Col11a1a Expression Is Required for Zebrafish Development

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Fish Maintenance, Care, and Staging
2.2. PCR
2.3. Cloning and Riboprobe Synthesis
2.4. In Situ Hybridization
2.5. Antisense Morpholino Oligonucleotide Injection
2.6. CRISPR/Cas9 Gene Editing
2.7. Statistical Analysis
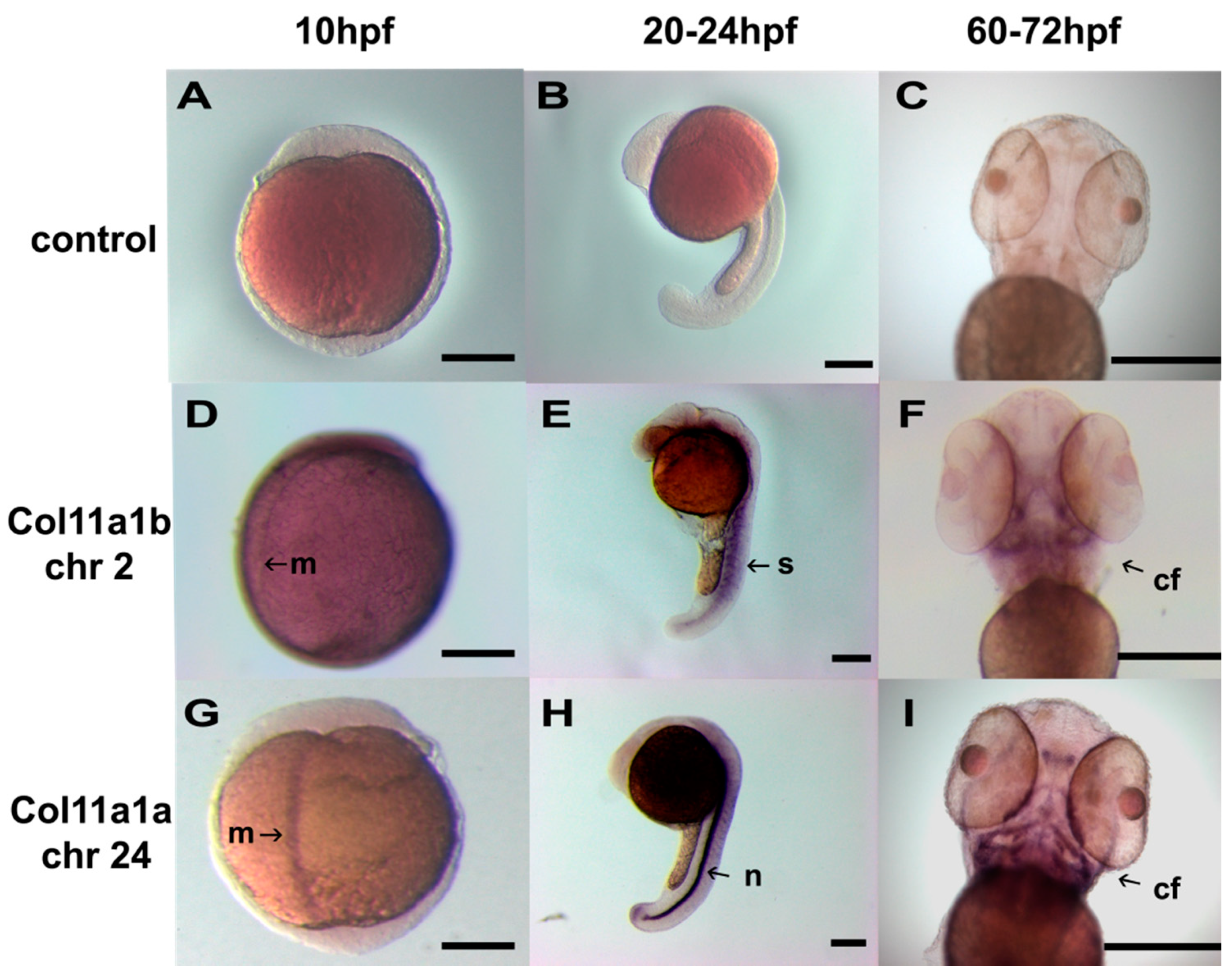
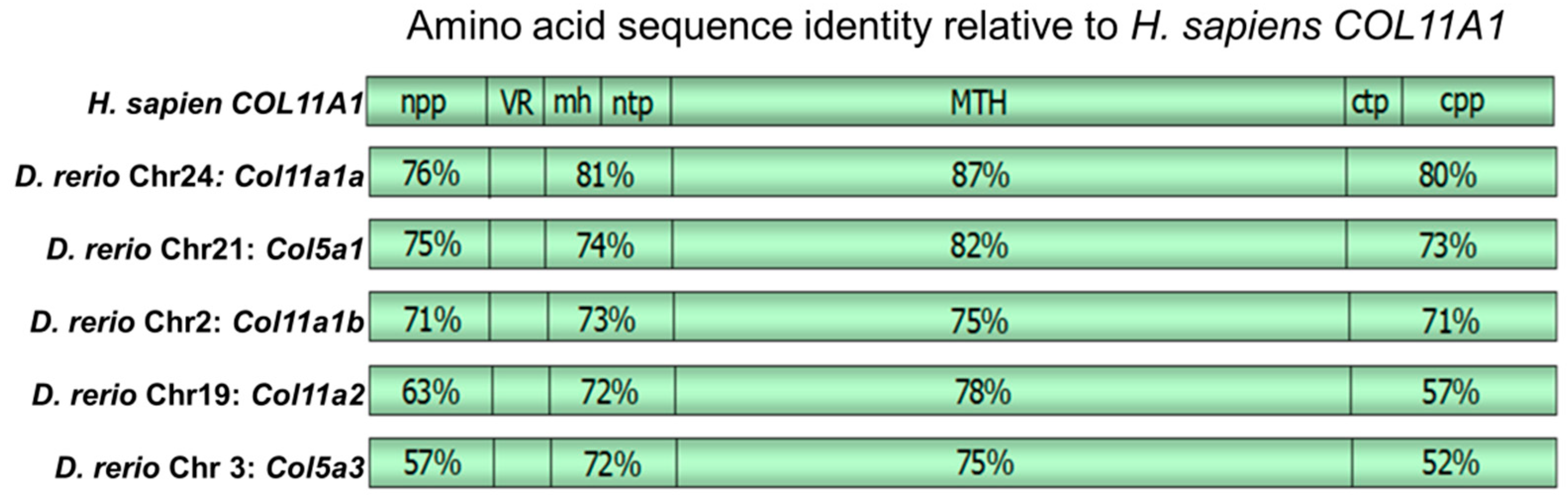
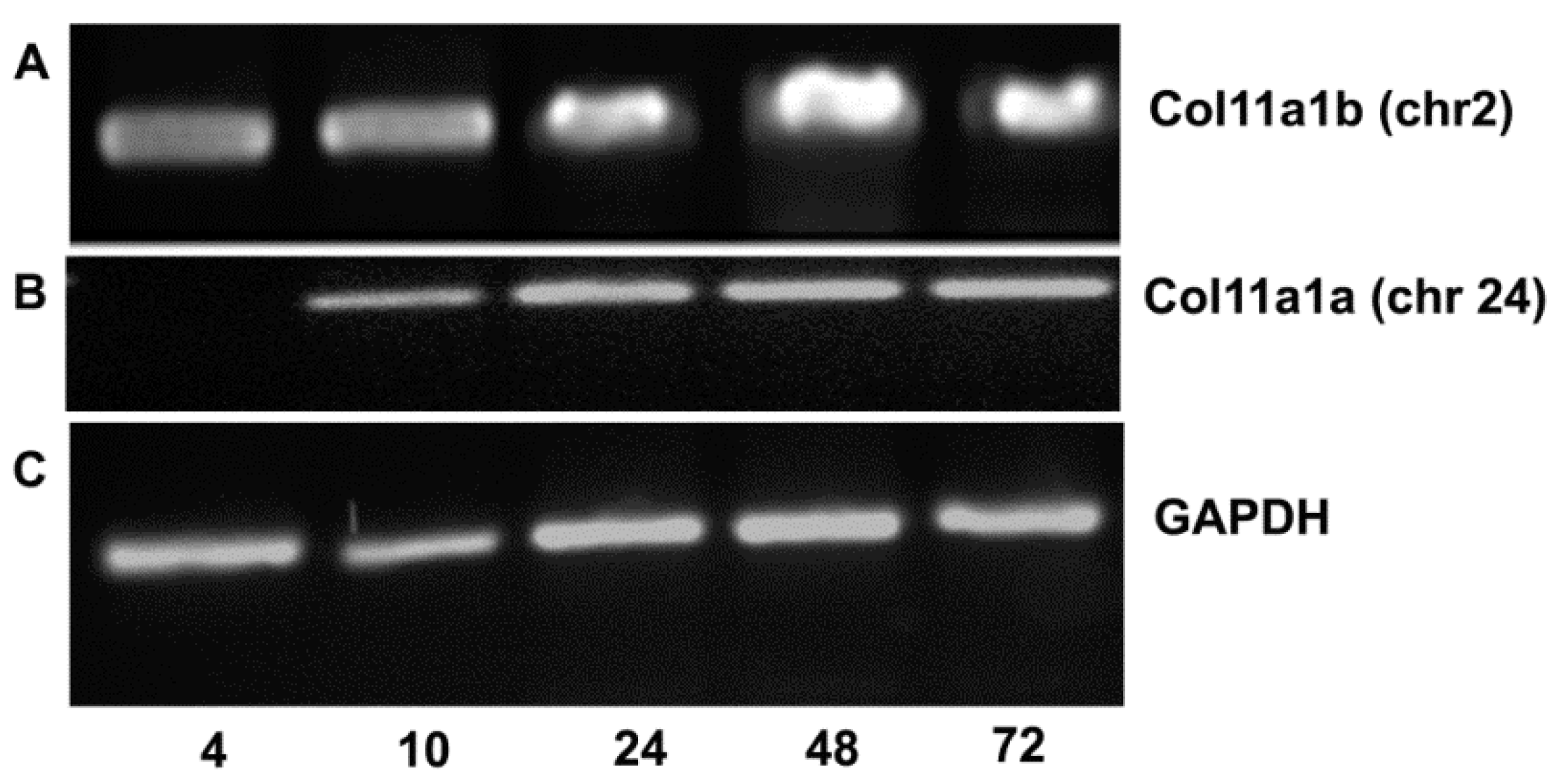
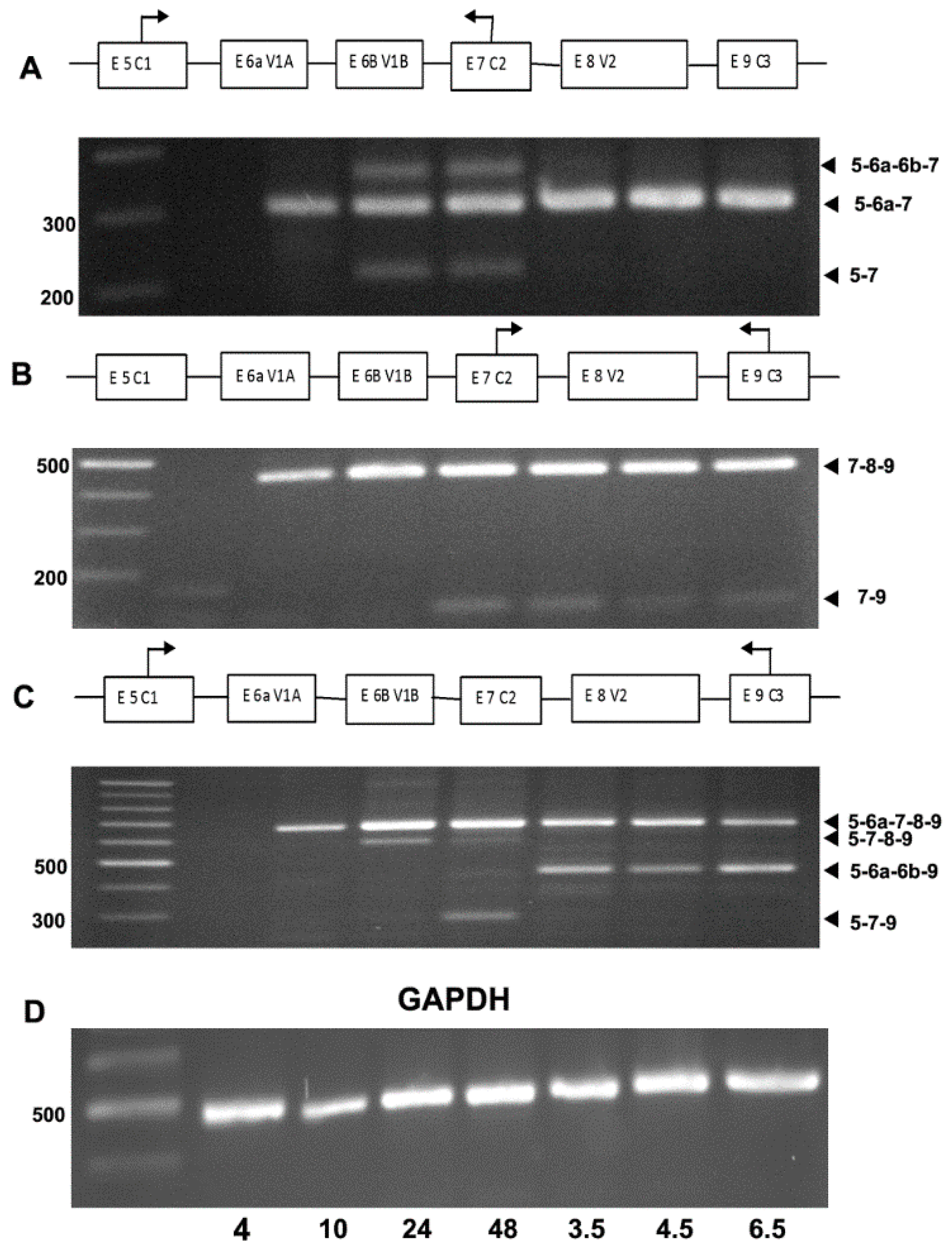
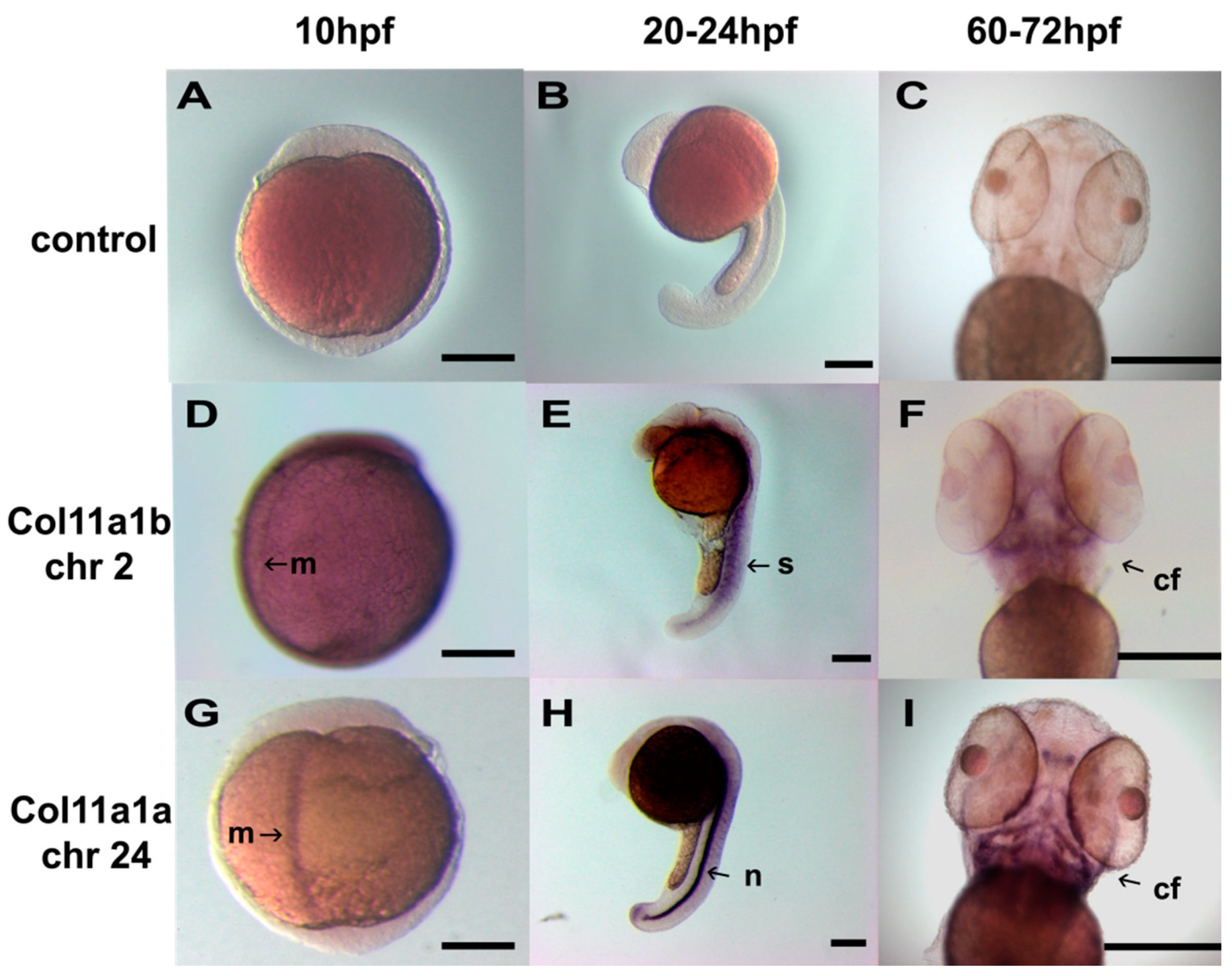
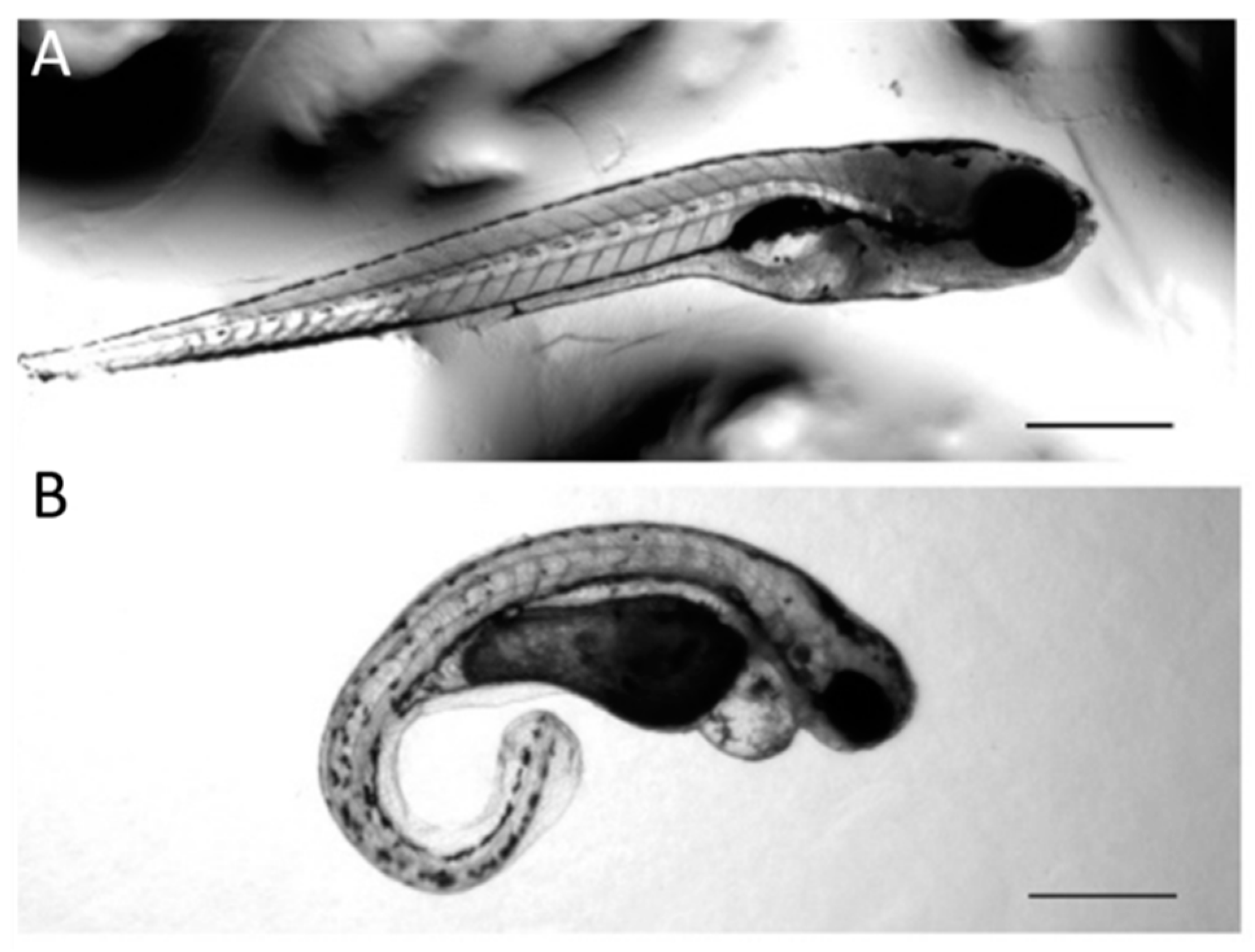
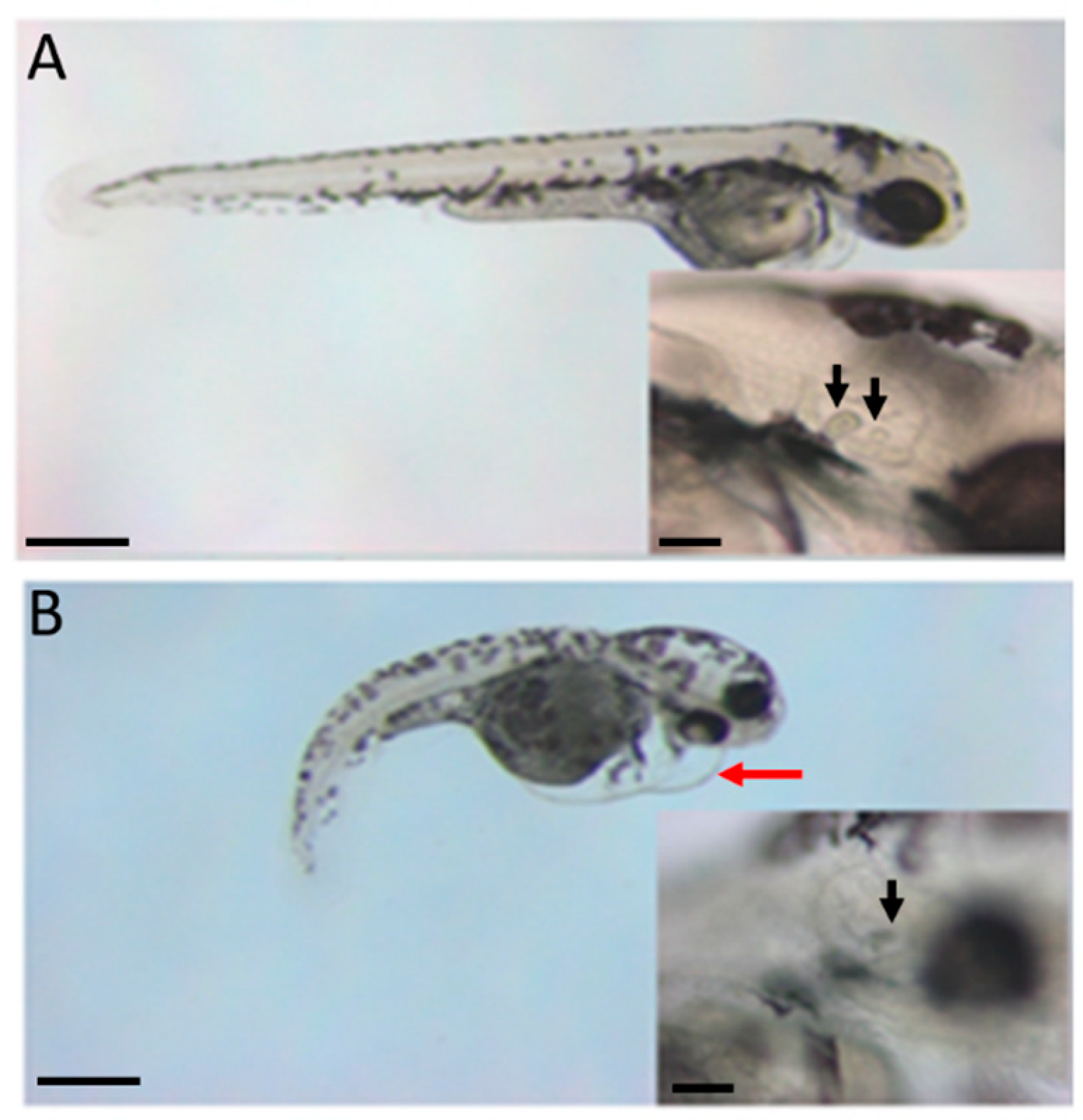
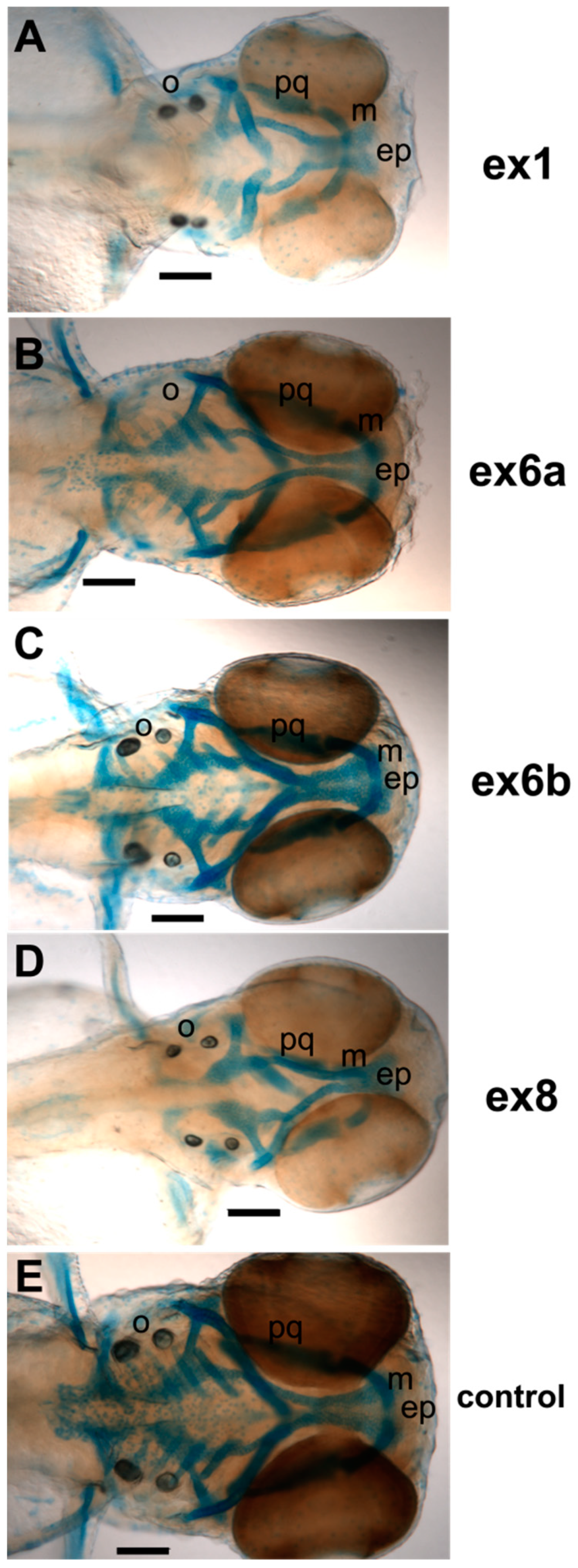
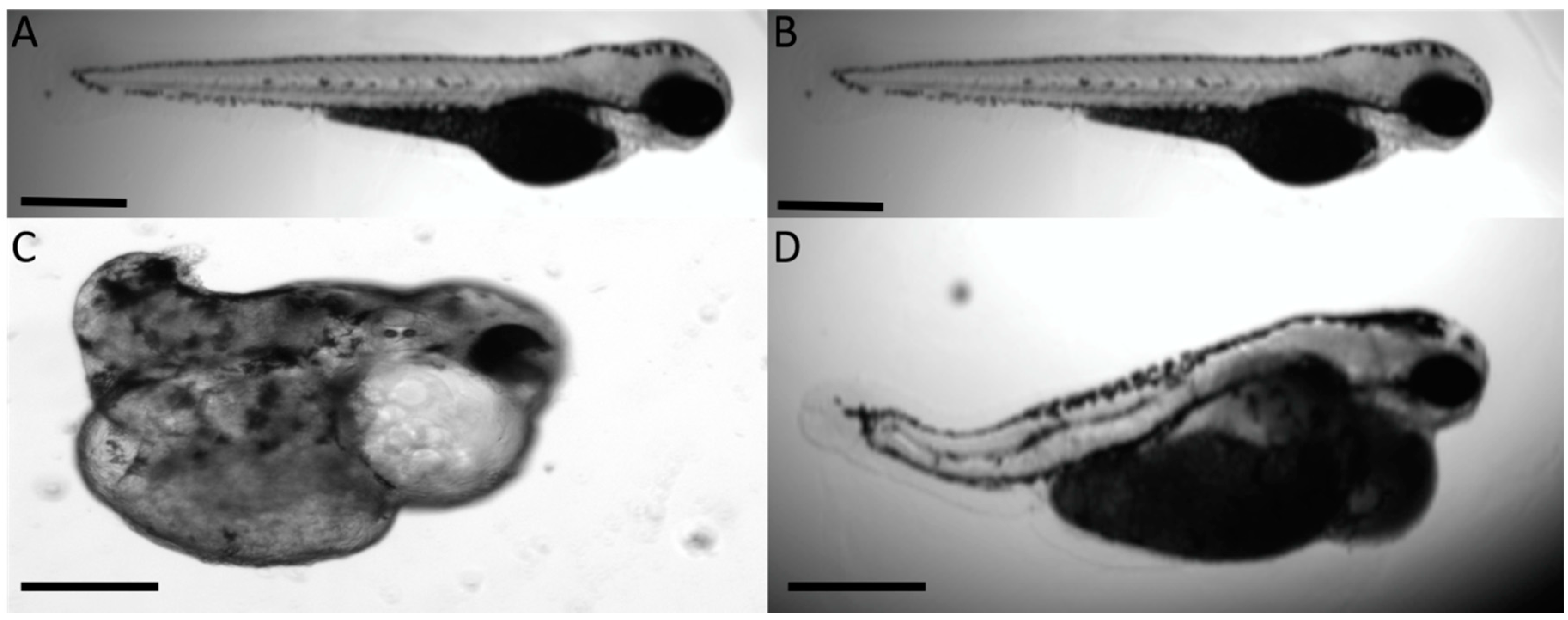
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Goldring, M.B.; Tsuchimochi, K.; Ijiri, K. The control of chondrogenesis. J. Cell. Biochem. 2006, 97, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.K.; Miyake, T. Divide, accumulate, differentiate: Cell condensation in skeletal development revisited. Int. J. Dev. Biol. 1995, 39, 881–893. [Google Scholar] [PubMed]
- Glenister, T.W. An embryological view of cartilage. J. Anat. 1976, 122, 323–330. [Google Scholar]
- Zylińska, B.; Silmanowicz, P.; Sobczyńska-Rak, A.; Jarosz, Ł.; Szponder, T. Treatment of articular cartilage defects: Focus on tissue engineering. In Vivo 2018, 32, 1289–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, B.B.; Phillips, B.T. Ringing in the new ear: Resolution of cell interactions in otic development. Dev. Biol. 2003, 261, 289–312. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, T.T.; Riley, B.B.; Chiang, M.Y.; Phillips, B. Development of the zebrafish inner ear. Dev. Dyn. 2002, 223, 427–458. [Google Scholar] [CrossRef]
- Whitfield, T.T.; Granato, M.; Van Eeden, F.J.M.; Schach, U.; Brand, M.; Furutani-Seiki, M.; Haffter, P.; Hammerschmidt, M.; Heisenberg, C.P.; Jiang, Y.J.; et al. Mutations affecting development of the zebrafish inner ear and lateral line. Development 1996, 123, 241–254. [Google Scholar]
