
Physical Mapping of QTL in Four Spring Wheat Populations under Conventional and Organic Management Systems. I. Earliness

 ,
,  ,
,
Abstract
1. Introduction
2. Results
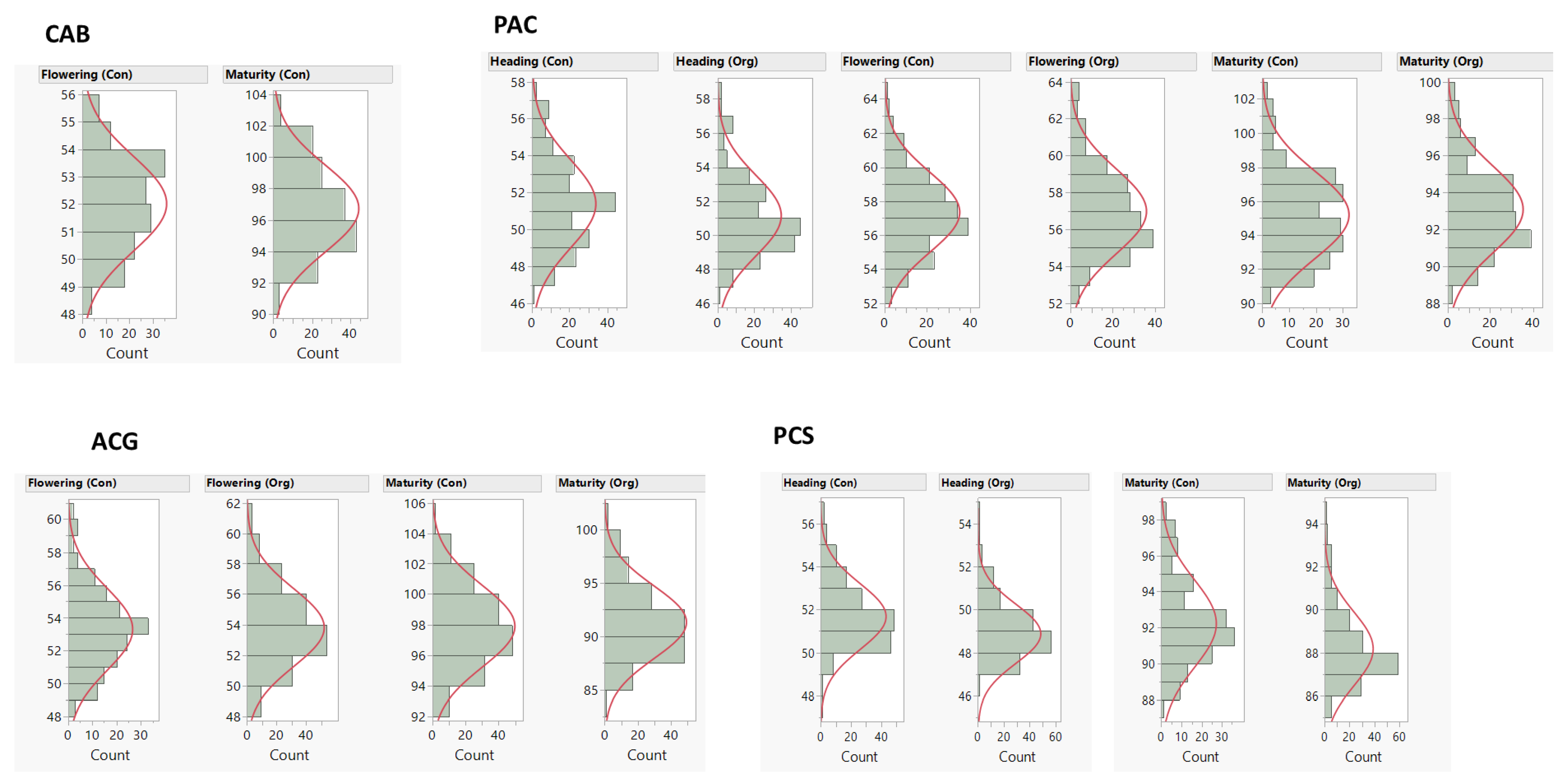
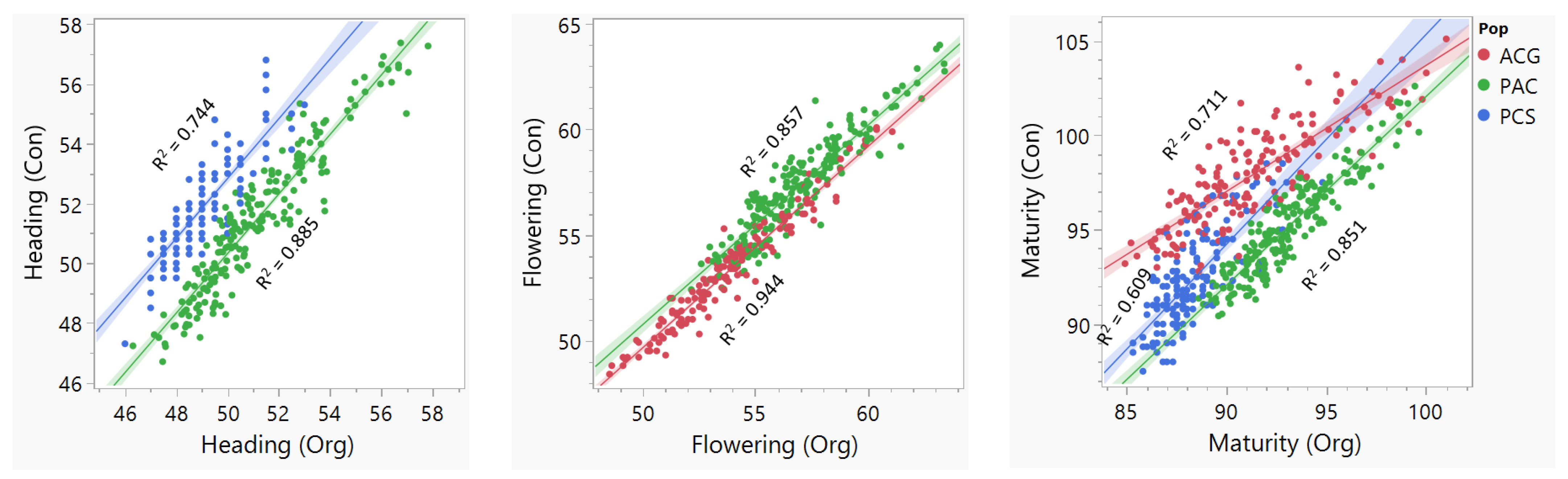
2.1. Phenotype and Genotype Data
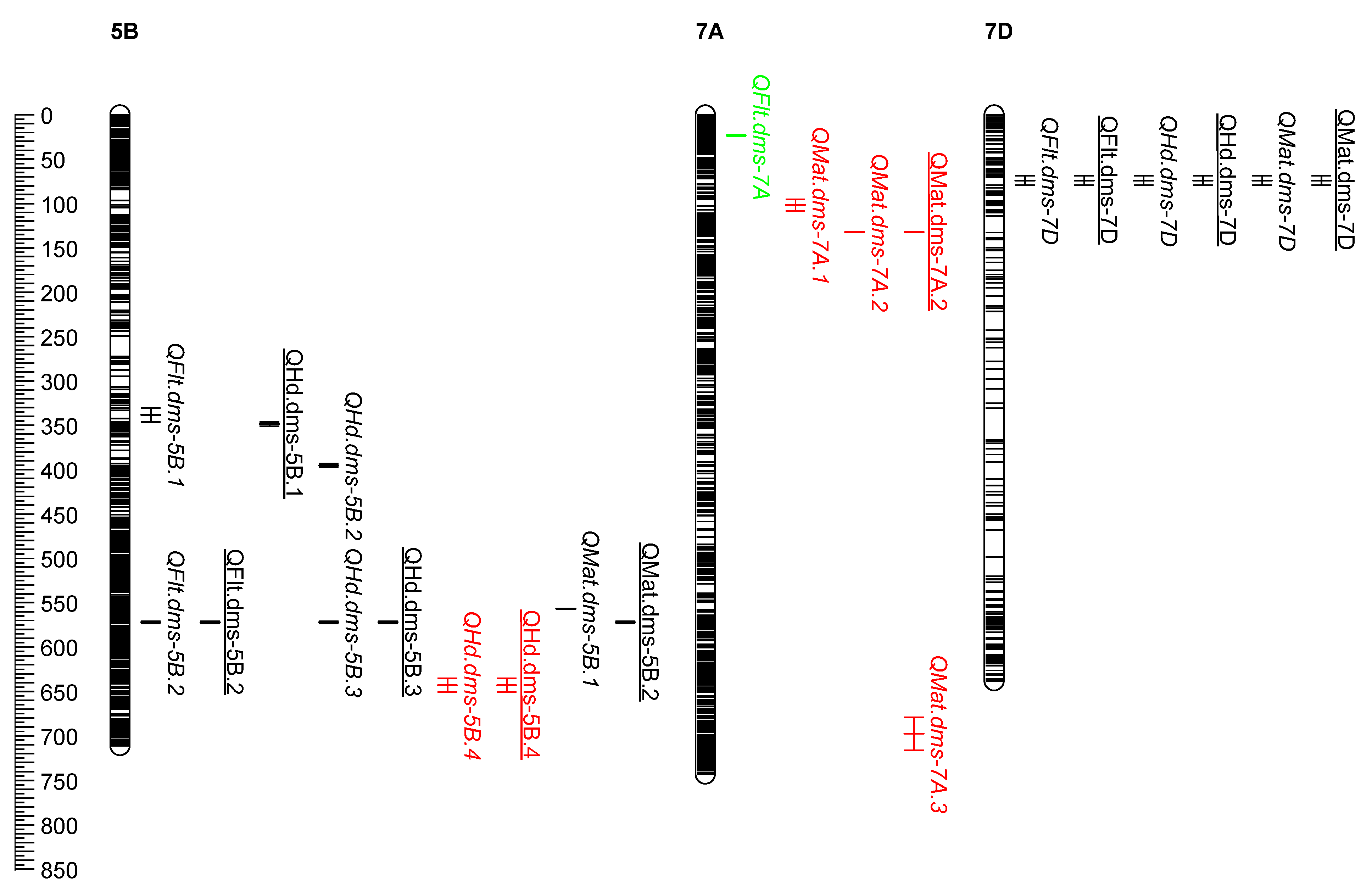
2.2. Physical Map of QTL
2.3. Coincident QTL
3. Discussion
3.1. QTL Based on Genetic and Physical Maps
3.2. Effects of Genetic Background and Management
4. Materials and Methods
4.1. Phenotyping and Genotyping
4.2. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACG | ‘Attila’ × ‘CDC Go’ |
| Bp | base pair |
| cM | CentiMorgan |
| CAB | ‘Cutler’ × ‘AC Barrie’ |
| CS | Chinese Spring |
| CWRS | Canada Western Red Spring |
| DArT | Diversity arrays Technology |
| DArTseq | Diversity Array-based genotyping by sequencing |
| ICIM | Inclusive composite interval mapping |
| IWGSC | International Wheat Genome Sequencing Consortium |
| LOD | The logarithm of the odds |
| Mb | Mega base pair |
| NUE | Nitrogen Use Efficiency |
| PAC | ‘Peace’ × ‘Carberry’ |
| PCS | ‘Peace’ × ‘CDC Stanley’ |
| QTL | Quantitative trait loci |
| RefSeq | Reference sequence |
| RIL | Recombinant inbred line |
| SNP | Single nucleotide polymorphism |
| SSR | Simple sequence repeat |
References
- Good, A.G.; Beatty, P.H. Fertilizing nature: A tragedy of excess in the commons. PLoS Biol. 2011, 9, e1001124. [Google Scholar] [CrossRef]
- Mason, H.E.; Navabi, A.; Frick, B.L.; O’Donovan, J.T.; Spaner, D.M. The weed-competitive ability of Canada western red spring wheat cultivars grown under organic management. Crop Sci. 2007, 47, 1167–1176. [Google Scholar] [CrossRef]
- Letourneau, D.K.; Bothwell, S.G. Comparison of organic and conventional farms: Challenging ecologists to make biodiversity functional. Front. Ecol. Environ. 2008, 6, 430–438. [Google Scholar] [CrossRef]
- Entz, M.H.; Guilford, R.; Gulden, R. Crop yield and soil nutrient status on 14 organic farms in the eastern portion of the northern Great Plains. Can. J. Plant Sci. 2001, 81, 351–354. [Google Scholar] [CrossRef]
- Pearson, D.; Henryks, J.; Jones, H. Organic food: What we know (and do not know) about consumers. Renew. Agric. Food Syst. 2011, 26, 171–177. [Google Scholar] [CrossRef]
- Murphy, K.M.; Campbell, K.G.; Lyon, S.R.; Jones, S.S. Evidence of varietal adaptation to organic farming systems. Field Crops Res. 2007, 102, 172–177. [Google Scholar] [CrossRef]
- Spaner, D.; Iqbal, M.; Navabi, A.; Strenzke, K.; Beres, B. Zealand hard red spring wheat. Can. J. Plant Sci. 2018, 98, 1409–1415. [Google Scholar] [CrossRef]
- Spaner, D.; Iqbal, M.; Navabi, A.; Strenzke, K.; Beres, B. Parata hard red spring wheat. Can. J. Plant Sci. 2016, 96, 517–524. [Google Scholar] [CrossRef]
- Spaner, D.; Iqbal, M.; Navabi, A.; Strenzke, K.; Beres, B. Jake hard red spring wheat. Can. J. Plant Sci. 2019, 100, 129–135. [Google Scholar] [CrossRef]
- Spaner, D.; Navabi, A.; Strenzke, K.; Iqbal, M.; Beres, B. Coleman hard red spring wheat. Can. J. Plant Sci. 2015, 95, 1037–1041. [Google Scholar] [CrossRef]
- Mason, H.; Navabi, A.; Frick, B.; O’Donovan, J.; Spaner, D. Cultivar and seeding rate effects on the competitive ability of spring cereals grown under organic production in Northern Canada. Agron. J. 2007, 99, 1199–1207. [Google Scholar] [CrossRef]
- Kaut, A.H.E.E.; Mason, H.E.; Navabi, A.; O’Donovan, J.T.; Spaner, D. Performance and stability of performance of spring wheat variety mixtures in organic and conventional management systems in western Canada. J. Agric. Sci. 2009, 147, 141–153. [Google Scholar] [CrossRef]
- Reid, T.A.; Yang, R.C.; Salmon, D.F.; Navabi, A.; Spaner, D. Realized gains from selection for spring wheat grain yield are different in conventional and organically managed systems. Euphytica 2011, 177, 253–266. [Google Scholar] [CrossRef]
- Kubota, H.; Yang, R.C.; Iqbal, M.; Spaner, D. There are different pathways to stable spring wheat grain yield and nitrogen utilization efficiency in conventional and organically-managed systems. Agron. J. 2019, 111, 2370–2377. [Google Scholar] [CrossRef]
