1. Introduction
In 2005, the question “How does a single somatic cell become a whole plant?” was among the 25 most important questions in the next quarter-century of biology [
1]. Somatic regeneration or embryogenesis is a synthetic process where a plant shoot or embryo (embryogenesis) or a root (rhizogenesis) is formed from a single somatic cell. What remains to be determined is whether the reprogramming of somatic cells into a new shoot or root is truly embryogenesis since only a partial plant is formed. Furthermore, whether this is a direct embryogenesis from a single cell or indirect process from a multicellular origin.
Plants have a remarkable developmental capacity to replicate via somatic cells without fertilization [
2,
3], leading to clonal proliferation. Steward et al. [
4] showed that segments of mature carrot tissue regenerated whole plants [
4], demonstrating the existence of totipotent plant somatic cells. In various plant tissues, regeneration occurs in response to applying exogenously applied phytohormones and plant growth regulators (PGRs), with auxins and cytokinins being the most significant. Somatic shoots can arise from several differentiated tissues in response to exogenous or endogenous stimuli [
5,
6]. Thus, somatic embryos and shoots are a powerful biotechnological tool for plant propagation and genetic improvement.
The ratio of externally applied phytohormones or PGRs determines the types of cells developed from the somatic tissue. Usually, a high cytokinin to auxin ratio results in shoots, while a low cytokinin to auxin ratio results in roots development. Intermediate cytokinin to auxin ratios form a callus. The regeneration of shoots and roots is divided into three stages; competence, induction, and development [
7]. Competence is the acquisition of the ability to regenerate (i.e., the tissues acquire the ability to respond to growth- regulators) and, thus, shoots or roots can be induced. The induction stage is the initiation of regeneration where the developmental fate of competent cells is determined (i.e., shoot, roots, or callus is formed from the induced tissue evoking the cells totipotent nature). The third stage is developing the induced organs determined in the second stage (i.e., shoots, roots, callus, or embryos under the regular developmental program of meristems). Ectopic expression of meristematic and embryonic genes (e.g., Shoot Meristemless (STM), Baby Boom (BBM), Enhancer of Shoot Regeneration (ESR1 and ESR2), Leafy Cotyledon (LEC), etc.) bypasses the early stages and forces somatic cells to produce shoots [
8].
The molecular events that lead somatic cells from leaf tissue to form a new shoot in tissue culture are not very well understood. Reprogramming competent somatic cells into totipotent cells is the initial step in somatic shoot formation and embryogenesis [
9].
Genetic analysis of the “shoot regeneration” trait in which the F
1 and F
2 progeny are analyzed for the “shoot regeneration” phenotype is based on the assumption that the Quantitiative Trait Loci (QTL) for the trait does not affect whole plant development. The fact that these QTL do not affect plant development is not a trivial assumption because it implies that the disturbance in the process of shoot regeneration in culture has no direct connection to normal plant development and embryogenesis, rather the disruption of genes that are thought to be involved in somatic embryogenesis [
10]. In other words, when plants are recalcitrant to regeneration, genes that are triggered during shoot regeneration or somatic embryogenesis, or at least during the initial stages of shoot regeneration before normal development occurs, are not part of normal plant development. It is possible that the genes that affect shoot regeneration could be redundant members of gene families that are activated by growth regulators, such as in the case of the cytokinin response gene family [
11]. These genes could be redundant genes from gene families involved in plant development, such as the family of KNOX genes [
12], WOX genes [
13], and NAC genes. Therefore, the spotlight should be on gene families and processes that may not be involved in normal development.
This supposition is also supported by empirical, experimental data showing that regeneration requires both auxin and cytokinin [
14]. The crosstalk between auxin and cytokinin and the genes affected by this crosstalk are good candidates for involvement in the induction of shoot regeneration. Plant cells are somehow able to measure both the concentration of auxin and cytokinin and the ratio between the two [
15,
16]. Differential circulation of auxin within plant tissues or organs functions as the main signal for auxin-dependent plant developmental processes and, thus, subject to tight regulation. Polar auxin transport is a fundamentally important regulatory process of auxin signaling [
17]. All of these information streams are then somehow integrated leading to the induction of shoot regeneration in the competent tissue.
Nitric oxide (NO) regulates growth processes such as vegetative and generative development, seed germination, root growth, gravitropism, flowering, and fruit ripening [
18]. The growth regulating effect of NO is caused by auxin–NO interplay and cytokinins interaction, regulating cell division [
19] and shoot regeneration [
20]. Additionally, NO participates in the abiotic stress responses of plants [
18].