- Nicolson, T. The Genetics of Hearing and Balance in Zebrafish. Annu. Rev. Genet. 2005, 39, 9–22. [Google Scholar] [CrossRef]
- Stickler, G.B.; Belau, P.G.; Farrell, F.J.; Jones, J.D.; Pugh, D.G.; Steinberg, A.G.; Ward, L.E. Hereditary progressive artho-opthalmopathy. Mayo Clin. Proc. 1965, 40, 433–455. [Google Scholar]
- Yelick, P.C.; Schilling, T.F. Molecular dissection of craniofacial development using zebrafish. Crit. Rev. Oral Biol. Med. 2002, 13, 308–322. [Google Scholar] [CrossRef]
- Mundlos, S.; Olsen, B.R. Heritable diseases of the skeleton. Part II: Molecular insights into skeletal development-matrix components and their homeostasis. FASEB J. 1997, 11, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Todhunter, R.J.; Garrison, S.J.; Jordan, J.; Hunter, L.; Castelhano, M.G.; Ash, K.; Meyers-Wallen, V.; Krotscheck, U.; Hayward, J.J.; Grenier, J. Gene expression in hip soft tissues in incipient canine hip dysplasia and osteoarthritis. J. Orthop. Res. 2019, 37, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Marshall, D. Ectodermal dysplasia. Report of kindred with ocular abnormalities and hearing defect. Am. J. Ophthalmol. 1958, 45, 143–156. [Google Scholar] [CrossRef]
- Chatterjee, S.; Lufkin, T. The Sound of Silence: Mouse Models for Hearing Loss. Genet. Res. Int. 2011, 2011, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Seegmiller, R.; Fraser, F.C.; Sheldon, H. A new chondrodystrophic mutant in mice. Electron microscopy of normal and abnormal chondrogenesis. J. Cell Biol. 1971, 48, 580–593. [Google Scholar] [CrossRef] [PubMed]
- Cremers, C.W.R.J.; Cornelius, W.R.J.; Smith, R. Genetic Hearing Impairment: Its Clinical Presentations; Karger: Basel, Switzerland, 2002; ISBN 9783805574495. [Google Scholar]
