



The Tumor Suppressor Adenomatous Polyposis Coli (apc) Is Required for Neural Crest-Dependent Craniofacial Development in Zebrafish

 , , ,
, , ,
Abstract
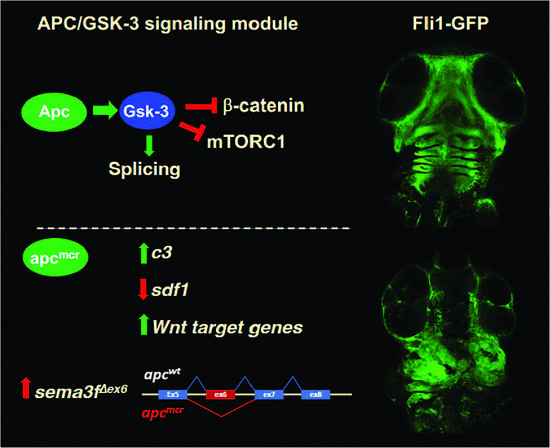
1. Introduction
2. Methods
2.1. Zebrafish Husbandry and Methods
2.2. RNA Isolation, RT-PCR, and Sequencing
2.3. Quantification of Alternative Spliced and Differentially Expressed Genes
2.4. Statistical Analysis and Downstream Bioinformatics
3. Results
3.1. The Tumor Suppressor Apc Is Required for the CNC Contribution to Craniofacial Structures
3.2. Increased Expression of Cytokines and Wnt Target Genes in apcmcr/mcr Larvae
3.3. Increased Expression of Complement c3 in apcmcr/mcr Larvae
3.4. An Oncogenic apc Mutation Alters mRNA Splicing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aybar, M.J.; Mayor, R. Early induction of neural crest cells: Lessons learned from frog, fish and chick. Curr. Opin. Genet. Dev. 2002, 12, 452–458. [Google Scholar] [CrossRef]
- Jain, R.; Rentschler, S.; Epstein, J.A. Notch and cardiac outflow tract development. Ann. N. Y. Acad. Sci. 2010, 1188, 184–190. [Google Scholar] [CrossRef]
- LaBonne, C.; Bronner-Fraser, M. Snail-related transcriptional repressors are required in Xenopus for both the induction of the neural crest and its subsequent migration. Dev. Biol. 2000, 221, 195–205. [Google Scholar] [CrossRef]
- Mayor, R.; Theveneau, E. The neural crest. Development 2013, 140, 2247–2251. [Google Scholar] [CrossRef]
- Shellard, A.; Mayor, R. Chemotaxis during neural crest migration. Semin. Cell Dev. Biol. 2016, 55, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Klymkowsky, M.W.; Rossi, C.C.; Artinger, K.B. Mechanisms driving neural crest induction and migration in the zebrafish and Xenopus laevis. Cell Adh. Migr. 2010, 4, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Bajanca, F.; Gouignard, N.; Colle, C.; Parsons, M.; Mayor, R.; Theveneau, E. In vivo topology converts competition for cell-matrix adhesion into directional migration. Nat. Commun. 2019, 10, 1518. [Google Scholar] [CrossRef]
- Barriga, E.H.; Franze, K.; Charras, G.; Mayor, R. Tissue stiffening coordinates morphogenesis by triggering collective cell migration in vivo. Nature 2018, 554, 523–527. [Google Scholar] [CrossRef]
- Canales Coutino, B.; Mayor, R. The mechanosensitive channel Piezo1 cooperates with semaphorins to control neural crest migration. Development 2021, 148, dev200001. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Fontaine, C.; Matthews, H.K.; Kuriyama, S.; Moreno, M.; Dunn, G.A.; Parsons, M.; Stern, C.D.; Mayor, R. Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature 2008, 456, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Fontaine, C.; Theveneau, E.; Tzekou, A.; Tada, M.; Woods, M.; Page, K.M.; Parsons, M.; Lambris, J.D.; Mayor, R. Complement fragment C3a controls mutual cell attraction during collective cell migration. Dev. Cell 2011, 21, 1026–1037. [Google Scholar] [CrossRef]
- Mancilla, A.; Mayor, R. Neural crest formation in Xenopus laevis: Mechanisms of Xslug induction. Dev. Biol. 1996, 177, 580–589. [Google Scholar] [CrossRef]
- Rabadan, M.A.; Herrera, A.; Fanlo, L.; Usieto, S.; Carmona-Fontaine, C.; Barriga, E.H.; Mayor, R.; Pons, S.; Marti, E. Delamination of neural crest cells requires transient and reversible Wnt inhibition mediated by Dact1/2. Development 2016, 143, 2194–2205. [Google Scholar] [CrossRef]