Here we describe a model system that is simple, reversible, and has a high-frequency shoot regeneration that allowed us to study the early events of shoot embryogenesis and shoot regeneration in cultured tobacco leaf segments at the cellular and transcriptomic level. These analyses show that no single factor determines the regeneration commitment of competent tobacco leaf segments.
2. Materials and Methods
2.1. Seed Sterilization and Plant Growth Conditions
Seeds of tobacco (Nicotiana tabacum L. cv. SR1) were placed in a 1.7 mL micro-tube (Eppendorf) filled with sodium hypochlorite (0.5% active material) and incubated for 5 min. The tube was shaken during the sterilization. After incubation, the seeds were rinsed three times with sterile water and spread on Petri dishes contained ½-strength MS medium (Duchefa Co., Haarlem, The Netherlands, Product number M0221.0050). After about two weeks, seedlings were planted in polypropylene Vitro Vent containers (Duchefa, NL; 9.6 cm × 9.6 cm and 9 cm in height) containing the same media to obtain disinfected plants. Plants were grown in sterile boxes in a growth room with 16 h of light and 8 h of darkness at 26 °C for several weeks until leaves were ready to be harvested.
2.2. Leaves Preparation and Regeneration
Leaves were detached from clean plants, and the midrib was removed. The leaf blade was cut into about 25 mm
2 (5 mm × 5 mm) segments and placed on regeneration (Reg) medium containing standard MS salts according to Evenor et al. [
15,
16], supplemented with 30 g l
−1 sucrose and 8 g L
−1 agar, and the following growth regulators: 4.57 μΜ IAA; 9.29 μΜ Kinetin, and 4.56 μM Zeatin (all from Duchefa Co.). The control medium was the same but lacking growth regulators. The medium was adjusted to pH 5.6. At least 20 leaf segments were placed on each Petri dish with at least four plates per treatment in all experiments. When GUS (ß-glucuronidase) activity was tested, leaf segments were incubated in GUS activity buffer described before by Evenor et al. [
16].
The effect of pre-incubation on Reg medium was tested by placing leaf segments on Reg medium for 1 to 8 days, with samples being move to MS medium after each indicated time period. Regeneration of leaf segments was scored 30–32 days after placing them on the MS medium.
Analyses of variance (ANOVA) was performed with the SAS/JMP software (SAS Institute Inc., Cary, NC, USA). Differences among means were calculated based on the Tukey–Kramer honestly significant difference (HSD) test for three or more treatments and T-test for two treatments [
21,
22].
2.3. Histological Studies of Leaf Segments
Leaf segments were removed from the medium and fixed in FAA mixture (5: 5: 90) of glacial acetic acid: formalin (40%
v/
v): ethanol (70%
v/
v). After fixation, the segments were dehydrated in stepwise ethanol series (30%, 50%, 70%, 90% (
v/
v) each for 24 h, followed by two 24 h incubations in fresh 100% ethanol) and embedded in paraffin. Paraffin sections (12 µm thick) were cut and then stained with Safranin and fast green FCF [
23,
24]. The thin sections were examined with a light microscope (Olympus BX50, 20 to 40 magnification).
2.4. RNA Preparation and Transcript Detection
RNA was isolated from leaf segment using PureLink midi kit (Invitrogen, Waltham, MA, USA) according to the manufacturer’s instructions and then treated with DNAse (Turbo DNA-free™, Ambion, Waltham, MA, USA). Single strand cDNA was prepared from total RNA using RevertAid
TM first-strand cDNA synthesis kit (Fermentas, Tel Aviv, Israel). PCR was conducted on cDNA samples isolated from each day using primers shown below [
21]. Microarray analysis using the total RNA preparations described above was carried out by a commercial vendor (Rouch-Nimblegen, Madison, WI, USA) using a proprietary microarray chip based on the TOBFAC tobacco gene database (
http://compsysbio.achs.virginia.edu/tobfac/). The RNA was hybridized to the custom microarray to identify genes whose expression changes during the seven days of the shoot induction period. RNA was isolated from day 0, day one, day four, and day seven after leaf segments were placed on Reg medium. The following comparisons were performed.
Treatment 1. 1 day in induction compared to 0 days in induction medium.
Treatment 2. 4 days in induction compared to 0 days in induction medium.
Treatment 3. 7 days in induction compared to 0 days in induction medium.