- Griffith, A.J.; Sprunger, L.K.; Sirko-Osadsa, D.A.; Tiller, G.E.; Meisler, M.H.; Warman, M.L. Marshall syndrome associated with a splicing defect at the COL11A1 locus. Am. J. Hum. Genet. 1998, 62, 816–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymko-Bennett, Y.M.; Kurima, K.; Olsen, B.; Seegmiller, R.; Griffith, A.J. Auditory function associated with Col11a1 haploinsufficiency in chondrodysplasia (cho) mice. Hear. Res. 2003, 175, 178–182. [Google Scholar] [CrossRef]
- Hufnagel, S.B.; Weaver, K.N.; Hufnagel, R.B.; Bader, P.I.; Schorry, E.K.; Hopkin, R.J. A novel dominant COL11A1 mutation resulting in a severe skeletal dysplasia. Am. J. Med. Genet. Part A 2014, 164, 2607–2612. [Google Scholar] [CrossRef]
- Acke, F.R.E.; Dhooge, I.J.M.; Malfait, F.; De Leenheer, E.M.R. Hearing impairment in Stickler syndrome: A systematic review. Orphanet J. Rare Dis. 2012, 7, 84. [Google Scholar] [CrossRef] [Green Version]
- Shoulders, M.D.; Raines, R.T. Collagen Structure and Stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef] [Green Version]
- Eyre, D. Collagen of articular cartilage. Arthritis Res. 2002, 4, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Nowlan, N.C. Initiation and emerging complexity of the collagen network during prenatal skeletal development. Eur. Cells Mater. 2020, 39, 136–155. [Google Scholar] [CrossRef] [PubMed]
- Jacenko, O.; Olsen, B.; LuValle, P. Organization and regulation of collagen genes. In Critical Reviews in Eukaryotic Gene Expression; Stein, G.S., Stein, J., Lians, J.B., Eds.; CRC Press: Boca Raton, FL, USA, 1991; Volume 1, pp. 327–353. [Google Scholar]
- Eyre, D.R.; Wu, J.J.; Fernandes, R.J.; Pietka, T.A.; Weis, M.A. Recent developments in cartilage research: Matrix biology of the collagen II/IX/XI heterofibril network. Biochem. Soc. Trans. 2002, 30, 893–899. [Google Scholar] [CrossRef]
- Mendler, M.; Eich-Bender, S.G.; Vaughan, L.; Winterhalter, K.H.; Bruckner, P. Cartilage contains mixed fibrils of collagen types II, IX, and XI. J. Cell Biol. 1989, 108, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Nah, H.D.; Barembaum, M.; Upholt, W.B. The chicken α1(XI) collagen gene is widely expressed in embryonic tissues. J. Biol. Chem. 1992, 267, 22581–22586. [Google Scholar]
