.; Marilena, C.; Andrea, C.; Lucia, N. Genome-wide analysis of LTR-retrotransposon diversity and its impact on the evolution of the genus Helianthus (L.). BMC Genom. 2017, 18, 634. [Google Scholar] [CrossRef]
- Barghini, E.; Natali, L.; Giordani, T.; Cossu, R.M.; Scalabrin, S.; Cattonaro, F.; Imkova, H.; Vrana, J.; Dole El, J.; Morgante, M. LTR retrotransposon dynamics in the evolution of the olive (Olea europaea) genome. DNA Res. 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Friesen, N.; Brandes, A.; Heslop-Harrison, J.S. Diversity, origin, and distribution of retrotransposons (gypsy and copia) in conifers. Mol. Biol. Evol. 2001, 18, 1176–1188. [Google Scholar] [CrossRef]
- Kalendar, R.; Flavell, A.J.; Ellis, T.H.N.; Sjakste, T.; Moisy, C.; Schulman, A.H. Analysis of plant diversity with retrotransposon-based molecular markers. Heredity 2011, 106, 520. [Google Scholar] [CrossRef]
- Schulman, A.H.; Flavell, A.J.; Paux, E.; Ellis, T.H.N. The Application of LTR Retrotransposons as Molecular Markers in Plants. Methods Mol. Biol. 2012, 859, 115–153. [Google Scholar] [CrossRef] [PubMed]
- Jing, R.; Vershinin, A.; Grzebyta, J.; Shaw, P.; Smykal, P.; Marshall, D.; Ambrose, M.J.; Ellis, T.N.; Flavell, A.J. The genetic diversity and evolution of field pea (Pisum) studied by high throughput retrotransposon based insertion polymorphism (RBIP) marker analysis. BMC Evol. Biol. 2010, 10, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Cong, J.M.; Chen, F.Q.; Sun, C.L. Study on Comprehensive Development of Melilotus suaverolens L. Anhui Agric. Sci. 2012, 2962–2963. [Google Scholar] [CrossRef]
- Ma, L. Talking about the Comprehensive Utilization of Plants. Xinjiang Anim. Husb. 2005, 56–57. [Google Scholar] [CrossRef]
- Cowan, R.S.; Allen, O.N.; Allen, E.K. The Leguminosae. A Source Book of Characteristics, Uses and Nodulation; University of Wisconsin Press: Madison, WI, USA, 1982; pp. 369–382. [Google Scholar]
- Sherif, E.A.A. Melilotus indicus (L.) All., a salt-tolerant wild leguminous herb with high potential for use as a forage crop in salt-affected soils. Flora 2009, 204, 737–746. [Google Scholar] [CrossRef]
- Stickler, F.C.; Johnson, I.J. Dry Matter and Nitrogen Production of Legumes and Legume Associations in the Fall of the Seeding Year1. Agron. J. 1959, 51, 135–137. [Google Scholar] [CrossRef]
- Zhang, H.X.; Bai, R.; Wu, F.; Guo, W.I.; Yan, Z.Z.; Yan, Q.; Zhang, Y.F.; Ma, J.X.; Zhang, J.Y. Genetic diversity, phylogenetic structure and development of core collections in Melilotus accessions from a Chinese gene bank. Sci. Rep. 2019, 9, 13017. [Google Scholar] [CrossRef]