- Iqbal, M.; Moakhar, N.P.; Strenzke, K.; Haile, T.; Pozniak, C.; Hucl, P.; Spaner, D. Genetic improvement in grain yield and other traits of wheat grown in Western Canada. Crop Sci. 2016, 56, 613–624. [Google Scholar] [CrossRef]
- Kamran, A.; Kubota, H.; Yang, R.C.; Randhawa, H.S.; Spaner, D. Relative performance of Canadian spring wheat cultivars under organic and conventional field conditions. Euphytica 2014, 196, 13–24. [Google Scholar] [CrossRef]
- Chen, H.; Iqbal, M.; Perez-Lara, E.; Yang, R.C.; Pozniak, C.; Spaner, D. Earliness per se quantitative trait loci and their interaction with Vrn-B1 locus in a spring wheat population. Mol. Breed. 2015, 35, 182. [Google Scholar] [CrossRef]
- Iqbal, M.; Navabi, A.; Salmon, D.F.; Yang, R.C.; Murdoch, B.M.; Moore, S.S.; Spaner, D. Genetic analysis of flowering and maturity time in high latitude spring wheat: Genetic analysis of earliness in spring wheat. Euphytica 2007, 154, 207–218. [Google Scholar] [CrossRef]
- Kamran, A.; Iqbal, M.; Navabi, A.; Randhawa, H.; Pozniak, C.; Spaner, D. Earliness per se QTLs and their interaction with the photoperiod insensitive allele Ppd-D1a in the Cutler × AC Barrie spring wheat population. Appl. Genet. 2013, 126, 1965–1976. [Google Scholar] [CrossRef]
- Kamran, A.; Randhawa, H.S.; Pozniak, C.; Spaner, D. Phenotypic effects of the flowering gene complex in Canadian spring wheat germplasm. Crop Sci. 2013, 53, 84–94. [Google Scholar] [CrossRef]
- Chen, H.; Bemister, D.H.; Iqbal, M.; Strelkov, S.E.; Spaner, D.M. Mapping genomic regions controlling agronomic traits in spring wheat under conventional and organic managements. Crop Sci. 2020, 60, 2038–2052. [Google Scholar] [CrossRef]
- Chen, H.; Iqbal, M.; Yang, R.C.; Spaner, D. Effect of Lr34/Yr18 on agronomic and quality traits in a spring wheat mapping population and implications for breeding. Mol. Breed. 2016, 36. [Google Scholar] [CrossRef]
- Perez-Lara, E.; Semagn, K.; Chen, H.; Iqbal, M.; N’Diaye, A.; Kamran, A.; Navabi, A.; Pozniak, C.; Spaner, D. QTLs Associated with agronomic traits in the Cutler × AC Barrie spring wheat mapping population using single nucleotide polymorphic markers. PLoS ONE 2016, 11, e0160623. [Google Scholar] [CrossRef]
- Zou, J.; Semagn, K.; Iqbal, M.; N’Diaye, A.; Chen, H.; Asif, M.; Navabi, A.; Perez-Lara, E.; Pozniak, C.; Yang, R.C.; et al. Mapping QTLs controlling agronomic traits in the Attila x CDC Go spring wheat population under organic management using 90K SNP array. Crop Sci. 2017, 365–377. [Google Scholar] [CrossRef]
- Zou, J.; Semagn, K.; Iqbal, M.; N’Diaye, A.; Chen, H.; Asif, M.; Navabi, A.; Perez-Lara, E.; Pozniak, C.; Yang, R.C.; et al. QTLs associated with agronomic traits in the Attila × CDC Go spring wheat population evaluated under conventional management. PLoS ONE 2017, 12, e0171528. [Google Scholar] [CrossRef] [PubMed]
- Bemister, D.H.; Semagn, K.; Iqbal, M.; Randhawa, H.; Strelkov, S.E.; Spaner, D.M. Mapping QTL associated with stripe rust, leaf rust, and leaf spotting in a Canadian spring wheat population. Crop Sci. 2019, 59, 650–658. [Google Scholar] [CrossRef]
- Asif, M.; Yang, R.C.; Navabi, A.; Iqbal, M.; Kamran, A.; Lara, E.P.; Randhawa, H.; Pozniak, C.; Spaner, D. Mapping QTL, selection differentials, and the effect of Rht-B1 under organic and conventionally managed systems in the Attila × CDC Go spring wheat mapping population. Crop Sci. 2015, 55, 1129–1142. [Google Scholar] [CrossRef]
- Perez-Lara, E.; Semagn, K.; Tran, A.N.; Ciechanowska, I.; Chen, H.; Iqbal, M.; N’Diaye, A.; Pozniak, C.; Strelkov, S.E.; Hucl, P.J.; et al. Population structure and genomewide association analysis of resistance to disease and insensitivity to Ptr toxins in Canadian spring wheat using 90K SNP array. Crop Sci. 2017, 57, 1522–1539. [Google Scholar] [CrossRef]
- Chen, H.; Semagn, K.; Iqbal, M.; Moakhar, N.P.; Haile, T.; N’Diaye, A.; Yang, R.C.; Hucl, P.J.; Pozniak, C.; Spaner, D. Genome-wide association mapping of genomic regions associated with phenotypic traits in Canadian western spring wheat. Mol. Breed. 2017, 37, 141. [Google Scholar] [CrossRef]