- Mayne, R.; Brewton, R.G.; Mayne, P.M.; Baker, J.R. Isolation and characterization of the chains of type V/type XI collagen present in bovine vitreous. J. Biol. Chem. 1993, 268, 9381–9386. [Google Scholar] [PubMed]
- Fang, M.; Adams, J.S.; McMahan, B.L.L.; Brown, R.J.R.J.; Oxford, J.T. The expression patterns of minor fibrillar collagens during development in zebrafish. Gene Expr. Patterns 2010, 10, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, J.A.; Valen, E.; Thyme, S.B.; Huang, P.; Ahkmetova, L.; Pauli, A.; Montague, T.G.; Zimmerman, S.; Richter, C.; Schier, A.F. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 2014, 9, e98186. [Google Scholar] [CrossRef]
- Moradi-Améli, M.; Deléage, G.; Geourgjon, C.; van der Rest, M. Common topology within a non-collagenous domain of several different collagen types. Matrix Biol. 1994, 14, 233–239. [Google Scholar] [CrossRef]
- Tisi, D.; Talts, J.; Timpl, R.; Hohenester, E. Structure of the C-terminal laminin G-like domain pair of the laminin α2 chain harbouring binding sites for α-dystroglycan and heparin. EMBO J. 2009, 19, 1432–1440. [Google Scholar] [CrossRef]
- Timpl, R.; Tisi, D.; Talts, J.F.; Andac, Z.; Sasaki, T.; Hohenester, E. Structure and function of laminin LG modules. Matrix Biol. 2000, 19, 309–317. [Google Scholar] [CrossRef]
- Hohenester, E.; Tisi, D.; Talts, J.F.; Timpl, R. The crystal structure of a laminin G-like module reveals the molecular basis of α-dystroglycan binding to laminins, perlecan, and agrin. Mol. Cell 1999, 4, 783–792. [Google Scholar] [CrossRef]
- Fallahi, A.; Kroll, B.; Warner, L.R.; Oxford, R.J.; Irwin, K.M.; Mercer, L.M.; Shadle, S.E.; Oxford, J.T. Structural model of the amino propeptide of collagen XI alpha1 chain with similarity to the LNS domains. Protein Sci. 2005, 14, 1526–1537. [Google Scholar] [CrossRef] [Green Version]