- Wu, F.; Luo, K.; Yan, Z.Z.; Zhang, D.Y.; Yan, Q.; Zhang, Y.F.; Yi, X.; Zhang, J.Y. Analysis of miRNAs and their target genes in five Melilotus albus NILs with different coumarin content. Sci. Rep. 2018. [Google Scholar] [CrossRef]
- Wang, H.; Liu, J.S. LTR retrotransposon landscape in Medicago truncatula: More rapid removal than in rice. BMC Genom. 2008, 9, 382. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H. Karyotype compartive studies of nine species on Melilotus. Bull. Bot. Res. 1984, 4, 145–157. [Google Scholar]
- Grzebelus, D. Class II transposable elements in Medicago truncatula. Model Legume Med. Truncatula 2019, 888–892. [Google Scholar] [CrossRef]
- Di, H.Y.; Duan, Z.; Luo, K.; Zhang, D.Y.; Wu, F.; Zhang, J.Y.; Liu, W.X.; Wang, Y.R.; Sun, G.L. Interspecific phylogenic relationships within genus Melilotus based on Nuclear and Chloroplast DNA. PLoS ONE 2015, 10, e0132596. [Google Scholar] [CrossRef]
- Wu, F.; Zhang, D.Y.; Ma, J.X.; Luo, K.; Di, H.Y.; Liu, Z.P.; Zhang, J.Y.; Wang, Y.R. Analysis of genetic diversity and population structure in accessions of the genus Melilotus. Ind. Crop. Prod. 2016, 85, 84–92. [Google Scholar] [CrossRef]
- Yan, Z.Z.; Wu, F.; Luo, K.; Zhao, Y.F.; Yan, Q.; Zhang, Y.F.; Wang, Y.R.; Zhang, J.Y. Cross-species transferability of EST-SSR markers developed from the transcriptome of Melilotus and their application to population genetics research. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Özer, G.; Sameeullah, M.; Bayraktar, H.; Göre, M.E. Genetic diversity among phytopathogenic Sclerotiniaceae, based on retrotransposon molecular markers. Phytopatholo. Mediterr. 2017, 56, 251–258. [Google Scholar] [CrossRef]
- Ishikawa, H.; Watano, Y.; Kano, K.; Ito, M.; Kurita, S. Development of primer sets for PCR amplification of the PgiC gene in ferns. J. Plant Res. 2002, 115, 0065–0070. [Google Scholar] [CrossRef]
- Konovalov, F.A.; Goncharov, N.P.; Goryunova, S.; Shaturova, A.; Proshlyakova, T.; Kudryavtsev, A. Molecular markers based on LTR retrotransposons BARE-1 and Jeli uncover different strata of evolutionary relationships in diploid wheats. Mol. Genet. Genom. 2010, 283, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.H.; Wang, P.; Jin, D.M.; Qu, X.P.; Wang, B. The application of RAPD markers in diversity detection and variety identification of Porphyra. Acta. Bot. Sin. 2000, 42, 403–407. [Google Scholar] [CrossRef]
- Aneja, B.; Yadav, N.R.; Chawla, V.; Yadav, R.C. Sequence-related amplified polymorphism (SRAP) molecular marker system and its applications in crop improvement. Mol. Breed. 2012, 30, 1635–1648. [Google Scholar] [CrossRef]