- Xiang, R.; Semagn, K.; Iqbal, M.; Hua, C.; Yang, R.-C.; Spaner, D. Phenotypic performance and associated QTLs of ‘Peace’ × ‘CDC Stanley’ mapping population under conventional and organic managements. Crop Sci. 2021. under review. [Google Scholar]
- Goffinet, B.; Gerber, S. Quantitative Trait Loci: A meta-analysis. Genetics 2000, 155, 463–473. [Google Scholar]
- Liu, H.; Mullan, D.; Zhang, C.; Zhao, S.; Li, X.; Zhang, A.; Lu, Z.; Wang, Y.; Yan, G. Major genomic regions responsible for wheat yield and its components as revealed by meta-QTL and genotype–phenotype association analyses. Planta 2020, 252. [Google Scholar] [CrossRef]
- Zheng, T.; Hua, C.; Li, L.; Sun, Z.; Yuan, M.; Bai, G.; Humphreys, G.; Li, T. Integration of meta-QTL discovery with omics: Towards a molecular breeding platform for improving wheat resistance to Fusarium head blight. Crop J. 2020. [Google Scholar] [CrossRef]
- Venske, E.; dos Santos, R.S.; Farias, D.d.R.; Rother, V.; da Maia, L.C.; Pegoraro, C.; Costa de Oliveira, A. Meta-analysis of the QTLome of Fusarium head blight resistance in bread wheat: Refining the current puzzle. Front. Plant Sci. 2019, 10, 727. [Google Scholar] [CrossRef] [PubMed]
- Soriano, J.M.; Alvaro, F. Discovering consensus genomic regions in wheat for root-related traits by QTL meta-analysis. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Hanocq, E.; Laperche, A.; Jaminon, O.; Laine, A.L.; Gouis, J.l. Most significant genome regions involved in the control of earliness traits in bread wheat, as revealed by QTL meta-analysis. Appl. Genet. 2007, 114, 569–584. [Google Scholar] [CrossRef]
- Semagn, K.; Beyene, Y.; Warburton, M.; Tarekegne, A.; Mugo, S.; Meisel, B.; Sehabiague, P.; Prasanna, B. Meta-analyses of QTL for grain yield and anthesis silking interval in 18 maize populations evaluated under water-stressed and well-watered environments. BMC Genom. 2013, 14, 313. [Google Scholar] [CrossRef]
- Semagn, K.; Bjornstad, A.; Skinnes, H.; Maroy, A.G.; Tarkegne, Y.; William, M. Distribution of DArT, AFLP, and SSR markers in a genetic linkage map of a doubled-haploid hexaploid wheat population. Genome 2006, 49, 545–555. [Google Scholar] [CrossRef]
- Dossa, K. A physical map of important QTLs, functional markers and genes available for sesame breeding programs. Physiol. Mol. Biol. Plants 2016, 22, 613–619. [Google Scholar] [CrossRef]
- Schönhals, E.M.; Ding, J.; Ritter, E.; Paulo, M.J.; Cara, N.; Tacke, E.; Hofferbert, H.-R.; Lübeck, J.; Strahwald, J.; Gebhardt, C. Physical mapping of QTL for tuber yield, starch content and starch yield in tetraploid potato (Solanum tuberosum L.) by means of genome wide genotyping by sequencing and the 8.3 K SolCAP SNP array. BMC Genom. 2017, 18, 642. [Google Scholar] [CrossRef]
- Yu, K.; Wang, X.; Li, W.; Sun, L.; Peng, Q.; Chen, F.; Zhang, W.; Guan, R.; Zhang, J. Identification and physical mapping of QTLs associated with flowering time in Brassica napus L. Euphytica 2019, 215. [Google Scholar] [CrossRef]
- Okada, T.; Jayasinghe, J.E.A.R.M.; Eckermann, P.; Watson-Haigh, N.S.; Warner, P.; Hendrikse, Y.; Baes, M.; Tucker, E.J.; Laga, H.; Kato, K.; et al. Effects of Rht-B1 and Ppd-D1 loci on pollinator traits in wheat. Appl. Genet. 2019, 132, 1965–1979. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.S.; Zhu, J.; Mansueto, L.; Bruskiewich, R. Identification of candidate genes for drought stress tolerance in rice by the integration of a genetic (QTL) map with the rice genome physical map. J. Zhejiang Univ. Sci. 2005, 6B, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Lecouls, A.C.; Reighard, G.L.; Abbott, A.G.; Dirlewanger, E. Physical mapping and integration of QTL intervals involved in fruit quality on peach fruit variety and rootstock molecular maps. Acta Hortic. 2002, 592, 273–278. [Google Scholar] [CrossRef]
