Contributions of Noncanonical Smoothened Signaling During Embryonic Development


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Noncanonical Smoothened Signaling
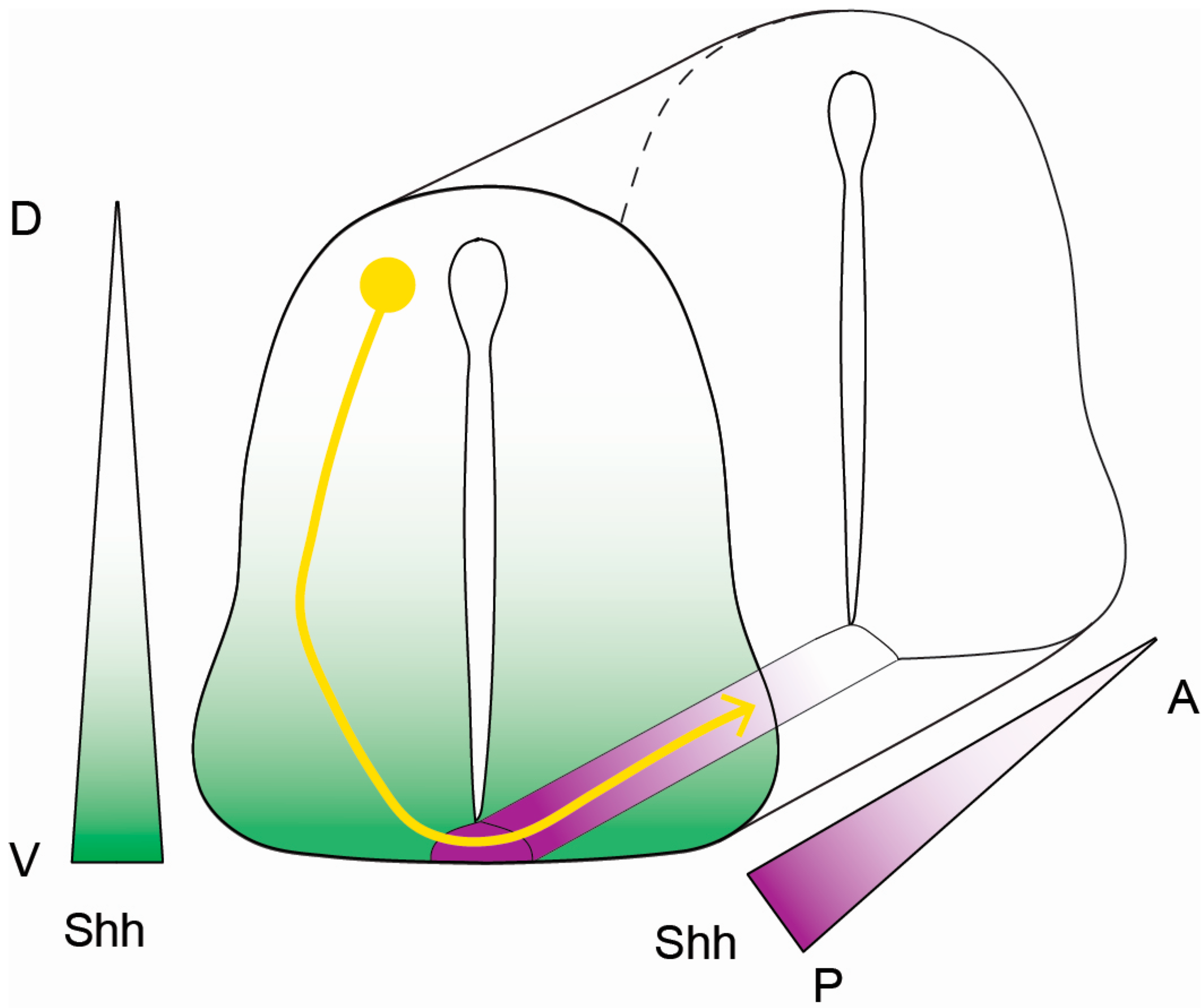
3. Cytoskeletal Dynamics, Cellular Migration, and Axon Guidance
4. Axon Fasciculation
5. Neurotransmitter Selection
6. Cellular Proliferation
7. Metabolism
8. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cohen, M.M., Jr. Hedgehog signaling update. Am. J. Med. Genet. A 2010, 152A, 1875–1914. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Marigo, V.; Davey, R.A.; Zuo, Y.; Cunningham, J.M.; Tabin, C.J. Biochemical evidence that patched is the Hedgehog receptor. Nature 1996, 384, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, J.; Takabatake, T.; Takeshima, K.; Hui, C. Ptch2, a second mouse Patched gene is co-expressed with Sonic hedgehog. Nat. Genet. 1998, 18, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Arensdorf, A.M.; Marada, S.; Ogden, S.K. Smoothened Regulation: A Tale of Two Signals. Trends Pharmacol. Sci. 2016, 37, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Strutt, H.; Thomas, C.; Nakano, Y.; Stark, D.; Neave, B.; Taylor, A.M.; Ingham, P.W. Mutations in the sterol-sensing domain of Patched suggest a role for vesicular trafficking in Smoothened regulation. Curr. Biol. 2001, 11, 608–613. [Google Scholar] [CrossRef]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, G.; Sircar, R.; Kong, J.H.; Nachtergaele, S.; Sagner, A.; Byrne, E.F.; Covey, D.F.; Siebold, C.; Rohatgi, R. Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Nachtergaele, S.; Mydock, L.K.; Krishnan, K.; Rammohan, J.; Schlesinger, P.H.; Covey, D.F.; Rohatgi, R. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat. Chem. Biol. 2012, 8, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Byrne, E.F.; Sircar, R.; Miller, P.S.; Hedger, G.; Luchetti, G.; Nachtergaele, S.; Tully, M.D.; Mydock-McGrane, L.; Covey, D.F.; Rambo, R.P.; et al. Structural basis of Smoothened regulation by its extracellular domains. Nature 2016, 535, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Nachtergaele, S.; Whalen, D.M.; Mydock, L.K.; Zhao, Z.; Malinauskas, T.; Krishnan, K.; Ingham, P.W.; Covey, D.F.; Siebold, C.; Rohatgi, R. Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. eLife 2013, 2, e01340. [Google Scholar] [CrossRef] [PubMed]
- Nedelcu, D.; Liu, J.; Xu, Y.; Jao, C.; Salic, A. Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nat. Chem. Biol. 2013, 9, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Myers, B.R.; Sever, N.; Chong, Y.C.; Kim, J.; Belani, J.D.; Rychnovsky, S.; Bazan, J.F.; Beachy, P.A. Hedgehog pathway modulation by multiple lipid binding sites on the smoothened effector of signal response. Dev. Cell 2013, 26, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Khaliullina, H.; Bilgin, M.; Sampaio, J.L.; Shevchenko, A.; Eaton, S. Endocannabinoids are conserved inhibitors of the Hedgehog pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 3415–3420. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Young, K.E.; Maiti, T.; Beachy, P.A. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. USA 2002, 99, 14071–14076. [Google Scholar] [CrossRef] [PubMed]