- Shellard, A.; Mayor, R. Collective durotaxis along a self-generated stiffness gradient in vivo. Nature 2021, 600, 690–694. [Google Scholar] [CrossRef]
- Shellard, A.; Szabo, A.; Trepat, X.; Mayor, R. Supracellular contraction at the rear of neural crest cell groups drives collective chemotaxis. Science 2018, 362, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Theveneau, E.; Marchant, L.; Kuriyama, S.; Gull, M.; Moepps, B.; Parsons, M.; Mayor, R. Collective chemotaxis requires contact-dependent cell polarity. Dev. Cell 2010, 19, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.; Mayor, R. Mechanisms of Neural Crest Migration. Annu. Rev. Genet. 2018, 52, 43–63. [Google Scholar] [CrossRef]
- Theveneau, E.; Mayor, R. Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol. 2012, 366, 34–54. [Google Scholar] [CrossRef]
- Theveneau, E.; Duband, J.L.; Altabef, M. Ets-1 confers cranial features on neural crest delamination. PLoS ONE 2007, 2, e1142. [Google Scholar] [CrossRef]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed]
- Abercrombie, M.; Heaysman, J.E. Observations on the social behaviour of cells in tissue culture. I. Speed of movement of chick heart fibroblasts in relation to their mutual contacts. Exp. Cell Res. 1953, 5, 111–131. [Google Scholar] [CrossRef]
- Ji, Y.; Hao, H.; Reynolds, K.; McMahon, M.; Zhou, C.J. Wnt Signaling in Neural Crest Ontogenesis and Oncogenesis. Cells 2019, 8, 1173. [Google Scholar] [CrossRef]
- Hutchins, E.J.; Bronner, M.E. Draxin acts as a molecular rheostat of canonical Wnt signaling to control cranial neural crest EMT. J. Cell Biol. 2018, 217, 3683–3697. [Google Scholar] [CrossRef]
- Maj, E.; Kunneke, L.; Loresch, E.; Grund, A.; Melchert, J.; Pieler, T.; Aspelmeier, T.; Borchers, A. Controlled levels of canonical Wnt signaling are required for neural crest migration. Dev. Biol. 2016, 417, 77–90. [Google Scholar] [CrossRef]
- Shull, L.C.; Lencer, E.S.; Kim, H.M.; Goyama, S.; Kurokawa, M.; Costello, J.C.; Jones, K.; Artinger, K.B. PRDM paralogs antagonistically balance Wnt/beta-catenin activity during craniofacial chondrocyte differentiation. Development 2022, 149, dev200082. [Google Scholar] [CrossRef] [PubMed]
- Valvezan, A.J.; Huang, J.; Lengner, C.J.; Pack, M.; Klein, P.S. Oncogenic mutations in adenomatous polyposis coli (Apc) activate mechanistic target of rapamycin complex 1 (mTORC1) in mice and zebrafish. Dis. Model. Mech. 2014, 7, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Valvezan, A.J.; Klein, P.S. GSK-3 and Wnt Signaling in Neurogenesis and Bipolar Disorder. Front. Mol. Neurosci. 2012, 5, 1. [Google Scholar] [CrossRef]
- Valvezan, A.J.; Zhang, F.; Diehl, J.A.; Klein, P.S. Adenomatous polyposis coli (APC) regulates multiple signaling pathways by enhancing glycogen synthase kinase-3 (GSK-3) activity. J. Biol. Chem. 2012, 287, 3823–3832. [Google Scholar] [CrossRef]
- Ji, L.; Lu, B.; Wang, Z.; Yang, Z.; Reece-Hoyes, J.; Russ, C.; Xu, W.; Cong, F. Identification of ICAT as an APC Inhibitor, Revealing Wnt-Dependent Inhibition of APC-Axin Interaction. Mol. Cell 2018, 72, 37–47.e34. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Polakis, P. Reversible modification of adenomatous polyposis coli (APC) with K63-linked polyubiquitin regulates the assembly and activity of the beta-catenin destruction complex. J. Biol. Chem. 2012, 287, 28552–28563. [Google Scholar] [CrossRef]