- Winton, L.M.; Krohn, A.L.; Conn, J.S. Microsatellite markers for the invasive plant species white sweetclover (Melilotus alba) and yellow sweetclover (Melilotus officinalis). Mol. Ecol. Notes 2007, 7, 1296–1298. [Google Scholar] [CrossRef]
- Yan, Z.Z.; Yan, R.; Fan, W.; Kai, L.; Zhang, D.Y.; Qi, Y.; Zhang, Y.F.; Zhao, Y.F.; Zhang, J.Y. The development and screening of EST-SSR markers in Melilotus albus. Pratacultural Sci. 2017. [Google Scholar] [CrossRef]
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Annu. Rev. Genet. 1999, 33, 479. [Google Scholar] [CrossRef]
- Casacuberta, J.M.; Vernhettes, S.; Audeon, C.; Grandbastien, M.A. Quasispecies in retrotransposons: A role for sequence variability in Tnt1 evolution. Genetica 1997, 100, 109–117. [Google Scholar] [CrossRef]
- Ramakrishnan, M.; Yrjl, K.; Mingbing, Z.; Kurungara, V.K.; Kalendar, R. Development and deployment of High-Throughput retrotransposon-based markers reveal genetic diversity and population structure of Asian Bamboo. Forests 2019, 11, 31. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, T.L.; Wang, Y.R.; Liu, Z.P. The development of 204 novel EST-SSRs and their use for genetic diversity analyses in cultivated alfalfa. Biochem. Syst. Ecol. 2014, 57, 227–230. [Google Scholar] [CrossRef]
- Liang, Y.H.; Lenz, R.R.; Dai, W. Development of retrotransposon-based molecular markers and their application in genetic mapping in chokecherry (Prunus virginiana L). Mol. Breed. 2016, 36, 109. [Google Scholar] [CrossRef]
- Nadeem, M.A.; Habyarimana, E.; Çiftçi, V.; Nawaz, M.A.; Karaköy, T.; Comertpay, G.; Shahid, M.Q.; Hatipoğlu, R.; Yeken, M.Z.; Ali, F. Characterization of genetic diversity in Turkish common bean gene pool using phenotypic and whole-genome DArTseq-generated silicoDArT marker information. PLoS ONE 2018, 13, e0205363. [Google Scholar] [CrossRef]
- Zhang, M. The genetic diversity of geograhical popualtions of the migratory locust analyzed with the percent of polymorphic loci and Shannon’s Index. Chin. Agric. Sci. Bull. 2008, 9, 376–381. [Google Scholar]
- Luikart, G.; England, P.R.; Tallmon, D.; Jordan, S.; Taberlet, P. The power and promise of population genomics: From genotyping to genome typing. Nat. Rev. Genet. 2003, 4, 981–994. [Google Scholar] [CrossRef]
- Estoup, A.; Jarne, P.; Cornuet, J.M. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol. Ecol. 2010, 11, 1591–1604. [Google Scholar] [CrossRef]
- Shan, Z. Improved SDS method for general plant genomic DNA extraction. Guangdong Agric. Sci. 2011, 38, 3. [Google Scholar] [CrossRef]