Each of the above hybridizations was conducted in three biological replications. cDNA was prepared from total RNA isolated and qPCR analysis was performed as described [
22] using the following primers:
NtWusforward-GTACGAGGTGGACACCCACAAC; NtWusreverse-GCAGCAGCAGCAATAAGCCTC; NtKnotted1forward–GGACAACAACAACAATAATCCAC; NtKnotted1reverse-CTTCCTCTTCTTCATGAACTCC; NtKnotted2forward-GTGAAGGCGTCGGATCGTCCGAAG; NtKnotted2reverse-CCCACCAACTGAGCAGCTTCTGGCGT; Nt18Sfor-GCGACGCATCATTCAAATTTC; Nt18Srev-TCCGGAATCGAACCCTAATTC; NtPDLP2for-ATTATCCTAATGGTGTGCCCG; NtPDLP2rev-AGCAACTCCTAAACCCACAC; NtPDLP3for-TCAGCACCAGATTACACTAAGTTAG; NtPDLP3rev-ACTTAGATTTTGAGGATTGTGCAAC; NtPDLP6for-GTTTATGACCAAATGCTACGCG; NtPDLP6rev-CTTTTCAACATCTTCATCTCCGC; SlANT1for-AGTTGTAGATTGAGGTGGCTGA; SlANT1rev-CCGGGAAGTCTACCAGCAAT.
All PCR primers used throughout this study were designed based on the tobacco genome in Sol Genomics Network (Sol Genomics Network) and purchased from Hy Labs Ltd. (Rehovot, Israel). Real-Time PCR (qRT-PCR) analyses were done as described by Schreiber et al. [
22]. The PCR reactions were performed in a T-GRADIENT thermal cycler (Biometra, Analytik Jena, and Gottingen, Germany).
Total RNA was extracted from sepals and flowers of tobacco using the TRIzol reagent system (Invitrogen Corp, Carlsbad, CA, USA). Genomic DNA contaminants were digested with TURBO DNA-free DNAase (Ambion Inc., Austin, TX, USA). The remaining RNA was then used as the template for cDNA synthesis using the Masterscript cDNA synthesis kit with random hexamer primers (KAPA Biosystems, Woburn, MA, USA).
The qRT-PCR analysis was performed using the KAPA SYBER FAST Master Mix (KAPA Biosystems, Woburn, MA, USA). DNA sequences complementary to tobacco 18S RIBOSOMAL RNA were used as control. Three technical replicates were performed for every biological repeat, three biological repeats for each condition. The qRT-PCR analyses were done using the Rotor-Gene Q detection system analyzed with the Rotor-Gene 6000 software (Qiagen Corbett Life Science, Dusseldorf, Germany). The relative abundance of the examined gene transcripts was calculated by the formula: 2(CT_examined gene-CT_reference gene), where CT represents the fractional cycle number at which the fluorescence crosses a fixed threshold.
2.5. Data Processing and Digital Tag Profiling
By removing 3′ adaptor fragments and several types of impurities from the raw reads, we obtained clean sequences. Then, sequences were mapped to the tobacco genome sequences in the Sol genomics database. No more than two mismatches were allowed in the alignment [
25]. A rigorous algorithm described previously [
26] was used for statistical analysis to identify differentially expressed genes (DEGs). We determined each gene expression level by the reads number uniquely mapped to the specific gene and the total number of uniquely mapped reads in the library. The threshold P-values were adjusted by the multiple testing procedures described by Benjamini and Yekutieli [
27] by controlling false discovery rate (FDR). In this study, FDR ≤ 0.01 and the absolute value of |log2Ratio|≥ 2 were used as the threshold for judging the gene expression significance. The DEGs were subjected to Gene Ontology (GO) database (
http://www.geneontology.org/) and mapped to the reference canonical pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG). The target sequences were allocated to the corresponding functional categories based on the BLAST searches by GO annotation using default parameters. The gene expression patterns of each pairwise comparison (0_DAP-vs-1_DAP, 0_DAP-vs-4_DAP, 0_DAP-vs-7_DAP, 4_DAP-vs-7_DAP, 0_DAP-vs-7_DAP) was analyzed, and genes were clustered according to their expression level using a self-organizing map using Cluster 3.0 with all the default parameters except the Euclidean distance of similarity metric. Additionally, the expression values were log2-transformed. Heat-maps with cluster data were then constructed using Java Tree View (
http://jtreeview.sourceforge.net/) for visualization of the hierarchical clustering results [
28].