- Wälchi, C.; Trueb, J.; Kessler, B.; Winterhalter, K.H.; Trueb, B. Complete primary structure of chicken collagen XIV. Eur. J. Biochem. 1993, 212, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Bork, P. The modular architecture of vertebrate collagens. FEBS Lett. 1992, 307, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Tillet, E.; Mann, K.; Nischt, R.; Pan, T.-C.; Chu, M.-L.; Timpl, R. Recombinant Analysis of Human α1(XVI) Collagen: Evidence for Processing of the N-Terminal Globular Domain. Eur. J. Biochem. 1995, 228, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Inoguchi, K.; Yoshioka, H.; Khaleduzzaman, M.; Ninomiya, Y. The mrna for α1(XIX) collagen chain, a new member of FACITs, contains a long unusual 3′ untranslated region and displays many unique splicing variants. J. Biochem. 1995, 117, 137–146. [Google Scholar] [CrossRef]
- Van Der Rest, M.; Garrone, R. Collagen family of proteins. FASEB J. 1991, 5, 2814–2823. [Google Scholar] [CrossRef] [Green Version]
- Fang, M.; Jacob, R.; McDougal, O.; Oxford, J.T. Minor fibrillar collagens, variable regions alternative splicing, intrinsic disorder, and tyrosine sulfation. Protein Cell 2012, 3, 419–433. [Google Scholar] [CrossRef] [Green Version]
- Tsumaki, N.; Kimura, T. Differential expression of an acidic domain in the amino-terminal propeptide of mouse pro-α2(XI) collagen by complex alternative splicing. J. Biol. Chem. 1995, 270, 2372–2378. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, H.; Ramirez, F. Pro-alpha 1(XI) collagen. Structure of the amino-terminal propeptide and expression of the gene in tumor cell lines. J. Biol. Chem. 1990, 265, 6423–6426. [Google Scholar] [PubMed]
- Gregory, K.E.; Oxford, J.T.; Chen, Y.; Gambee, J.E.; Gygi, S.P.; Aebersold, R.; Neame, P.J.; Mechling, D.E.; Bächinger, H.P.; Morris, N.P. Structural organization of distinct domains within the non-collagenous N-terminal region of collagen type XI. J. Biol. Chem. 2000, 275, 11498–11506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhidkova, N.I.; Justice, S.K.; Mayne, R. Alternative mRNA processing occurs in the variable region of the pro- α1(XI) and pro-α2(XI) collagen chains. J. Biol. Chem. 1995, 270, 9486–9493. [Google Scholar] [CrossRef] [Green Version]