- Hurlstone, A.F.; Haramis, A.P.; Wienholds, E.; Begthel, H.; Korving, J.; Van Eeden, F.; Cuppen, E.; Zivkovic, D.; Plasterk, R.H.; Clevers, H. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature 2003, 425, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Goessling, W.; North, T.E.; Lord, A.M.; Ceol, C.; Lee, S.; Weidinger, G.; Bourque, C.; Strijbosch, R.; Haramis, A.P.; Puder, M.; et al. APC mutant zebrafish uncover a changing temporal requirement for wnt signaling in liver development. Dev. Biol. 2008, 320, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Westerfield, M. The Zebrafish Book a Guide for the Laboratory Use of Zebrafish Danio (Brachydanio) Rerio; Institute of Neuroscience, University of Oregon: Eugene, OR, USA, 1993. [Google Scholar]
- Thisse, C.; Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008, 3, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Shinde, M.Y.; Sidoli, S.; Kulej, K.; Mallory, M.J.; Radens, C.M.; Reicherter, A.L.; Myers, R.L.; Barash, Y.; Lynch, K.W.; Garcia, B.A.; et al. Phosphoproteomics reveals that glycogen synthase kinase-3 phosphorylates multiple splicing factors and is associated with alternative splicing. J. Biol. Chem. 2017, 292, 18240–18255. [Google Scholar] [CrossRef]
- Nguyen-McCarty, M.; Klein, P.S. Autophagy is a signature of a signaling network that maintains hematopoietic stem cells. PLoS ONE 2017, 12, e0177054. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Lawson, N.D.; Weinstein, B.M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 2002, 248, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Schulte-Merker, S.; Hammerschmidt, M.; Beuchle, D.; Cho, K.W.; De Robertis, E.M.; Nusslein-Volhard, C. Expression of zebrafish goosecoid and no tail gene products in wild-type and mutant no tail embryos. Development 1994, 120, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. The Wnt Gene Homepage. 1999. Available online: http://www.stanford.edu/~rnusse/wntwindow.html (accessed on 15 February 2019).
- Olesnicky Killian, E.C.; Birkholz, D.A.; Artinger, K.B. A role for chemokine signaling in neural crest cell migration and craniofacial development. Dev. Biol. 2009, 333, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Belmadani, A.; Tran, P.B.; Ren, D.; Assimacopoulos, S.; Grove, E.A.; Miller, R.J. The chemokine stromal cell-derived factor-1 regulates the migration of sensory neuron progenitors. J. Neurosci. 2005, 25, 3995–4003. [Google Scholar] [CrossRef]
- Heyd, F.; Lynch, K.W. Phosphorylation-dependent regulation of PSF by GSK3 controls CD45 alternative splicing. Mol. Cell 2010, 40, 126–137. [Google Scholar] [CrossRef]
- Hernandez, F.; Perez, M.; Lucas, J.J.; Mata, A.M.; Bhat, R.; Avila, J. Glycogen synthase kinase-3 plays a crucial role in tau exon 10 splicing and intranuclear distribution of SC35. Implications for Alzheimer’s disease. J. Biol. Chem. 2004, 279, 3801–3806. [Google Scholar] [CrossRef]
- Wang, S.B.; Venkatraman, V.; Crowgey, E.L.; Liu, T.; Fu, Z.; Holewinski, R.; Ranek, M.; Kass, D.A.; O’Rourke, B.; Van Eyk, J.E. Protein S-Nitrosylation Controls Glycogen Synthase Kinase 3beta Function Independent of Its Phosphorylation State. Circ. Res. 2018, 122, 1517–1531. [Google Scholar] [CrossRef]