- Somers, D.J.; Isaac, P.; Edwards, K. A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.). Appl. Genet. 2004, 109, 1105–1114. [Google Scholar] [CrossRef]
- Hanocq, E.; Niarquin, M.; Heumez, E.; Rousset, M.; Le Gouis, J. Detection and mapping of QTL for earliness components in a bread wheat recombinant inbred lines population. Appl. Genet. 2004, 110, 106–115. [Google Scholar] [CrossRef]
- Snape, J.W.; Butterworth, K.; Whitechurch, E.; Worland, A.J. Waiting for fine times: Genetics of flowering time in wheat. Euphytica 2001, 119, 185–190. [Google Scholar] [CrossRef]
- Zhao, C.H.; Sun, H.; Liu, C.; Yang, G.M.; Liu, X.J.; Wang, Y.P.; Lv, F.X.; Wu, C.Y.; Xu, J.W.; Wu, Y.Z.; et al. Detection of quantitative trait loci for wheat (Triticum aestivum L.) heading and flowering date. J. Agric. Sci. 2019, 157, 20–30. [Google Scholar] [CrossRef]
- Distelfeld, A.; Li, C.; Dubcovsky, J. Regulation of flowering in temperate cereals. Curr. Opin. Plant Biol. 2009, 12, 178–184. [Google Scholar] [CrossRef]
- Worland, A.J.; Börner, A.; Korzun, V.; Li, W.M.; Petrovíc, S.; Sayers, E.J. The influence of photoperiod genes on the adaptability of European winter wheats. Euphytica 1998, 100, 385–394. [Google Scholar] [CrossRef]
- Chen, F.; Gao, M.; Zhang, J.; Zuo, A.; Shang, X.; Cui, D. Molecular characterization of vernalization and response genes in bread wheat from the Yellow and Huai Valley of China. BMC Plant Biol. 2013, 13, 199. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, E.P.; Turner, A.S.; Laurie, D.A. Photoperiod insensitive Ppd-A1a mutations in tetraploid wheat (Triticum durum Desf.). Appl. Genet. 2009, 118, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Laurie, D.A. Comparative genetics of flowering time. Plant Mol. Biol. 1997, 35, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.M.; Turner, A.S.; Herry, L.; Griffiths, S.; Laurie, D.A. Mutant alleles of photoperiod-1 in wheat (Triticum aestivum L.) that confer a late flowering phenotype in long days. PLoS ONE 2013, 8, e79459. [Google Scholar] [CrossRef] [PubMed]
- Daoura, B.G.; Chen, L.; Du, Y.; Hu, Y. Genetic effects of dwarfing gene Rht-5 on agronomic traits in common wheat (Triticum aestivum L.) and QTL analysis on its linked traits. Field Crops Res. 2014, 156, 22–29. [Google Scholar] [CrossRef]
- Kroupin, P.Y.; Karlov, G.I.; Bespalova, L.A.; Salina, E.A.; Chernook, A.G.; Watanabe, N.; Bazhenov, M.S.; Panchenko, V.V.; Nazarova, L.A.; Kovtunenko, V.Y.; et al. Effects of Rht17 in combination with Vrn-B1 and Ppd-D1 alleles on agronomic traits in wheat in black earth and non-black earth regions. BMC Plant Biol. 2020, 20, 304. [Google Scholar] [CrossRef]
- Chen, L.; Du, Y.; Lu, Q.; Chen, H.; Meng, R.; Cui, C.; Lu, S.; Yang, Y.; Chai, Y.; Li, J.; et al. The photoperiod-insensitive allele Ppd-D1a promotes earlier flowering in Rht12 dwarf plants of bread wheat. Front. Plant Sci. 2018, 9, 1312. [Google Scholar] [CrossRef]
- Yao, N.; Lee, C.-R.; Semagn, K.; Sow, M.; Nwilene, F.; Kolade, O.; Bocco, R.; Oyetunji, O.; Mitchell-Olds, T.; Ndjiondjop, M.-N. QTL mapping in three rice populations uncovers major genomic regions associated with African rice gall midge resistance. PLoS ONE 2016, 11, e0160749. [Google Scholar] [CrossRef]
- Garin, V.; Wimmer, V.; Borchardt, D.; Malosetti, M.; van Eeuwijk, F. The influence of QTL allelic diversity on QTL detection in multi-parent populations: A simulation study in sugar beet. bioRxiv 2020. [Google Scholar] [CrossRef]
- Symonds, V.V.; Godoy, A.V.; Alconada, T.; Botto, J.F.; Juenger, T.E.; Casal, J.J.; Lloyd, A.M. Mapping quantitative trait loci in multiple populations of Arabidopsis thaliana identifies natural allelic variation for trichome density. Genetics 2005, 169, 1649–1658. [Google Scholar] [CrossRef]
- Kang, S.-T.; Kwak, M.; Kim, H.-K.; Choung, M.-G.; Han, W.-Y.; Baek, I.-Y.; Kim, M.Y.; Van, K.; Lee, S.-H. Population-specific QTLs and their different epistatic interactions for pod dehiscence in soybean [Glycine max (L.) Merr.]. Euphytica 2008, 166, 15. [Google Scholar] [CrossRef]