- Oxford, J.T.; Doege, K.J.; Morris, N.P. Alternative exon splicing within the amino-terminal nontriple-helical domain of the rat pro-α1(XI) collagen chain generates multiple forms of the mRNA transcript which exhibit tissue-dependent variation. J. Biol. Chem. 1995, 270, 9478–9485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, G.G.; Branam, A.M.; Huang, G.; Pelegri, F.; Cole, W.G.; Wenstrup, R.M.; Greenspan, D.S. Characterization of the six zebrafish clade B fibrillar procollagen genes, with evidence for evolutionarily conserved alternative splicing within the pro-α1(V) C-propeptide. Matrix Biol. 2010, 29, 261–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, G.B.; Oxford, J.T.; Hausafus, L.C.; Smoody, B.F.; Morris, N.P. Temporal and spatial expression of alternative splice-forms of the alpha1(XI) collagen gene in fetal rat cartilage. Dev. Dyn. 1998, 213, 12–26. [Google Scholar] [CrossRef]
- Richards, A.J.; Martin, S.; Nicholls, A.C.; Harrison, J.B.; Pope, F.M.; Burrows, N.P. A single base mutation in COL5A2 causes Ehlers-Danlos syndrome type II. J. Med. Genet. 1998, 35, 846–848. [Google Scholar] [CrossRef] [Green Version]
- Annunen, S.; Körkkö, J.; Czarny, M.; Warman, M.L.; Brunner, H.G.; Kääriäinen, H.; Mulliken, J.B.; Tranebjærg, L.; Brooks, D.G.; Cox, G.F.; et al. Splicing Mutations of 54-bp Exons in the COL11A1 Gene Cause Marshall Syndrome, but Other Mutations Cause Overlapping Marshall/Stickler Phenotypes. Am. J. Hum. Genet. 1999, 65, 974–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robin, N.H.; Moran, R.T.; Ala-Kokko, L. Gene Reviews: Stickler Syndrome; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Seegmiller, R.E.; Foster, C.; Burnham, J.L. Understanding chondrodysplasia (cho): A comprehensive review of cho as an animal model of birth defects, disorders, and molecular mechanisms. Birth Defects Res. 2019, 111, 237–247. [Google Scholar] [CrossRef]