- Jong-Wook, C.; Tae-Sung, K.; Sundan, S.; Sok-Young, L.; Gyu-Taek, C. Development of 65 Novel Polymorphic cDNA-SSR Markers in Common Vetch (Vicia sativa subsp. sativa) using next generation sequencing. Molecules 2013, 8376–8392. [Google Scholar] [CrossRef]
- Gaggiotti, F.O. A Genome-Scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian Perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef]
- Yeh, F.; Yang, R.C.; Boyle, T. Software Microsoft Window-Based Freeware for Population Genetic Analysis, Popgene Version 1.32 ed.; University of Alberta: Edmonton, AB, Canada, 1997. [Google Scholar]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Pavlícek, A.; Hrdá, S.; Flegr, J. Free-Tree--freeware program for construction of phylogenetic trees on the basis of distance data and bootstrap/jackknife analysis of the tree robustness. Application in the RAPD analysis of genus Frenkelia. Folia Bio. 1999, 45, 97–99. [Google Scholar]
- Page, R.D.M. Tree View: An application to display phylogenetic trees on personal computers. Bioinformatics 1996, 12. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Pritcharda, J.K.; Wena, X.; Falush, D. Documentation for Structure Software: Version 2.3; University of Chicago: Chicago, IL, USA, 2010. [Google Scholar]
- Evanno, G.S.; Regnaut, S.J.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]





| Total Length of LTR-RTs Screened (bp) | No. of LTR-RTs Identified | No. of Primers Designed for Each Type of LTR-RTs | ||
|---|---|---|---|---|
| 585,894,349 | Ty1/Copia | 77,935 | RBIP | 232 |
| Ty3/gypsy | 101,240 | IRAP | 105 | |
| Unknown | 1865 | ISBP | 10 | |
| REMAP | 3 | |||
| Primer Code | Type | Primer Sequence | Allele Size Range (kp) | NA | He | PIC |
|---|---|---|---|---|---|---|
| 25 | RBIP | F:GAGAACTGAGAAGAGGGTC R:CTCCACCTTGACTTGAATC | 0.2–0.3 | 5 | 0.76 | 0.72 |
| 40 | RBIP | F:GAAAGGATTCTGAGCGTAG R:ATACTCTCCACCACTGTCA | 0.5–0.7 | 3 | 0.67 | 0.6 |
| 41 | RBIP | F:GAAAGGATTCTGAGCGTAG R:GTAATACTCTCCACCACTGTC | 0.4–0.7 | 5 | 0.67 | 0.63 |
| 45 | IRAP | F:TATGCTTCAACCTGAGGG R:GTTCATTTCTGCTCGCTC | 0.3–0.4 | 4 | 0.62 | 0.56 |
| 46 | RBIP | F:GAAAGTCTAATGCCGAGG R:AATACTCTCCACCACGGT | 0.4–0.5 | 5 | 0.45 | 0.43 |
| 54 | RBIP | F:TCTCAGACATAGAACCCG R:AGTGATGGTAACCCAACC | 0.3–0.4 | 4 | 0.59 | 0.53 |
| 55 | RBIP | F:GTGTCCACAAAGGATTCC R:TCTCCACAAGACCACTTC | 0.2–0.4 | 4 | 0.43 | 0.4 |
| 62 | IRAP | F:ATTTAGTGGCAGCCCTTC R:GACCTTTCTTTCCGCATC | 0.3–0.4 | 3 | 0.6 | 0.52 |
| 67 | RBIP | F:GACAACTTGAACGGACAAAC R:AGGGTAAAGGCTAAGGGAG | 0.15–0.2 | 3 | 0.57 | 0.48 |
| 68 | RBIP | F: GGGACAACTACATAACTTGG R: GCTGCCACTAAATCAGAG | 0.4–0.5 | 3 | 0.54 | 0.44 |
| 69 | RBIP | F: TCACTTACCTATTGCTCTCC R: TGCTTCCTTGACAGTCTTAG | 0.15–0.4 | 3 | 0.64 | 0.57 |
| 74 | RBIP | F:TTCATACCACTCCGAGAG R:GGATGTCCATTAGAGGCT | 0.15–0.3 | 4 | 0.68 | 0.61 |