2.6. Chlorophyll and Segment Size and NO Measurements
Chlorophyll content and chloroplast numbers were measured according to Kolotilin et al. [
29]. Each time point was the average of five replicas. The area of leaf segments on various media was measured using ImageJ free software [
30]. Each time point is the average of 10 measurements ± SE.
NO staining at the border of leaf segments placed on regeneration medium was done by incubating the leaf segments in 50 mM MES buffer, pH 5.6, and 1 mM DAF-2DA (4,5-diaminofluorescein-2 diacetate) for 30 min prior to visualization. Stain intensity per cell was analyzed by using ImageJ free software [
26]. The effect of NO on regeneration was done by placing leaf segments on Reg medium containing NO donors (20 μM, S-Nitroso-N-acetyl-DL-penicillamine; 5 μM, Molsidomine), NO scavenger (100 μM, PTIO), and NO synthesis inhibitor (5 μM, Diphenyleneiodonium)
. 2.7. Tobacco Plant Transformation and Measurements of GUS Activity
Tobacco (
Nicotiana tabacum cv. SR1) plants were grown in sterile vessels and leaf segments excised as needed for transformation. Transgenic tobacco plants expressing a reporter gene for cytokinin (AtARR5::GUS or Aux/IAA3::GUS) that was a gift from Prof. Joe Kieber (The University of North Carolina at Chapel Hill) and 35S::PDLP5-GFP (a gift from Dr Jung-Youn Lee, University of Delaware) [
31] were generated via Agrobacterium-mediated transformation using standard cocultivation methods. Transgenic seedlings were grown to flowering, self-fertilized, and homozygous T2 plants expressing Aux/IAA::GUS were used in auxin responsiveness assay by observing GUS activity in response to plant growth regulators. For chimera analysis, transgenic tissues were generated using a tomato
ScANT1 anthocyanin-inducing plasmid (a gift from Dr. I. Levin Volcani Center). A total of 84 (5 mm
2) leaf segments were transformed and each analyzed for the presence of the colored cells.
4. Discussion
The above findings indicate the interplay among plant growth regulators and the complexity of the interactions involved in shoot regeneration. Placing tobacco leaf segments on a regeneration medium for up to 4 days and removing them to a medium without growth regulators does not lead to
de novo shoot regeneration, a longer period on regeneration medium induced shoot formation. During the first week on regeneration medium, tobacco leaf segments lost chlorophyll and increased in segment area similarly to leaf segments on medium without growth regulators. Placing the tobacco leaf segment for a longer period leads to de novo shoot regeneration on medium without growth regulators, to a further rise in segment area and increased chlorophyll. After seven to 8 days on regeneration medium, all tobacco leaf segments will regenerate shoots. There are minor changes in segment morphology, mostly expansion and thickening. During those seven days, cross-sections of the leaf segments show cellular multiplications and cytosolic thickening, indicating increased mitosis [
32]. However, after more than five days on Reg medium, leaf segments will still develop shoots when removed from the Reg medium and placed on medium without plant growth regulators. This observation indicated that exposure to growth regulators in the Reg medium for 5–7 days initiates a nonreversible chain of events that will result in the formation of a shoot some 20–30 days later in the absence of plant growth regulators. Exposure to growth regulators up to 4 days is reversible, indicating that although leaf cells are competent to produce de novo shoots, there is an induction period in which leaf cells proliferate but are not committed to regeneration. This induction period indicated that some other factors are needed to induce shoot regeneration and that these factors are activated after four days in the presence of plant growth regulators.
Regeneration of de novo shoots requires both auxin and cytokinin [
14], and the crosstalk between them is considered essential for the induction of shoot regeneration [
15,
16]. As we show above, plant cells must measure both the concentration of auxin and cytokinin and the ratio between them to decide whether to produce shoots roots or callus [
15,
16]. Polar auxin transport is a fundamental important regulatory process of auxin signaling [
17]. Thus, the differential gradient of auxin within the plant tissues or organs can function as the main signal for auxin-dependent plant developmental processes and, therefore, subject to tight regulation. For appropriate tissue formation during development, cells must know their position relative to other cells. In animals, it is well documented that gradients of signaling molecules (morphogens) that convey positional information determine cell fate [
37]. Positional information is remarkably missing from plant tissue culture studies as explants are always placed on agar plates or liquid floats or in liquid media. When we tested for positional information of the leaf segment, we found that there is a peak in auxin responsiveness before commitment to regeneration occurs. This peak in auxin responsiveness coincides with the start of plasmodesmata production and deposition in the cell wall.