- Brasier, K.; Ward, B.; Smith, J.; Seago, J.; Oakes, J.; Balota, M.; Davis, P.; Fountain, M.; Brown-Guedira, G.; Sneller, C.; et al. Identification of quantitative trait loci associated with nitrogen use efficiency in winter wheat. PLoS ONE 2020, 15, e0228775. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, R.; Tong, Y.; Zhao, H.; Xie, Q.; Liu, D.; Zhang, A.; Li, B.; Xu, H.; An, D. Mapping QTLs for yield and nitrogen-related traits in wheat: Influence of nitrogen and phosphorus fertilization on QTL expression. Appl. Genet. 2014, 127, 59–72. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Su, J.; Liu, Q.; Zhu, Y.; Tong, Y.; Li, J.; Jing, R.; Li, B.; Li, Z. Mapping QTLs for nitrogen uptake in relation to the early growth of wheat (Triticum aestivum L.). Plant Soil 2006, 284, 73–84. [Google Scholar] [CrossRef]
- Guo, Y.; Kong, F.m.; Xu, Y.f.; Zhao, Y.; Liang, X.; Wang, Y.y.; An, D.g.; Li, S.s. QTL mapping for seedling traits in wheat grown under varying concentrations of N, P and K nutrients. Appl. Genet. 2012, 124, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Laperche, A.; Brancourt-Hulmel, M.; Heumez, E.; Gardet, O.; Hanocq, E.; Devienne-Barret, F.D.; Gouis, J.L. Using genotype × nitrogen interaction variables to evaluate the QTL involved in wheat tolerance to nitrogen constraints. Appl. Genet. 2007, 115. [Google Scholar] [CrossRef] [PubMed]
- Mahender, A.; Ali, J.; Prahalada, G.D.; Sevilla, M.A.L.; Balachiranjeevi, C.H.; Md, J.; Maqsood, U.; Li, Z. Genetic dissection of developmental responses of agro-morphological traits under different doses of nutrient fertilizers using high-density SNP markers. PLoS ONE 2019, 14, e0220066. [Google Scholar] [CrossRef] [PubMed]
- Yue, F.; Rong-rong, Z.; Ze-chuan, L.; Li-yong, C.; Xing-hua, W.; Shi-hua, C. Quantitative Trait Locus Analysis for Rice Yield Traits under Two Nitrogen Levels. Rice Sci. 2015, 22, 108–115. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, C.; Wang, Y.; He, T.; Gao, R.; Xu, H.; Guo, G.; Li, Y.; Zhou, L.; Lu, R.; et al. Expression analysis of nitrogen metabolism-related genes reveals differences in adaptation to low-nitrogen stress between two different barley cultivars at seedling stage. Int. J. Genom. 2018, 2018, 8152860. [Google Scholar] [CrossRef]
- Gelli, M.; Mitchell, S.E.; Liu, K.; Clemente, T.E.; Weeks, D.P.; Zhang, C.; Holding, D.R.; Dweikat, I.M. Mapping QTLs and association of differentially expressed gene transcripts for multiple agronomic traits under different nitrogen levels in sorghum. BMC Plant Biol. 2016, 16, 16. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Zhao, Y.; Zhang, Y.; Zhang, J.; Ma, H.; Han, Y. Transcriptome analysis reveals nitrogen deficiency induced alterations in leaf and root of three cultivars of potato (Solanum tuberosum L.). PLoS ONE 2020, 15, e0240662. [Google Scholar] [CrossRef]
- Briggs, K.G.; Kibite, S.; Kutschera, K. Cutler red spring wheat. Can. J. Plant Sci 1991, 72, 229–233. [Google Scholar] [CrossRef]
- McCaig, T.N.; DePauw, R.M.; Clarke, J.M.; McLeod, J.G.; Fernandez, M.R.; Knox, R.E. AC Barrie hard red spring wheat. Can. J. Plant Sci. 1996, 76, 337–339. [Google Scholar] [CrossRef]
- Humphreys, D.G.; Townley-Smith, T.F.; Lukow, O.M.; McCallum, B.D.; Fetch, T.G.; Gilbert, J.A.; Menzies, J.G.; Tkachuk, V.; Brown, P.D.; Fox, S.L. Peace hard red spring wheat. Can. J. Plant Sci. 2014, 94, 1297–1302. [Google Scholar] [CrossRef]
- DePauw, R.M.; Knox, R.E.; McCaig, T.N.; Clarke, F.R.; Clarke, J.M. Carberry hard red spring wheat. Can. J. Plant Sci. 2011, 91, 529–534. [Google Scholar] [CrossRef]
- Tadesse, W.; Manes, Y.; Singh, R.P.; Payne, T.; Braun, H.J. Adaptation and performance of CIMMYT spring wheat genotypes targeted to high rainfall areas of the world. Crop Sci. 2010, 50, 2240–2248. [Google Scholar] [CrossRef]
- Chen, H.; Moakhar, N.P.; Iqbal, M.; Pozniak, C.; Hucl, P.; Spaner, D. Genetic variation for flowering time and height reducing genes and important traits in western Canadian spring wheat. Euphytica 2016, 208, 377–390. [Google Scholar] [CrossRef]