| 76 | RBIP | F: TGTGTGTGTGTGTCTGTTCT R: AACCTCGTAGTTCGGGTA | 0.4–0.5 | 3 | 0.51 | 0.45 |
| 78 | RBIP | F: CATCCTGAATAGAGTCCCT R: ATCGGTATCCCTTAGCAC | 0.2–0.3 | 3 | 0.49 | 0.43 |
| 83 | RBIP | F: CTGTAGTATTCAAGGGTGG R: GAAGCCATTCTAAGGGTC | 0.2–0.4 | 4 | 0.58 | 0.49 |
| 93 | RBIP | F: CTCCTTGACTGTTGCCATTA R: GGGAAGAAACCCTGGATT | 0.2–0.3 | 3 | 0.57 | 0.51 |
| 95 | RBIP | F: CCTGAAGAAGAATGGTCC R: GTGGTAAGAAGTTGAAGCC | 0.15–0.3 | 4 | 0.58 | 0.52 |
| 105 | RBIP | F: TCTCAACTCCAATGGCAG R: TTCAGAGGCAGAAGCATC | 0.2–0.4 | 4 | 0.52 | 0.43 |
| 138 | IRAP | F: GCATTGTTGTCACAGTCAAG R:GCAAGTTACTCTTCATACCTGG | 0.3–0.7 | 5 | 0.64 | 0.59 |
| 146 | RBIP | F: ATCCCTTCTCTCCTTCCCT R: TCACCTTGATACTTGCCG | 0.1–0.2 | 3 | 0.57 | 0.48 |
| 148 | IRAP | F: GGTGTGGACAGATAGTAAGG R: GAGTTGGTAGGTTGAGTTTG | 0.1–0.2 | 5 | 0.77 | 0.73 |
| 149 | RBIP | F: CTAAATGGAGGGAAGAGAGA R: GTGACAACTTGAGTGCCA | 0.2–0.3 | 3 | 0.56 | 0.46 |
| 152 | IRAP | F: CTTATCTCCCTCAACAAGC R: CTACAGAAATGGCGACTTC | 0.1–0.2 | 4 | 0.7 | 0.65 |
| 153 | IRAP | F: CAGCAACATAACGAGAACG R:CCGAGAGAAATGAGAGAGAAGT | 0.2–0.5 | 4 | 0.68 | 0.62 |
| 155 | RBIP | F: CTTGTTGCGTTAGTGTGC R: AACTGGGATGGTCCGTAT | 0.5–0.7 | 3 | 0.42 | 0.38 |
| 170 | RBIP | F: GTGACGAGAAGAAGAAAGG R:CACAGATTTACCACTGGC | 0.2–0.3 | 3 | 0.63 | 0.56 |
| 183 | RBIP | F: TTACTAATCCCACCACCC R: GACGAAGGAGAAGAGAATG | 0.2–0.4 | 3 | 0.56 | 0.47 |
| 196 | RBIP | F: GATTGTTCCGATTCAGGC R: AGGACTTGCTGGATTTGG | 0.3–0.5 | 5 | 0.66 | 0.6 |
| 209 | RBIP | F: GTCTCACACACAAGATTCC R: GGTGGTTAGGGAGGTTAT | 0.15–0.3 | 5 | 0.75 | 0.71 |
| 210 | RBIP | F: GTCTCACACACAAGATTCC R: GGTGGTTAGGGAGGTTAT | 0.15–0.3 | 5 | 0.76 | 0.72 |
| 226 | RBIP | F: GCTTCAAGTGTGGTGGAT R: AACGCAACCCTTCTCTCT | 0.15–0.3 | 5 | 0.64 | 0.59 |
| 229 | RBIP | F: ATCGGAATGGACTCTACC R: GTGTATGCGTATGTGTGAG | 0.15–0.3 | 7 | 0.8 | 0.77 |
| 277 | RBIP | F: TCAGATGGAGTTGTGAGG R: GAGGCTAAACCCTACGAT | 0.2–0.3 | 4 | 0.56 | 0.52 |
| 280 | RBIP | F: GAACTGTATGTGTCCAAGG R: CCAGGAAGAGAACAAGAC | 0.15–0.3 | 3 | 0.58 | 0.51 |
| 281 | RBIP | F: AGAGGAAGAAGACAACCG R:GTCACAAAGGATGAGGGT | 0.15–0.3 | 4 | 0.67 | 0.61 |
| 283 | RBIP | F: CCCGAATCTAAGGTCAAAGT R: CACGCAAGAAACACATCAC | 0.3–0.5 | 3 | 0.52 | 0.41 |
| 284 | IRAP | F: ATTTGGACCAGGCACACT R: AAGCACTCCGTCATCGTA | 0.2–0.4 | 4 | 0.63 | 0.55 |
| 286 | IRAP | F: CGGATGATACGAAAGTGAG R: GCTTCTGTTGTTAGCCCAT | 0.15–0.3 | 5 | 0.73 | 0.69 |
| 290 | RBIP | F: ACTAAGGTTCCAGGCTGT R: GACTCATCCAACAATCCC | 0.3–0.4 | 3 | 0.59 | 0.51 |
| 293 | RBIP | F: CGGCAAGGTAGAGAGAAGT R: AATGGGCTTTGGAGTAGG | 0.15–0.3 | 4 | 0.58 | 0.52 |
| 300 | IRAP | F: CTCTCACACATACACAAAGG R: ATCTGGAGTTCTGGAAGTC | 0.3–0.5 | 4 | 0.56 | 0.51 |