Another cellular messenger molecule is NO that regulates growth processes such as vegetative and generative development, seed germination, root growth, gravitropism, flowering, and fruit ripening [
18]. NO also participates in the abiotic stress responses of plants [
18]. The growth regulating effect of NO is caused by the auxin–NO interplay and cytokinin interaction, regulating cell division [
19] and shoot regeneration [
20]. Our data show that NO has an important role in shoot regeneration and that inhibiting NO production inhibits shoot regeneration. All the information that the leaf cells receive from starting to communicate with their neighbors is then somehow translated to induction of shoot regeneration in the competent tissue.
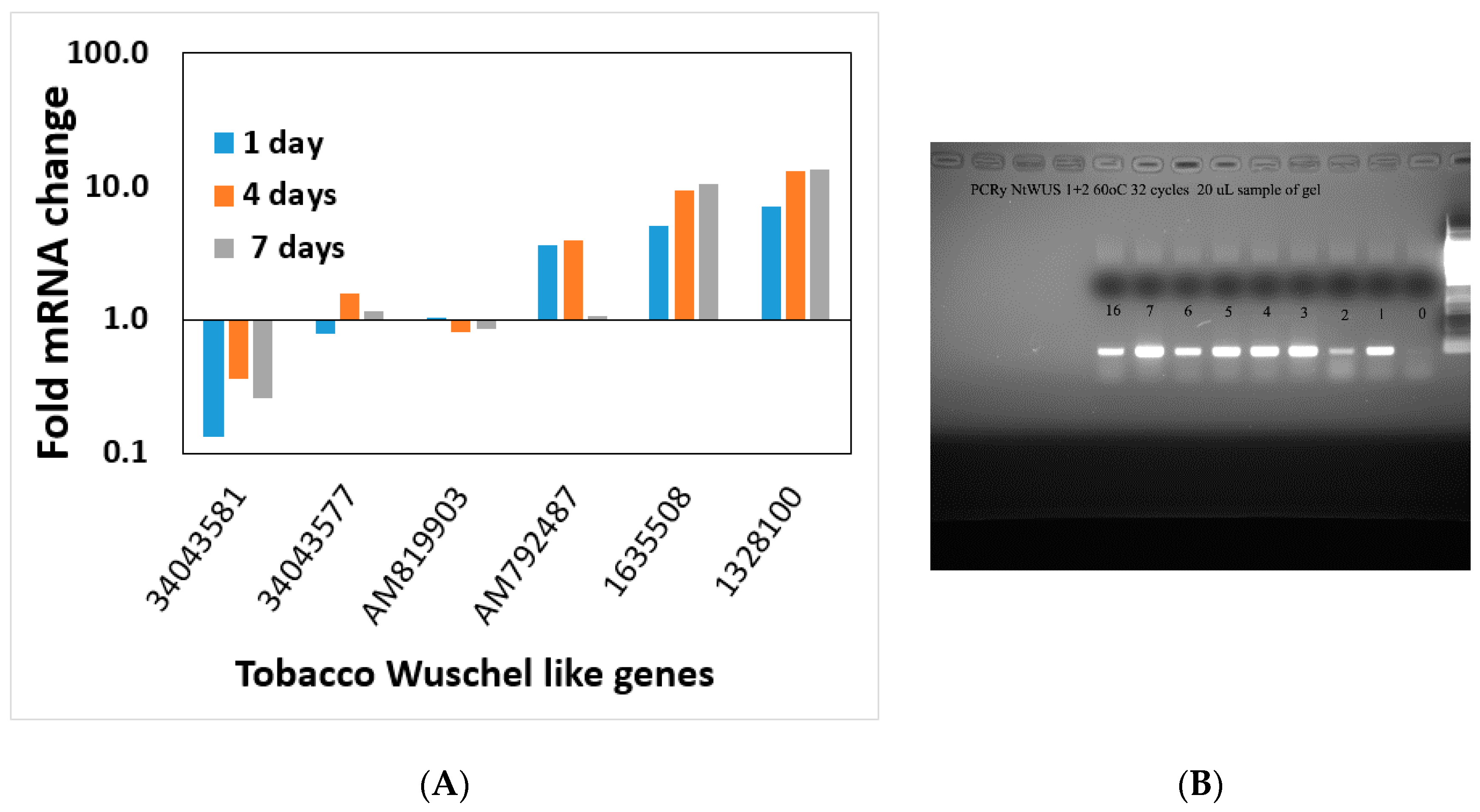
During the induction of shoot regeneration, cells in close proximity to each other divide and start exchanging information that triggers the differentiation and commitment into a meristem and eventually a shoot. Thus, we wanted to determine whether global changes in gene expression or changes in the expression of particular genes occur during this induction period without the complication of developmental genes. Meristematic genes (WUS or WOX and Knotted or KNOX gene families) are part of the maintenance genes linked to shoot regeneration and embryogenesis [
36]. Some of these genes increase during the shoot induction period, and some decrease. The altered expression of meristematic genes starts at day one on the regeneration medium and continues through the induction period.