- Arensdorf, A.M.; Dillard, M.E.; Menke, J.M.; Frank, M.W.; Rock, C.O.; Ogden, S.K. Sonic Hedgehog Activates Phospholipase A2 to Enhance Smoothened Ciliary Translocation. Cell Rep. 2017, 19, 2074–2087. [Google Scholar] [CrossRef] [PubMed]
- May, S.R.; Ashique, A.M.; Karlen, M.; Wang, B.; Shen, Y.; Zarbalis, K.; Reiter, J.; Ericson, J.; Peterson, A.S. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 2005, 287, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Aza-Blanc, P.; Ramirez-Weber, F.A.; Laget, M.P.; Schwartz, C.; Kornberg, T.B. Proteolysis that is inhibited by hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell 1997, 89, 1043–1053. [Google Scholar] [CrossRef]
- Ruiz i Altaba, A. Gli proteins encode context-dependent positive and negative functions: Implications for development and disease. Development 1999, 126, 3205–3216. [Google Scholar] [PubMed]
- Wang, B.; Fallon, J.F.; Beachy, P.A. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 2000, 100, 423–434. [Google Scholar] [CrossRef]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.T.; Zhao, Z.; Ingham, P.W. Hedgehog signalling. Development 2016, 143, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog signal transduction network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Rohatgi, R. G-protein-coupled receptors, Hedgehog signaling and primary cilia. Semin. Cell Dev. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J. Historical review: A brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol. Sci. 2004, 25, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.M.; Sung, J.Y.; Hebert, T.E. Gbetagamma subunits-Different spaces, different faces. Pharmacol. Res. Off. J. Ital. Pharmacol. Soc. 2016, 111, 434–441. [Google Scholar]
- Dupre, D.J.; Robitaille, M.; Rebois, R.V.; Hebert, T.E. The role of Gbetagamma subunits in the organization, assembly, and function of GPCR signaling complexes. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 31–56. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Alcedo, J.; Ayzenzon, M.; Von Ohlen, T.; Noll, M.; Hooper, J.E. The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell 1996, 86, 221–232. [Google Scholar] [CrossRef]
- Van den Heuvel, M.; Ingham, P.W. smoothened encodes a receptor-like serpentine protein required for hedgehog signalling. Nature 1996, 382, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, M.; McMahon, A.P. The effect of pertussis toxin on zebrafish development: A possible role for inhibitory G-proteins in hedgehog signaling. Dev. Biol. 1998, 194, 166–171. [Google Scholar] [CrossRef] [PubMed]
- DeCamp, D.L.; Thompson, T.M.; de Sauvage, F.J.; Lerner, M.R. Smoothened activates Galphai-mediated signaling in frog melanophores. J. Biol. Chem. 2000, 275, 26322–26327. [Google Scholar] [CrossRef] [PubMed]
- Low, W.C.; Wang, C.; Pan, Y.; Huang, X.Y.; Chen, J.K.; Wang, B. The decoupling of Smoothened from Galphai proteins has little effect on Gli3 protein processing and Hedgehog-regulated chick neural tube patterning. Dev. Biol. 2008, 321, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Ogden, S.K.; Fei, D.L.; Schilling, N.S.; Ahmed, Y.F.; Hwa, J.; Robbins, D.J. G protein Galphai functions immediately downstream of Smoothened in Hedgehog signalling. Nature 2008, 456, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Barzi, M.; Kostrz, D.; Menendez, A.; Pons, S. Sonic Hedgehog-induced proliferation requires specific Galpha inhibitory proteins. J. Biol. Chem. 2011, 286, 8067–8074. [Google Scholar] [CrossRef] [PubMed]