| 301 | RBIP | F: CAAGCACGGTAAGTTAGC R: CGAGTTCAAGAGCACCTT | 0.15–0.4 | 4 | 0.49 | 0.46 |
| 302 | RBIP | F: AAAGAGGACAAAGACCCG R: TAGTCAACGCACATACGC | 0.2–0.5 | 5 | 0.5 | 0.47 |
| 324 | IRAP | F: GGTATCAGAGCCTGGTTAG R: AAACAGTCCTCAGTTCCTC | 0.15–0.3 | 5 | 0.65 | 0.6 |
| 336 | IRAP | F: GAGGAAGTAGACGCTTATTG R: GTTGGTGGTGTCATTCAC | 0.2–0.3 | 3 | 0.57 | 0.51 |
| 350 | RBIP | F: TCACAGAGTTTGAGTCCC R: GAAGAAGAAGGTGGGTTC | 0.2–0.3 | 4 | 0.62 | 0.54 |
| Mean | 3.96 | 0.61 | 0.54 |
| Species | Accessions | NPL | PPL (%) | NA | Ne | h | I |
|---|---|---|---|---|---|---|---|
| M. albus | 3 | 79 | 27.05 | 1.2705 | 1.1679 | 0.0998 | 0.1493 |
| M. altissimus | 2 | 29 | 9.93 | 1.0993 | 1.0702 | 0.0411 | 0.0601 |
| M. dentatus | 3 | 50 | 17.12 | 1.1712 | 1.1128 | 0.0655 | 0.0970 |
| M. elegans | 2 | 45 | 15.41 | 1.1541 | 1.1090 | 0.0638 | 0.0932 |
| M. hirsutus | 2 | 43 | 14.73 | 1.1473 | 1.1041 | 0.0610 | 0.0891 |
| M. indicus | 3 | 70 | 23.97 | 1.2397 | 1.1583 | 0.0918 | 0.1360 |
| M. infestus | 3 | 61 | 20.89 | 1.2089 | 1.1452 | 0.0826 | 0.1213 |
| M. italicus | 3 | 98 | 33.56 | 1.3356 | 1.2282 | 0.1309 | 0.1929 |
| M. officinalis | 3 | 49 | 16.78 | 1.1678 | 1.1150 | 0.0658 | 0.0968 |
| M. polonicus | 2 | 24 | 8.22 | 1.0822 | 1.0581 | 0.0340 | 0.0497 |
| M. segetalis | 1 | 0 | 0.00 | 1 | 1 | 0 | 0 |
| M. siculus | 3 | 56 | 19.18 | 1.1918 | 1.1342 | 0.0762 | 0.1117 |
| M. speciosus | 2 | 38 | 13.01 | 1.1301 | 1.0920 | 0.0539 | 0.0787 |
| M. spicatus | 2 | 2 | 0.68 | 1.0068 | 1.0048 | 0.0028 | 0.0041 |
| M. suaveolens | 2 | 17 | 5.82 | 1.0582 | 1.0412 | 0.0241 | 0.0352 |
| M. sulcatus | 1 | 0 | 0.00 | 1 | 1 | 0 | 0 |
| M. tauricus | 2 | 17 | 5.82 | 1.0582 | 1.0412 | 0.0241 | 0.0352 |
| M. wolgicus | 1 | 0 | 0.00 | 1 | 1 | 0 | 0 |
| Mean | 37.67 | 12.90 | 1.1290 | 1.0879 | 0.0510 | 0.0750 |
| Code | Species | Accession Number | Origin | Latitude | Longitude |
|---|---|---|---|---|---|
| 1 | M. albus | PI 342739 | England, United Kingdom | N 52°26′ | W 19°06′ |
| PI 595327 | China | N 43°18′ | E 86°40′ | ||
| PI 662299 | Vienna, Austria | N 48°20′ | E 16°33′ | ||
| 2 | M. altissimus | Ames 18376 | Nebraska, United States | N 41°26′ | W 99°23′ |
| PI 420163 | France | N 46°13′ | E 2°12′ | ||
| 3 | M. dentatus | PI 108656 | Armenia | N 40°4′ | E 45°2′ |
| PI 90753 | China | N 35°51′ | E 104°11′ | ||
| PI 595334 | China | N 42°49′ | E 85°30′ | ||
| 4 | M. elegans | PI 260271 | Ethiopia | N 9°9′ | E 37°48′ |
| PI 250873 | Iran | N32°4′ | E 54°4′ | ||
| 5 | M. hirsutus | Ames 22882 | Russian Federation | — | — |
| PI 129697 | Sweden | N 60°7′ | E 18°38′ | ||
| 6 | M. indicus | Ames 21619 | Nebraska, United States | N 41°29′ | W 99°54′ |