- Schilling, T.F.; Walker, C.; Kimmel, C.B. The chinless mutation and neural crest cell interactions in zebrafish jaw development. Development 1996, 122, 1417–1426. [Google Scholar]
- Newgreen, D.F.; Erickson, C.A. The migration of neural crest cells. Int. Rev. Cytol. 1986, 103, 89–145. [Google Scholar] [PubMed]
- Perris, R.; Krotoski, D.; Bronner-Fraser, M. Collagens in avian neural crest development: Distribution in vivo and migration-promoting ability in vitro. Development 1991, 113, 969–984. [Google Scholar] [PubMed]
- Lallier, T.; Leblanc, G.; Artinger, K.B.; Bronner-Fraser, M. Cranial and trunk neural crest cells use different mechanisms for attachment to extracellular matrices. Development 1992, 116, 531–541. [Google Scholar] [PubMed]
- Maxwell, G.D. Substrate dependence of cell migration from explanted neural tubes in vitro. Cell Tissue Res. 1976, 172, 325–330. [Google Scholar] [CrossRef]
- Seufert, D.W.; Hanken, J.; Klymkowsky, M.W. Type II collagen distribution during cranial development in Xenopus laevis. Anat. Embryol. 1994, 189, 81–89. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Target | Guide Sequence 1 | Forward Primer | Reverse Primer |
|---|---|---|---|---|
| E101 | Exon 1 | ATTTAGGTGACACTATAGGCCAAGGTGGTCCCCAATGGTTTTAGAGCTAGAAATAGCAAG | GGCACTTTTGGGATTGTAGAAG | CATCTCCTCTTAGAAAGCCCCT |
| E201 | Exon 2 | ATTTAGGTGACACTATAAAGAGCATCACAGCCAGACGGTTTTAGAGCTAGAAATAGCAAG | CTGCTGACATTTTGCATGTCTT | CATTTAAACGCAGCTGAACGTA |
| E301 | Exon 3 | ATTTAGGTGACACTATAAGGCGTCCAGCAGCTGGGCGGTTTTAGAGCTAGAAATAGCAAG | GTAAGAAGAAGCTGACCAAGCC | CCCGTTTATTTCTACCTCATGC |
| E401 | Exon 4 | ATTTAGGTGACACTATATGGCACCAGGATCCTGGATGGTTTTAGAGCTAGAAATAGCAAG | GTAAGAAGAAGCTGACCAAGCC | CCCGTTTATTTCTACCTCATGC |
| E501 | Exon 5 | ATTTAGGTGACACTATAGCCTGCAGTGTGTCCTTGTGGTTTTAGAGCTAGAAATAGCAAG | GCTCTGTTTTTGGTCTCCTCAG | AGACGTCCAGAAGCGTTTAGTC |
| E2701 | Exon 27 | ATTTAGGTGACACTATAGGTGTCCGTGGTCTAAAGGGGTTTTAGAGCTAGAAATAGCAAG | TTCACTGTTGTCATTTTCAGGG | ACGTGTGACGATTTCTCCATTA |