- Iqbal, M.; Navabi, A.; Yang, R.C.; Salmon, D.F.; Spaner, D. Molecular characterization of vernalization response genes in Canadian spring wheat. Genome 2007, 50, 511–516. [Google Scholar] [CrossRef]
- Wang, S.; Wong, D.; Forrest, K.; Allen, A.; Chao, S.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L.; et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 2014, 12, 787–796. [Google Scholar] [CrossRef]
- Egea, L.A.; Mérida-García, R.; Kilian, A.; Hernandez, P.; Dorado, G. Assessment of genetic diversity and structure of large Garlic (Allium sativum) germplasm bank, by diversity arrays technology “genotyping-by-sequencing” platform (DArTseq). Front. Genet. 2017, 8, 98. [Google Scholar] [CrossRef]
- Ndjiondjop, M.N.; Semagn, K.; Gouda, A.C.; Kpeki, S.B.; Dro Tia, D.; Sow, M.; Goungoulou, A.; Sie, M.; Perrier, X.; Ghesquiere, A.; et al. Genetic variation and population structure of Oryza glaberrima and development of a mini-core collection using DArTseq. Front. Plant Sci. 2017, 8, 1748. [Google Scholar] [CrossRef]
- Beales, J.; Turner, A.; Griffiths, S.; Snape, J.W.; Laurie, D.A. A pseudo-response regulator is misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.). Appl. Genet. 2007, 115, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.H.; Spielmeyer, W.; Gale, K.R.; Rebetzke, G.J.; Richards, R.A. Perfect markers for the Rht-B1b and Rht-D1b dwarfing genes in wheat. Appl. Genet. 2002, 105, 1038–1042. [Google Scholar] [CrossRef]
- Zimin, A.V.; Puiu, D.; Hall, R.; Kingan, S.; Clavijo, B.J.; Salzberg, S.L. The first near-complete assembly of the hexaploid bread wheat genome, Triticum aestivum. GigaScience 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Heffelfinger, C.; Fragoso, C.A.; Lorieux, M. Constructing linkage maps in the genomics era with MapDisto 2.0. Bioinformatics 2017, 33, 2224–2225. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population Code | Initial Cross | Population Size | Phenotyping | Genotyping * | References |
|---|---|---|---|---|---|
| CAB | ‘Cutler’ × ‘AC Barrie’ | 158 | Evaluated five times in 2007–2008 and 2010–2012 under the conventional management system. | Genotyped with the wheat 90K Illumina iSelect SNP array and two functional markers (Ppd-D1 and Rht-D1). | [23] |
| ACG | ‘Attila’ × ‘CDC Go’ | 167 | Evaluated three times (2008–2010) under organic and seven times (2008–2014) under conventional management system. | Genotyped with the wheat 90K Illumina iSelect SNP array and three functional markers (Ppd-D1, Vrn-A1 and Rht-B1). | [24,25] |
| PAC | ‘Peace’ × ‘Carberry’ | 208 | Evaluated four times from 2016 to 2020 under organic and conventional management systems. | Genotyped with 36,226 markers (22,741 SilicoDArT and 13,885 SNPs) using DArTseq-based genotyping by sequencing method and three functional markers (Vrn-B3, Rht-B1, and Glu-A3). | [21,26] |
| PCS | ‘Peace’ × ‘CDC Stanley’ | 165 | Evaluated twice (2016–2017) under organic and conventional management systems. | Genotyped with the wheat 90K Illumina iSelect SNP array and three SSR markers (DuPw004, barc170, and wmc650). | [30] |
| Trait | Management 1 | ‘Cutler’ × ‘AC Barrie’ | ‘Attila’ × ‘CDC Go’ | ‘Peace’ × ‘Carberry’ | ‘Peace’ × ‘CDC Stanley’ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Range | H 2 | Mean | Range | H 2 | Mean | Range | H 2 | Mean | Range | H 2 | ||
| Heading (days) | Con | 51.4 | 46.7–57.4 | 0.72 | 51.7 | 47.3–56.8 | 0.64 | ||||||
| Org | 51.2 | 46.3–58.6 | 0.77 | 48.9 | 46.0–54.0 | 0.70 | |||||||
| Flowering (days) | Con | 52.0 | 48.3–55.7 | 0.43 | 53.3 | 48.4–60.1 | 0.73 | 57.3 | 52.4–64 | 0.75 | |||
| Org | 53.8 | 48.5–61.1 | 0.72 | 57.0 | 52.1–63.4 | 0.71 | |||||||
| Maturity (days) | Con | 96.7 | 91.6–103.7 | 0.50 | 97.9 | 92.8–105.1 | 0.45 | 95.2 | 90.4–102.9 | 0.53 | 92.2 | 87.5–98.5 | 0.62 |
| Org | 91.3 | 84.9–101 | 0.44 | 93.1 | 88.6–99.6 | 0.40 | 88.2 | 85.3–94.8 | 0.53 | ||||