- Vuolo, L.; Herrera, A.; Torroba, B.; Menendez, A.; Pons, S. Ciliary adenylyl cyclases control the Hedgehog pathway. J. Cell Sci. 2015, 128, 2928–2937. [Google Scholar] [CrossRef] [PubMed]
- Riobo, N.A.; Saucy, B.; Dilizio, C.; Manning, D.R. Activation of heterotrimeric G proteins by Smoothened. Proc. Natl. Acad. Sci. USA 2006, 103, 12607–12612. [Google Scholar] [CrossRef] [PubMed]
- Polizio, A.H.; Chinchilla, P.; Chen, X.; Kim, S.; Manning, D.R.; Riobo, N.A. Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J. Biol. Chem. 2011, 286, 19589–19596. [Google Scholar] [CrossRef] [PubMed]
- Meyer, G.; Feldman, E.L. Signaling mechanisms that regulate actin-based motility processes in the nervous system. J. Neurochem. 2002, 83, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; Damhofer, H.; Roelink, H. Hedgehog-stimulated chemotaxis is mediated by smoothened located outside the primary cilium. Sci. Signal. 2012, 5, ra60. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; Borensztajn, K.S.; Roelink, H.; Peppelenbosch, M.P.; Spek, C.A. Sonic hedgehog induces transcription-independent cytoskeletal rearrangement and migration regulated by arachidonate metabolites. Cell Signal. 2007, 19, 2596–2604. [Google Scholar] [CrossRef] [PubMed]
- Chinchilla, P.; Xiao, L.; Kazanietz, M.G.; Riobo, N.A. Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle 2010, 9, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Cao, J.; He, X.; Serra, R.; Qu, J.; Cao, X.; Yang, S. Ciliary IFT80 balances canonical versus non-canonical hedgehog signalling for osteoblast differentiation. Nat. Commun. 2016, 7, 11024. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; Peppelenbosch, M.P.; Spek, C.A.; Roelink, H. Leukotriene synthesis is required for hedgehog-dependent neurite projection in neuralized embryoid bodies but not for motor neuron differentiation. Stem Cells 2008, 26, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Merchan, P.; Bribian, A.; Sanchez-Camacho, C.; Lezameta, M.; Bovolenta, P.; de Castro, F. Sonic hedgehog promotes the migration and proliferation of optic nerve oligodendrocyte precursors. Mol. Cell. Neurosci. 2007, 36, 355–368. [Google Scholar] [CrossRef] [PubMed]
- De Ramon Francas, G.; Zuniga, N.R.; Stoeckli, E.T. The spinal cord shows the way—How axons navigate intermediate targets. Dev. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Charron, F.; Stein, E.; Jeong, J.; McMahon, A.P.; Tessier-Lavigne, M. The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell 2003, 113, 11–23. [Google Scholar] [CrossRef]
- Yam, P.T.; Langlois, S.D.; Morin, S.; Charron, F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron 2009, 62, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Okada, A.; Charron, F.; Morin, S.; Shin, D.S.; Wong, K.; Fabre, P.J.; Tessier-Lavigne, M.; McConnell, S.K. Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature 2006, 444, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, D.K.; Luttrell, L.M. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene 2004, 23, 7969–7978. [Google Scholar] [CrossRef] [PubMed]