WUSCHEL is linked to shoot regeneration [
35,
36] and our data show that increasing
WUSCHEL expression in the first four days of the shoot induction period is not sufficient to induce shoot regeneration since removal from Reg media during those four days reverses the process. Ectopic expression of
STM and
WUSCHEL activated a subset of meristem functions, including cell division, and ultimately organogenesis [
38]. This overexpression suggests that
WUSCHEL, combined with STM, initiates certain leaf cells’ progression to organ initiation [
38]. Global gene expression of particular genes of interest analyses could not pin down a single or specific process necessary to induce shoot regeneration. This observation that a multitude of genes change when global gene expression is studied during regeneration was shown in Arabidopsis as well [
8]. All the gene families check in this study show a similar pattern. Several gene family members increase during the induction period and several decreases, and most do not change. It seems that shoot regeneration is a complex phenomenon that involves many processes and is not triggered by a single gene. There was no single process within the cells regenerating into a shoot at the induction period.
Transformation of tobacco with
SlANT1 (a tomato MYB transcription factor that induces anthocyanin synthesis in tomato and tobacco plants) [
18] showed visual chimerism. Using this phenotype, we monitored the process of shoot regeneration. We observed that without selection pressure, tobacco shoots are formed from a cluster of cells that do not originate from a single cell, thus, leading to chimerism. Chimerism in transformed plants can explain variations in transgene expression of the regenerated plant as well as the non-mendelian distribution of transgenic progeny that occurs sometimes. It seems that the way to overcome the chimerism problem is using at least T1 seeds and in targets that do not produce seeds, be aware that chimerism can obscure the results.
The process of tissue regeneration is present in most multicellular organisms but is restricted to certain organs or tissues in different organisms [
39]. In most cases, wounding is required to induce regeneration in all plants and animals. Compared to animals, plants retain a high degree of developmental plasticity and display various tissues or organ regeneration [
40]. Regeneration can range from repairing a small wound or amputation to new organs or individual organisms and varies markedly between taxa. The ability of cells to determine their position in space and communicate with their neighbors is crucial to establishing proper patterning in a developing or regenerating organ. While no stem cell movement is present in plants, we show that auxin flow direction is a determinant in shoot regeneration. Thus, cells’ capability to establish their environment and position is essential to the regeneration process through taxa [
37]. It was shown that with the positional change of the root cells’ fate, the chromatin state is remodeled [
41]. Thus, positional information and phytochemical crosstalk is essential to plant cell regeneration, not because each cell needs to know its environment, but because plant cells regenerate as a group.
The ability to clonally propagate various plants, modify plants by introducing foreign genes (like Bt protein), or modify existing genes by genomic editing in crop plants, is an important component of modern agricultural biotechnology. The plasticity of plant cells and, thus, tissue regeneration, is paramount to all these current bio-agricultural industries. Understanding the regeneration process can enhance the ability to control various aspects of tissue regeneration, making the process more efficient and target oriented. Our findings indicate that leaf tissue regeneration requires positional information between the cells and their cellular neighbors. What is unknown is how they are connected, and the signals they exchange. Modifying or controlling these parameters can enhance or decrease tissue regeneration. For example, adding NO enhances shoot regeneration from leaf segment.
Our findings indicate to complexity and the interplay among plant growth regulators and the interactions involved in shoot regeneration. During the very early stages of regeneration cell divisions and cellular communication is essential for regeneration that thereafter occurs from not from a single cell but a group of cells. We observed cell divisions just as leaf segments are placed on regeneration medium, only groups of cells become shoot primordia but these are not discernable during the first days and early stages of regeneration.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