| Chrom | ‘Cutler’ × ‘AC Barrie’ | ‘Attila’ × ‘CDC Go’ | ‘Peace’ × ‘Carberry’ | ‘Peace’ × ‘CDC Stanley’ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of Markers | Genetic Map Length (cM) | Physical Map Length (bp) | No. of Markers | Genetic Map Length (cM) | Physical Map Length (bp) | No. of Markers | Genetic Map Length (cM) | Physical Map Length (bp) | No. of Markers | Genetic Map Length (cM) | Physical Map Length (bp) | |
| 1A | 302 | 129 | 596,457,062 | 273 | 78 | 595,208,262 | 371 | 535 | 595,860,808 | 59 | 147 | 593,443,946 |
| 1B | 237 | 244 | 698,233,083 | 54 | 25 | 698,181,188 | 457 | 1485 | 692,480,911 | 111 | 538 | 683,188,703 |
| 1D | 134 | 34 | 353,726,668 | 32 | 6 | 410,643,066 | 77 | 158 | 451,725,315 | 33 | 164 | 479,789,480 |
| 2A | 353 | 321 | 787,699,648 | 255 | 115 | 786,363,784 | 269 | 1591 | 759,862,164 | 81 | 230 | 770,603,608 |
| 2B | 869 | 567 | 795,242,489 | 391 | 163 | 665,646,566 | 347 | 181 | 450,448,794 | 119 | 371 | 812,709,904 |
| 2D | 93 | 35 | 598,126,556 | 30 | 40 | 555,087,626 | 129 | 4234 | 654,057,050 | 29 | 143 | 645,341,017 |
| 3A | 378 | 249 | 752,968,879 | 173 | 121 | 737,952,205 | 380 | 265 | 753,051,285 | 16 | 234 | 745,304,963 |
| 3B | 444 | 268 | 851,724,030 | 80 | 224 | 851,724,030 | 241 | 5223 | 848,161,565 | 12 | 122 | 847,814,062 |
| 3D | 25 | 28 | 579,990,391 | - | - | - | 119 | 176 | 617,655,900 | 9 | 31 | 606,042,121 |
| 4A | 424 | 180 | 725,639,518 | 194 | 62 | 681,910,513 | 222 | 1845 | 751,532,475 | 45 | 158 | 722,926,434 |
| 4B | 320 | 85 | 669,758,748 | 44 | 42 | 655,593,292 | 210 | 125 | 672,766,284 | 38 | 195 | 644,427,716 |
| 4D | 35 | 31 | 482,822,161 | - | - | - | 45 | 63 | 513,418,898 | 15 | 88 | 456,267,708 |
| 5A | 415 | 132 | 710,849,404 | 124 | 106 | 667,797,441 | 223 | 414 | 705,303,717 | 64 | 454 | 684,682,180 |
| 5B | 957 | 626 | 714,558,337 | 269 | 292 | 697,707,787 | 579 | 523 | 713,404,985 | 147 | 172 | 714,258,884 |
| 5D | 16 | 4 | 557,692,932 | 38 | 3 | 569,677,699 | 160 | 4049 | 568,959,847 | 15 | 407 | 568,666,242 |
| 6A | 374 | 94 | 601,826,399 | 169 | 59 | 498,653,429 | 275 | 395 | 622,068,866 | 58 | 140 | 619,171,526 |
| 6B | 187 | 66 | 730,597,670 | 525 | 151 | 719,416,487 | 423 | 1002 | 731,066,251 | 42 | 60 | 729,835,185 |
| 6D | 32 | 16 | 494,665,522 | 38 | 7 | 7,562,690 | 130 | 177 | 492,458,353 | 15 | 317 | 494,670,623 |
| 7A | 559 | 166 | 744,464,513 | 252 | 130 | 744,464,355 | 602 | 1999 | 743,593,777 | 86 | 351 | 733,173,741 |
| 7B | 338 | 290 | 763,315,508 | 200 | 92 | 739,367,419 | 334 | 496 | 759,405,691 | 57 | 531 | 722,136,838 |
| 7D | 34 | 33 | 577,952,590 | 17 | 18 | 575,785,725 | 138 | 573 | 642,704,389 | 7 | 69 | 606,914,235 |
| Total | 6526 | 3596 | 13,788,312,108 | 3158 | 1734 | 11,858,743,564 | 5731 | 25,508 | 13,739,987,325 | 1058 | 4922 | 13,881,369,116 |
| Trait | QTL Name | Population 1 | Management 2 | Chromosome | QTL Location 3 | Confidence Interval (Mb) | R2 (%): Conventional | R2 (%): Organic |
|---|---|---|---|---|---|---|---|---|
| Heading | QHd.dms-1D | PAC | Org | 1D | 1D:325876831-336302522 | 10.4 | 2.8 | |
| Heading | QHd.dms-2B | PAC | Con & Org | 2B | 2B:55797422-62651542 | 6.9 | 11.5 | 5.6 |
| Heading | QHd.dms-3B | PCS | Org | 3B | 3B:45204750-49026001 | 3.8 | 5.8 | |
| Heading | QHd.dms-4A | PCS | Con | 4A | 4A:17686877-17787027 | 0.1 | 8.8 | |
| Heading | QHd.dms-5A.1 | PAC | Con & Org | 5A | 5A:60279363-76684980 | 16.4 | 9.6 | 3.7 |
| Heading | QHd.dms-5A.2 | PAC | Org | 5A | 5A:620541477-621338663 | 0.8 | 5.9 | |
| Heading | QHd.dms-5B.1 | PAC | Org | 5B | 5B:349752769-354808447 | 5.1 | 4.1 | |
| Heading | QHd.dms-5B.2 | PAC | Con | 5B | 5B:396826652-400681156 | 3.9 | 5.6 | |
| Heading | QHd.dms-5B.3 | PAC | Con & Org | 5B | 5B:574535307-577015908 | 2.5 | 16.2 | 19.3 |
| Heading | QHd.dms-5B.4 | PCS | Con & Org | 5B | 5B:639044446-653916583 | 14.9 | 8.1 | 1.8 |
| Heading | QHd.dms-7D | PAC | Con & Org | 7D | 7D:73333549-83998578 | 10.7 | 16.6 | 10.9 |
| Flowering | QFlt.dms-2A.1 | PAC | Con | 2A | 2A:37479415-37904660 | 0.4 | 2.0 | |
| Flowering | QFlt.dms-2A.2 | PAC | Org | 2A | 2A:700598519-700903870 | 0.3 | 2.3 | |
| Flowering | QFlt.dms-2B | PAC | Con & Org | 2B | 2B:55797422-62651542 | 6.9 | 5.7 | 2.9 |