- Bourikas, D.; Pekarik, V.; Baeriswyl, T.; Grunditz, A.; Sadhu, R.; Nardo, M.; Stoeckli, E.T. Sonic hedgehog guides commissural axons along the longitudinal axis of the spinal cord. Nat. Neurosci. 2005, 8, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Parra, L.M.; Zou, Y. Sonic hedgehog induces response of commissural axons to Semaphorin repulsion during midline crossing. Nat. Neurosci. 2010, 13, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Song, S.K.; Choi, S.Y.; Kim, K.T. Opposing effects of protein kinase A and C on capacitative calcium entry into HL-60 promyelocytes. Biochem. Pharmacol. 1998, 56, 561–567. [Google Scholar] [CrossRef]
- Yam, P.T.; Kent, C.B.; Morin, S.; Farmer, W.T.; Alchini, R.; Lepelletier, L.; Colman, D.R.; Tessier-Lavigne, M.; Fournier, A.E.; Charron, F. 14–3-3 proteins regulate a cell-intrinsic switch from sonic hedgehog-mediated commissural axon attraction to repulsion after midline crossing. Neuron 2012, 76, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Martinelli, D.C.; Zheng, X.; Tessier-Lavigne, M.; Fan, C.M. Gas1 is a receptor for sonic hedgehog to repel enteric axons. Proc. Natl. Acad. Sci. USA 2015, 112, E73–E80. [Google Scholar] [CrossRef] [PubMed]
- Learte, A.R.; Hidalgo, A. The role of glial cells in axon guidance, fasciculation and targeting. Adv. Exp. Med. Biol. 2007, 621, 156–166. [Google Scholar] [PubMed]
- Trousse, F.; Marti, E.; Gruss, P.; Torres, M.; Bovolenta, P. Control of retinal ganglion cell axon growth: A new role for Sonic hedgehog. Development 2001, 128, 3927–3936. [Google Scholar] [PubMed]
- Guo, D.; Standley, C.; Bellve, K.; Fogarty, K.; Bao, Z.Z. Protein kinase Calpha and integrin-linked kinase mediate the negative axon guidance effects of Sonic hedgehog. Mol. Cell. Neurosci. 2012, 50, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Belgacem, Y.H.; Borodinsky, L.N. Sonic hedgehog signaling is decoded by calcium spike activity in the developing spinal cord. Proc. Natl. Acad. Sci. USA 2011, 108, 4482–4487. [Google Scholar] [CrossRef] [PubMed]
- Borodinsky, L.N.; Root, C.M.; Cronin, J.A.; Sann, S.B.; Gu, X.; Spitzer, N.C. Activity-dependent homeostatic specification of transmitter expression in embryonic neurons. Nature 2004, 429, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Marek, K.W.; Kurtz, L.M.; Spitzer, N.C. cJun integrates calcium activity and tlx3 expression to regulate neurotransmitter specification. Nat. Neurosci. 2010, 13, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.S.; Stepanchick, A.N.; Tewson, P.H.; Hartle, C.M.; Zhang, J.; Quinn, A.M.; Hughes, T.E.; Mirshahi, T. Cilia have high cAMP levels that are inhibited by Sonic Hedgehog-regulated calcium dynamics. Proc. Natl. Acad. Sci. USA 2016, 113, 13069–13074. [Google Scholar] [CrossRef] [PubMed]
- Delling, M.; DeCaen, P.G.; Doerner, J.F.; Febvay, S.; Clapham, D.E. Primary cilia are specialized calcium signalling organelles. Nature 2013, 504, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Ulloa, F.; Briscoe, J. Morphogens and the control of cell proliferation and patterning in the spinal cord. Cell Cycle 2007, 6, 2640–2649. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, H.; Visbal, A.P.; Obeid, N.F.; Ta, A.Q.; Faruki, A.A.; Wu, M.-F.; Hilsenbeck, S.G.; Shaw, C.A.; Yu, P.; Plummer, N.W.; et al. An essential role for G-alpha-i2 in Smoothened-stimulated epithelial cell proliferation in the mammary gland. Sci. Signal. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Teperino, R.; Aberger, F.; Esterbauer, H.; Riobo, N.; Pospisilik, J.A. Canonical and non-canonical Hedgehog signalling and the control of metabolism. Semin. Cell Dev. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.M.; Gao, X.; McKay, J.; McKay, R.; Salo, Z.; Graff, J.M. Hedgehog signaling plays a conserved role in inhibiting fat formation. Cell Metab. 2006, 3, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Kopinke, D.; Roberson, E.C.; Reiter, J.F. Ciliary Hedgehog Signaling Restricts Injury-Induced Adipogenesis. Cell 2017, 170, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Pospisilik, J.A.; Schramek, D.; Schnidar, H.; Cronin, S.J.; Nehme, N.T.; Zhang, X.; Knauf, C.; Cani, P.D.; Aumayr, K.; Todoric, J.; et al. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell 2010, 140, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Matz-Soja, M.; Rennert, C.; Schonefeld, K.; Aleithe, S.; Boettger, J.; Schmidt-Heck, W.; Weiss, T.S.; Hovhannisyan, A.; Zellmer, S.; Kloting, N.; et al. Hedgehog signaling is a potent regulator of liver lipid metabolism and reveals a GLI-code associated with steatosis. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Teperino, R.; Amann, S.; Bayer, M.; McGee, S.L.; Loipetzberger, A.; Connor, T.; Jaeger, C.; Kammerer, B.; Winter, L.; Wiche, G.; et al. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell 2012, 151, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Marada, S.; Navarro, G.; Truong, A.; Stewart, D.P.; Arensdorf, A.M.; Nachtergaele, S.; Angelats, E.; Opferman, J.T.; Rohatgi, R.; McCormick, P.J.; et al. Functional Divergence in the Role of N-Linked Glycosylation in Smoothened Signaling. PLoS Genet. 2015, 11, e1005473. [Google Scholar] [CrossRef] [PubMed]
- Boscher, C.; Dennis, J.W.; Nabi, I.R. Glycosylation, galectins and cellular signaling. Curr. Opin. Cell Biol. 2011, 23, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Franco, I.; Gulluni, F.; Campa, C.C.; Costa, C.; Margaria, J.P.; Ciraolo, E.; Martini, M.; Monteyne, D.; De Luca, E.; Germena, G.; et al. PI3K class II alpha controls spatially restricted endosomal PtdIns3P and Rab11 activation to promote primary cilium function. Dev. Cell 2014, 28, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Barakat, M.T.; Humke, E.W.; Scott, M.P. Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol. Med. 2011, 16, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandit, T.; Ogden, S.K. Contributions of Noncanonical Smoothened Signaling During Embryonic Development. J. Dev. Biol. 2017, 5, 11. https://doi.org/10.3390/jdb5040011
Pandit T, Ogden SK. Contributions of Noncanonical Smoothened Signaling During Embryonic Development. Journal of Developmental Biology. 2017; 5(4):11. https://doi.org/10.3390/jdb5040011
Chicago/Turabian StylePandit, Tanushree, and Stacey K. Ogden. 2017. "Contributions of Noncanonical Smoothened Signaling During Embryonic Development" Journal of Developmental Biology 5, no. 4: 11. https://doi.org/10.3390/jdb5040011
APA StylePandit, T., & Ogden, S. K. (2017). Contributions of Noncanonical Smoothened Signaling During Embryonic Development. Journal of Developmental Biology, 5(4), 11. https://doi.org/10.3390/jdb5040011