- Beauchamp, M.C.; Alam, S.S.; Kumar, S.; Jerome-Majewska, L.A. Spliceosomopathies and neurocristopathies: Two sides of the same coin? Dev. Dyn. 2020, 249, 924–945. [Google Scholar] [CrossRef]
- Griffin, C.; Saint-Jeannet, J.P. Spliceosomopathies: Diseases and mechanisms. Dev. Dyn. 2020, 249, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Cherry, S.; Lynch, K.W. Alternative splicing and cancer: Insights, opportunities, and challenges from an expanding view of the transcriptome. Genes Dev. 2020, 34, 1005–1016. [Google Scholar] [CrossRef]
- Vaquero-Garcia, J.; Barrera, A.; Gazzara, M.R.; Gonzalez-Vallinas, J.; Lahens, N.F.; Hogenesch, J.B.; Lynch, K.W.; Barash, Y. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife 2016, 5, e11752. [Google Scholar] [CrossRef] [PubMed]
- Gammill, L.S.; Gonzalez, C.; Bronner-Fraser, M. Neuropilin 2/semaphorin 3F signaling is essential for cranial neural crest migration and trigeminal ganglion condensation. Dev. Neurobiol. 2007, 67, 47–56. [Google Scholar] [CrossRef]
- Osborne, N.J.; Begbie, J.; Chilton, J.K.; Schmidt, H.; Eickholt, B.J. Semaphorin/neuropilin signaling influences the positioning of migratory neural crest cells within the hindbrain region of the chick. Dev. Dyn. 2005, 232, 939–949. [Google Scholar] [CrossRef]
- Parry, D.A.; Logan, C.V.; Stegmann, A.P.; Abdelhamed, Z.A.; Calder, A.; Khan, S.; Bonthron, D.T.; Clowes, V.; Sheridan, E.; Ghali, N.; et al. SAMS, a syndrome of short stature, auditory-canal atresia, mandibular hypoplasia, and skeletal abnormalities is a unique neurocristopathy caused by mutations in Goosecoid. Am. J. Hum. Genet. 2013, 93, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Perez, J.A.; Mallo, M.; Gendron-Maguire, M.; Gridley, T.; Behringer, R.R. Goosecoid is not an essential component of the mouse gastrula organizer but is required for craniofacial and rib development. Development 1995, 121, 3005–3012. [Google Scholar] [CrossRef]
- Yamada, G.; Mansouri, A.; Torres, M.; Stuart, E.T.; Blum, M.; Schultz, M.; De Robertis, E.M.; Gruss, P. Targeted mutation of the murine goosecoid gene results in craniofacial defects and neonatal death. Development 1995, 121, 2917–2922. [Google Scholar] [CrossRef]
- Gonzalez Malagon, S.G.; Lopez Munoz, A.M.; Doro, D.; Bolger, T.G.; Poon, E.; Tucker, E.R.; Adel Al-Lami, H.; Krause, M.; Phiel, C.J.; Chesler, L.; et al. Glycogen synthase kinase 3 controls migration of the neural crest lineage in mouse and Xenopus. Nat. Commun. 2018, 9, 1126. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.E.; Jones, K.L.; Smith, F.J.; Williams, T.; Li, H. An Alternative Splicing Program for Mouse Craniofacial Development. Front. Physiol. 2020, 11, 1099. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.M.; Cho, M.T.; Telegrafi, A.; Wilson, A.; Brooks, S.; Botti, C.; Gowans, G.; Autullo, L.A.; Krishnamurthy, V.; Willing, M.C.; et al. Variants in HNRNPH2 on the X Chromosome Are Associated with a Neurodevelopmental Disorder in Females. Am. J. Hum. Genet. 2016, 99, 728–734. [Google Scholar] [CrossRef]