| AMO | N | Lethality |
|---|---|---|
| Col11a1b-MOe1 | 54 | 14 (26%) |
| Col11a1a-MOe1 | 87 | 50 (57%) |
| Col11a1a-MOe6a | 80 | 24 (30%) |
| Col11a1a-MOe8 | 56 | 19 (34%) |
| Std. AMO control | 32 | 11 (34%) |
| AMO | Length (mm) | % Decrease |
|---|---|---|
| Col11a1b-MOe1 | 2.67 ± 0.16 | −13.1% |
| Col11a1a-MOe1 | 2.81 ± 0.12 | −7.5% |
| Col11a1a-MOe6a | 2.85 ± 0.12 | −6.0% |
| Col11a1a-MOe8 | 2.85 ± 0.18 | −6.0% |
| Std. AMO control | 3.02 ± 0.17 | 0 |
| AMO | n | Missing or Extra Otoliths | Pericardial Edema | Curved Notochord | Smaller Meckel’s Cartilage |
|---|---|---|---|---|---|
| Col11a1b-MOe1 | 40 | 40 (100%) | 39 (98%) | 30 (75%) | 40 (100%) |
| Col11a1a-MOe1 | 34 | 18 (53%) | 28 (82%) | 25 (74%) | 33 (97%) |
| Col11a1a-MOe6a | 44 | 30 (68%) | 42 (95%) | 41 (93%) | 44 (100%) |
| Col11a1a-MOe8 | 18 | 0 | 0 | 1 | 18 (100%) |
| Std. AMO control | 32 | 0 | 0 | 0 | 0 |
| CRISPR/Cas9 | N | Lethality |
|---|---|---|
| Col11a1a −/− | 303 | 299 (98%) |
| Col11a1a +/− | 299 | 152 (50%) |
| Wild-type control | 311 | 25 (8%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hardy, M.J.; Reeck, J.C.; Fang, M.; Adams, J.S.; Oxford, J.T. Col11a1a Expression Is Required for Zebrafish Development. J. Dev. Biol. 2020, 8, 16. https://doi.org/10.3390/jdb8030016
Hardy MJ, Reeck JC, Fang M, Adams JS, Oxford JT. Col11a1a Expression Is Required for Zebrafish Development. Journal of Developmental Biology. 2020; 8(3):16. https://doi.org/10.3390/jdb8030016
Chicago/Turabian StyleHardy, Makenna J., Jonathon C. Reeck, Ming Fang, Jason S. Adams, and Julia Thom Oxford. 2020. "Col11a1a Expression Is Required for Zebrafish Development" Journal of Developmental Biology 8, no. 3: 16. https://doi.org/10.3390/jdb8030016
APA StyleHardy, M. J., Reeck, J. C., Fang, M., Adams, J. S., & Oxford, J. T. (2020). Col11a1a Expression Is Required for Zebrafish Development. Journal of Developmental Biology, 8(3), 16. https://doi.org/10.3390/jdb8030016