| PI 107562 | Uzbekistan | N 41°23′ | E 69°4′ | ||
| PI 43595 | — | — | — | ||
| 7 | M. infestus | PI 306326 | Algeria | N 27°13′ | E 2°29′ |
| PI 306327 | Italy | N 41°52′ | E12°34′ | ||
| PI 306328 | Hungary | N 47°9′ | E 19°30′ | ||
| 8 | M. italicus | PI 317635 | Czechoslovakia | N 14°28′ | E 121°2′ |
| PI 317638 | Israel | N 31°2′ | E 34°51′ | ||
| PI 317634 | Manitoba Canada | N 56°11′ | W 97°4′ | ||
| 9 | M. offcinalis | PI 132270 | Romania | N 45°49′ | E 24°29′ |
| PI 342889 | Germany | N 51°13′ | E 10°23′ | ||
| PI 342898 | France | N 46°34′ | E 2°18′ | ||
| 10 | M. polonicus | PI 314386 | Former Soviet Union | N 45°5′ | E 41°50′ |
| PI 108647 | Former Soviet Union | N 45°5′ | E 41°50′ | ||
| 11 | M. segetalis | PI 317649 | Czechoslovakia | N 48°2′ | E 18°22′ |
| 12 | M. siculus | PI 129703 | Malta | N 35°56′ | E 14°22′ |
| PI 318508 | Greece | N 39°4′ | E 21°49′ | ||
| PI 33366 | Former Soviet Union | — | — | ||
| 13 | M. speciosus | PI 317650 | Manitoba, Canada | N 53°45′ | W 98°48′ |
| PI 64995 | Morocco | N 31°52′ | W 6°13′ | ||
| 14 | M. spicatus | Ames 18402 | Nebraska, United States | N 41°29′ | W 99°54′ |
| PI 314466 | Uzbekistan | — | — | ||
| 15 | M. suaveolens | Ames 23793 | Mongolia | N 48°10′ | E 91°45′ |
| PI 595395 | United States | N 41°52′ | W 93°5′ | ||
| 16 | M. sulcatus | PI 227595 | Tunisia | N 33°53′ | E 9°32′ |
| 17 | M. tauricus | PI 67510 | Ukraine | N 44°24′ | E 33°49′ |
| Ames 25789 | Ukraine | N 44°24′ | E 33°49′ | ||
| 18 | M. wolgicus | PI 317666 | Czechoslovakia | N 48°2′ | E 18°22′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouyang, Z.; Wang, Y.; Ma, T.; Kanzana, G.; Wu, F.; Zhang, J. Genome-Wide Identification and Development of LTR Retrotransposon-Based Molecular Markers for the Melilotus Genus. Plants 2021, 10, 890. https://doi.org/10.3390/plants10050890
Ouyang Z, Wang Y, Ma T, Kanzana G, Wu F, Zhang J. Genome-Wide Identification and Development of LTR Retrotransposon-Based Molecular Markers for the Melilotus Genus. Plants. 2021; 10(5):890. https://doi.org/10.3390/plants10050890
Chicago/Turabian StyleOuyang, Zifeng, Yimeng Wang, Tiantian Ma, Gisele Kanzana, Fan Wu, and Jiyu Zhang. 2021. "Genome-Wide Identification and Development of LTR Retrotransposon-Based Molecular Markers for the Melilotus Genus" Plants 10, no. 5: 890. https://doi.org/10.3390/plants10050890
APA StyleOuyang, Z., Wang, Y., Ma, T., Kanzana, G., Wu, F., & Zhang, J. (2021). Genome-Wide Identification and Development of LTR Retrotransposon-Based Molecular Markers for the Melilotus Genus. Plants, 10(5), 890. https://doi.org/10.3390/plants10050890