- Marques, F.; Tenney, J.; Duran, I.; Martin, J.; Nevarez, L.; Pogue, R.; Krakow, D.; Cohn, D.H.; Li, B. Altered mRNA Splicing, Chondrocyte Gene Expression and Abnormal Skeletal Development due to SF3B4 Mutations in Rodriguez Acrofacial Dysostosis. PLoS Genet. 2016, 12, e1006307. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Ewald, A.J. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Locker, J.; Sahai, E.; Segall, J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012, 14, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Simeonov, K.P.; Byrns, C.N.; Clark, M.L.; Norgard, R.J.; Martin, B.; Stanger, B.Z.; Shendure, J.; McKenna, A.; Lengner, C.J. Single-cell lineage tracing of metastatic cancer reveals selection of hybrid EMT states. Cancer Cell 2021, 39, 1150–1162.e1159. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Rupaimoole, R.; Choi, H.J.; Noh, K.; Chen, J.; Hu, Q.; Sood, A.K.; Afshar-Kharghan, V. Complement Component 3 Is Regulated by TWIST1 and Mediates Epithelial-Mesenchymal Transition. J. Immunol. 2016, 196, 1412–1418. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RT-qPCR | Forward | Reverse |
|---|---|---|
| CCL19 | CCCCATTGCAGCTACTGTATTC | AGGTGTTTTTCTCTGGTGGGG |
| IL-11a | CCGGTTCAAGTCTCTTCCAG | AGGTTTGCATGGAGCTGAGA |
| IL-11b | CATCTTATCCAAGCTATCATCCAG | GATCTCGGGTGCTGTCTGTC |
| LEPA | GGAACACATTGACGGGCAAA | ATGGGTTTGTCAGCGGGAAT |
| CBX7a | TGCGGTGGAGTCAATAACGAA | CAGGTGCTGTACTTAGGCGA |
| DKK1a | GTGGAGTTTGTCTGTCGTGC | AAGCTGACACACACCGTTGA |
| DKK1b | CACCGCAGCAGCCTTTAATC | CGCGAGACTGGAAGCAAAAC |
| NOTUM1a | CACCTGTAACGACGGGACTC | CCAGCCGCCCTCAAGAAATA |
| LFT1 | ACGCACGAGTGAGCATCTAC | CGTGAATGGGAATCAACCTGG |
| SALL1b | CTTCAGGGAGATAACCCGGC | CCATCGTATGATCGAGGAGAACA |
| SOCS3a | AAGCAGGGAAGACAAGAGCC | AGAGCTGGTCAAAAGAGCCTAT |
| GSC | CCTACAGGTTATGACAGCGCC | GACAAGGTGCCCACGTTCAT |
| EMILIN3a | GGCACAAGAACCACTGTGCAT | CCAAGCACACTTCATTGCCT |
| GAPDH | GTGGAGTCTACTGGTGTCTTC | GTGCAGGAGGCATTGCTTACA |
| C3A.1 | GACGCCCAACTTGAAACCAC | GCAACCTCAGGGATGGCATA |
| C3A.6 | ACCAGGAATGCCCTTCAGTG | ACCTTTGACTCCTCCGGGAT |
| RT-qPCR for splice forms: | ||
| SEMA3fa_ALL | ACTCAACAGCGTCTCAGCTT | TACTGGTCGGTCCTCATTGC |
| SEMA3fa_Long splice form | ACAACCCCATCTGCACCTATG | TCTGGTGAGGCACTAGGGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Jones, W.D.; Quesnel-Vallières, M.; Devadiga, S.A.; Lorent, K.; Valvezan, A.J.; Myers, R.L.; Li, N.; Lengner, C.J.; Barash, Y.; et al. The Tumor Suppressor Adenomatous Polyposis Coli (apc) Is Required for Neural Crest-Dependent Craniofacial Development in Zebrafish. J. Dev. Biol. 2023, 11, 29. https://doi.org/10.3390/jdb11030029
Liu X, Jones WD, Quesnel-Vallières M, Devadiga SA, Lorent K, Valvezan AJ, Myers RL, Li N, Lengner CJ, Barash Y, et al. The Tumor Suppressor Adenomatous Polyposis Coli (apc) Is Required for Neural Crest-Dependent Craniofacial Development in Zebrafish. Journal of Developmental Biology. 2023; 11(3):29. https://doi.org/10.3390/jdb11030029
Chicago/Turabian StyleLiu, Xiaolei, William D. Jones, Mathieu Quesnel-Vallières, Sudhish A. Devadiga, Kristin Lorent, Alexander J. Valvezan, Rebecca L. Myers, Ning Li, Christopher J. Lengner, Yoseph Barash, and et al. 2023. "The Tumor Suppressor Adenomatous Polyposis Coli (apc) Is Required for Neural Crest-Dependent Craniofacial Development in Zebrafish" Journal of Developmental Biology 11, no. 3: 29. https://doi.org/10.3390/jdb11030029
APA StyleLiu, X., Jones, W. D., Quesnel-Vallières, M., Devadiga, S. A., Lorent, K., Valvezan, A. J., Myers, R. L., Li, N., Lengner, C. J., Barash, Y., Pack, M., & Klein, P. S. (2023). The Tumor Suppressor Adenomatous Polyposis Coli (apc) Is Required for Neural Crest-Dependent Craniofacial Development in Zebrafish. Journal of Developmental Biology, 11(3), 29. https://doi.org/10.3390/jdb11030029