| Flowering | QFlt.dms-3B | CAB | Con | 3B | 3B:432437212-432750764 | 0.3 | 7.9 | |
| Flowering | QFlt.dms-5A.1 | CAB | Con | 5A | 5A:582841379-583000992 | 0.2 | 8.2 | |
| Flowering | QFlt.dms-5A.2 | ACG | Con & Org | 5A | 5A:587346439-587412126 | 0.1 | 19.2 | 20.8 |
| Flowering | QFlt.dms-5B.1 | PAC | Con | 5B | 5B:333880729-349752769 | 15.9 | 9.0 | |
| Flowering | QFlt.dms-5B.2 | PAC | Con & Org | 5B | 5B:574535307-577015908 | 2.5 | 8.4 | 13.6 |
| Flowering | QFlt.dms-7A | CAB | Con | 7A | 7A:24248719-25391676 | 1.1 | 8.6 | |
| Flowering | QFlt.dms-7D | PAC | Con & Org | 7D | 7D:73333549-83998578 | 10.7 | 12.6 | 12.4 |
| Maturity | QMat.dms-1A.1 | CAB | Con | 1A | 1A:35556032-39776886 | 4.2 | 3.7 | |
| Maturity | QMat.dms-1A.2 | CAB | Con | 1A | 1A:399444508-426644827 | 27.2 | 3.7 | |
| Maturity | QMat.dms-1A.3 | CAB | Con | 1A | 1A:443059667-462755618 | 19.7 | 3.7 | |
| Maturity | QMat.dms-3A.1 | PAC | Con | 3A | 3A:392012081-406623288 | 14.6 | 2.9 | |
| Maturity | QMat.dms-3A.2 | PAC | Org | 3A | 3A:513855127-553098513 | 39.2 | 3.6 | |
| Maturity | QMat.dms-3B.1 | PCS | Org | 3B | 3B:45204750-49026001 | 3.8 | 2.1 | |
| Maturity | QMat.dms-3B.2 | CAB | Con | 3B | 3B:821149227-835005886 | 13.9 | 3.3 | |
| Maturity | QMat.dms-4A.1 | PCS | Con | 4A | 4A:16086950-17686877 | 1.6 | 2.9 | |
| Maturity | QMat.dms-4A.2 | CAB | Con | 4A | 4A:65464862-68459336 | 3.0 | 1.7 | |
| Maturity | QMat.dms-4A.3 | PAC | Con | 4A | 4A:580400959-582726391 | 2.3 | 2.5 | |
| Maturity | QMat.dms-4B.1 | PAC | Con & Org | 4B | 4B:30510315-31959109 | 1.4 | 5.5 | 4.0 |
| Maturity | QMat.dms-4B.2 | ACG | Con | 4B | 4B:569184188-599613837 | 30.4 | 11.0 | |
| Maturity | QMat.dms-4D | CAB | Con | 4D | 4D:21025268-32965037 | 11.9 | 3.9 | |
| Maturity | QMat.dms-5A.1 | CAB | Con | 5A | 5A:569871147-570121744 | 0.3 | 2.0 | |
| Maturity | QMat.dms-5A.2 | ACG | Con & Org | 5A | 5A:587346439-587412126 | 0.1 | 3.7 | 16.7 |
| Maturity | QMat.dms-5A.3 | PAC | Con & Org | 5A | 5A:619697105-620189324 | 0.5 | 2.5 | 1.8 |
| Maturity | QMat.dms-5A.4 | PAC | Con & Org | 5A | 5A:689113847-692552481 | 3.4 | 3.7 | 3.0 |
| Maturity | QMat.dms-5B.1 | PAC | Con | 5B | 5B:559880753-560779737 | 0.9 | 4.5 | |
| Maturity | QMat.dms-5B.2 | PAC | Org | 5B | 5B:574535307-577015908 | 2.5 | 3.7 | |
| Maturity | QMat.dms-7A.1 | PCS | Con | 7A | 7A:96812406-110000000 | 13.2 | 7.1 | |
| Maturity | QMat.dms-7A.2 | PCS | Con & Org | 7A | 7A:133489405-134226943 | 0.7 | 7.1 | 2.0 |
| Maturity | QMat.dms-7A.3 | PCS | Con | 7A | 7A:680676790-717910027 | 37.2 | 5.3 | |
| Maturity | QMat.dms-7D | PAC | Con & Org | 7D | 7D:73333549-83998578 | 10.7 | 11.4 | 14.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semagn, K.; Iqbal, M.; Chen, H.; Perez-Lara, E.; Bemister, D.H.; Xiang, R.; Zou, J.; Asif, M.; Kamran, A.; N’Diaye, A.; et al. Physical Mapping of QTL in Four Spring Wheat Populations under Conventional and Organic Management Systems. I. Earliness. Plants 2021, 10, 853. https://doi.org/10.3390/plants10050853
Semagn K, Iqbal M, Chen H, Perez-Lara E, Bemister DH, Xiang R, Zou J, Asif M, Kamran A, N’Diaye A, et al. Physical Mapping of QTL in Four Spring Wheat Populations under Conventional and Organic Management Systems. I. Earliness. Plants. 2021; 10(5):853. https://doi.org/10.3390/plants10050853
Chicago/Turabian StyleSemagn, Kassa, Muhammad Iqbal, Hua Chen, Enid Perez-Lara, Darcy H. Bemister, Rongrong Xiang, Jun Zou, Muhammad Asif, Atif Kamran, Amidou N’Diaye, and et al. 2021. "Physical Mapping of QTL in Four Spring Wheat Populations under Conventional and Organic Management Systems. I. Earliness" Plants 10, no. 5: 853. https://doi.org/10.3390/plants10050853
APA StyleSemagn, K., Iqbal, M., Chen, H., Perez-Lara, E., Bemister, D. H., Xiang, R., Zou, J., Asif, M., Kamran, A., N’Diaye, A., Randhawa, H., Pozniak, C., & Spaner, D. (2021). Physical Mapping of QTL in Four Spring Wheat Populations under Conventional and Organic Management Systems. I. Earliness. Plants, 10(5), 853. https://doi.org/10.3390/plants10050853