
The Core Splicing Factors EFTUD2, SNRPB and TXNL4A Are Essential for Neural Crest and Craniofacial Development

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Xenopus Embryos, Constructs and Microinjections
2.2. Whole-Mount In Situ Hybridization
2.3. RT-PCR Analysis
2.4. Alcian Blue Staining
2.5. TUNEL Assay
2.6. TNT Reaction
2.7. Statistical Method
3. Results
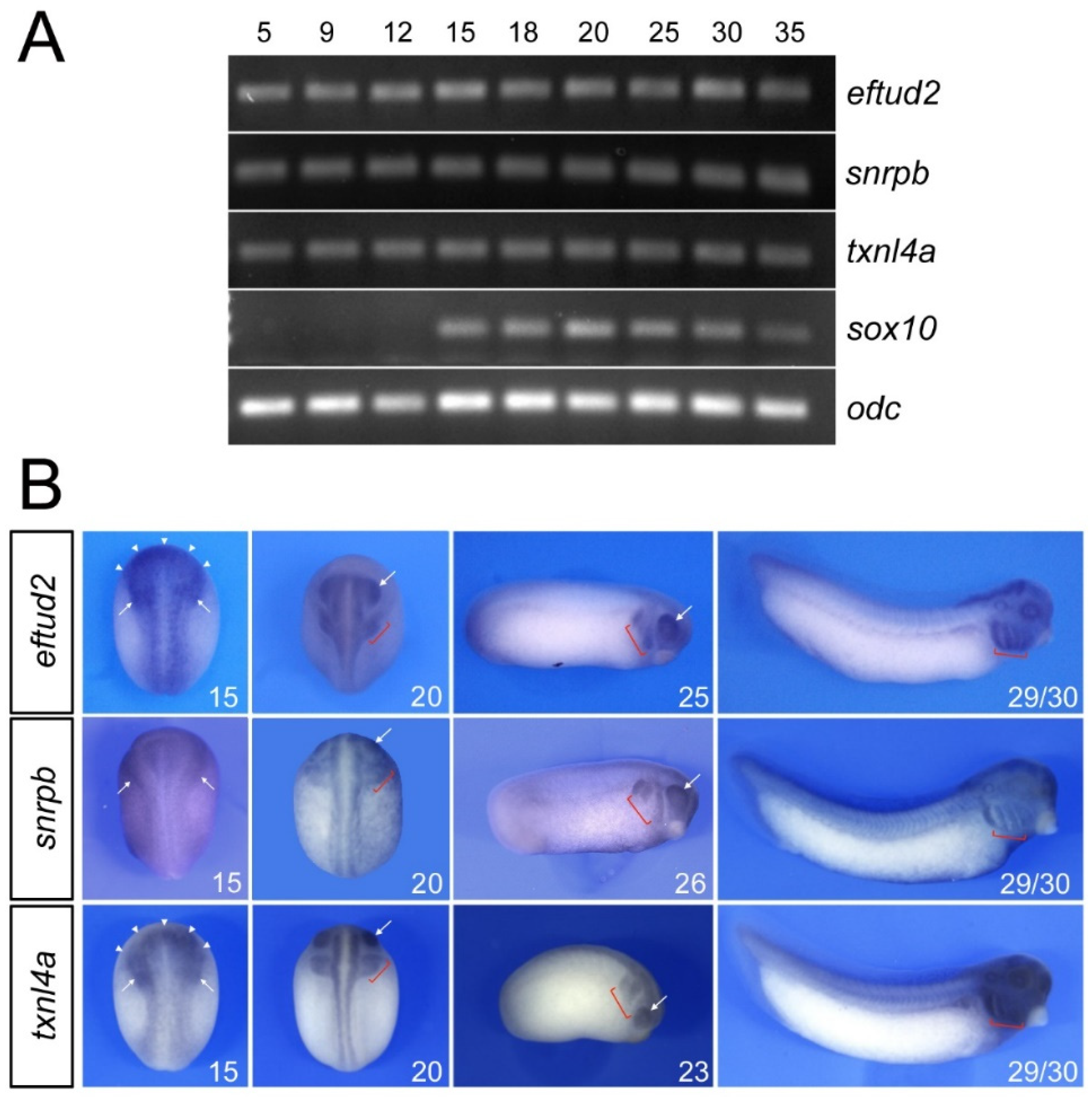
3.1. Developmental Expression of Eftud2, Snrpb and Txnl4a
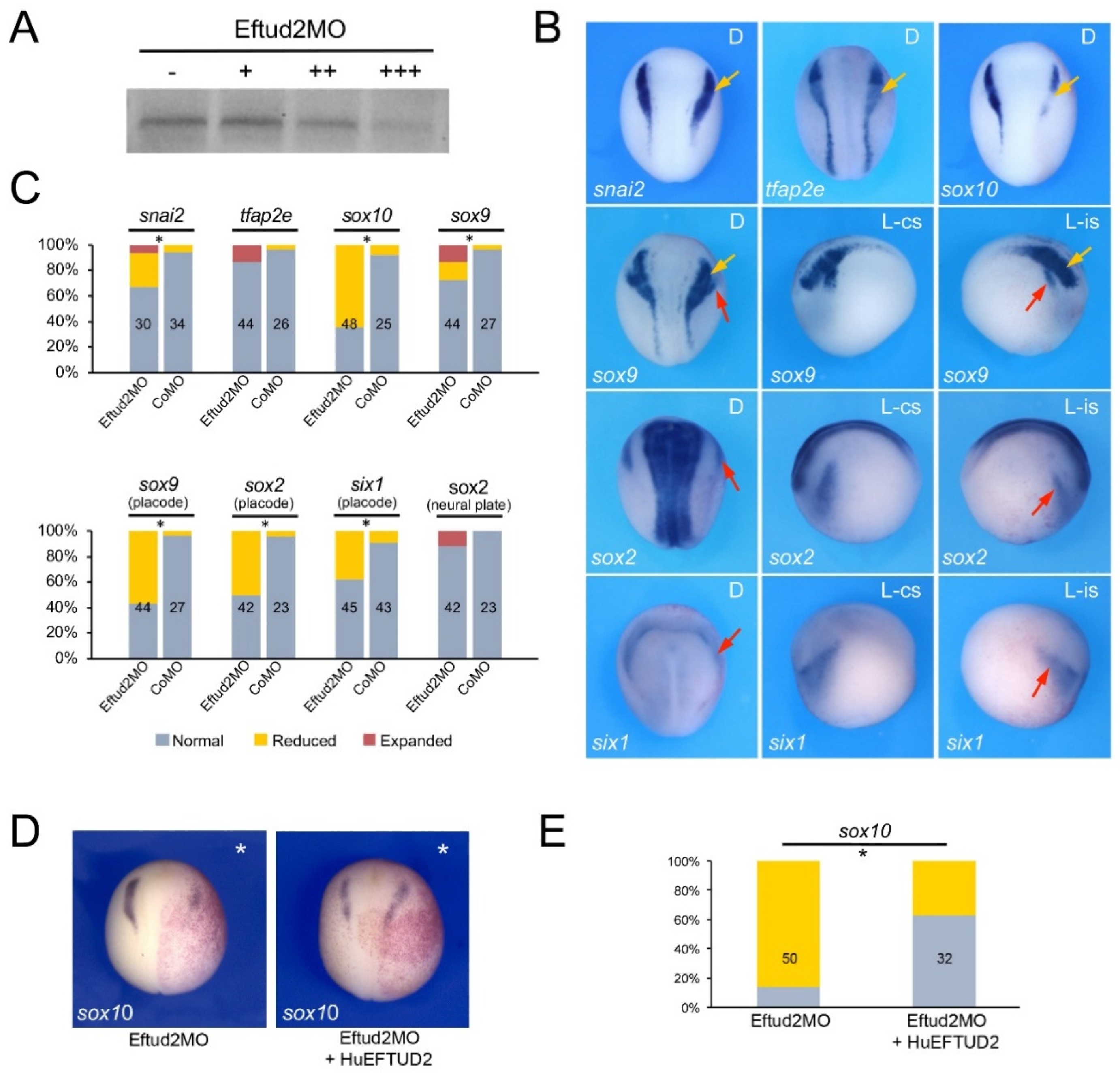
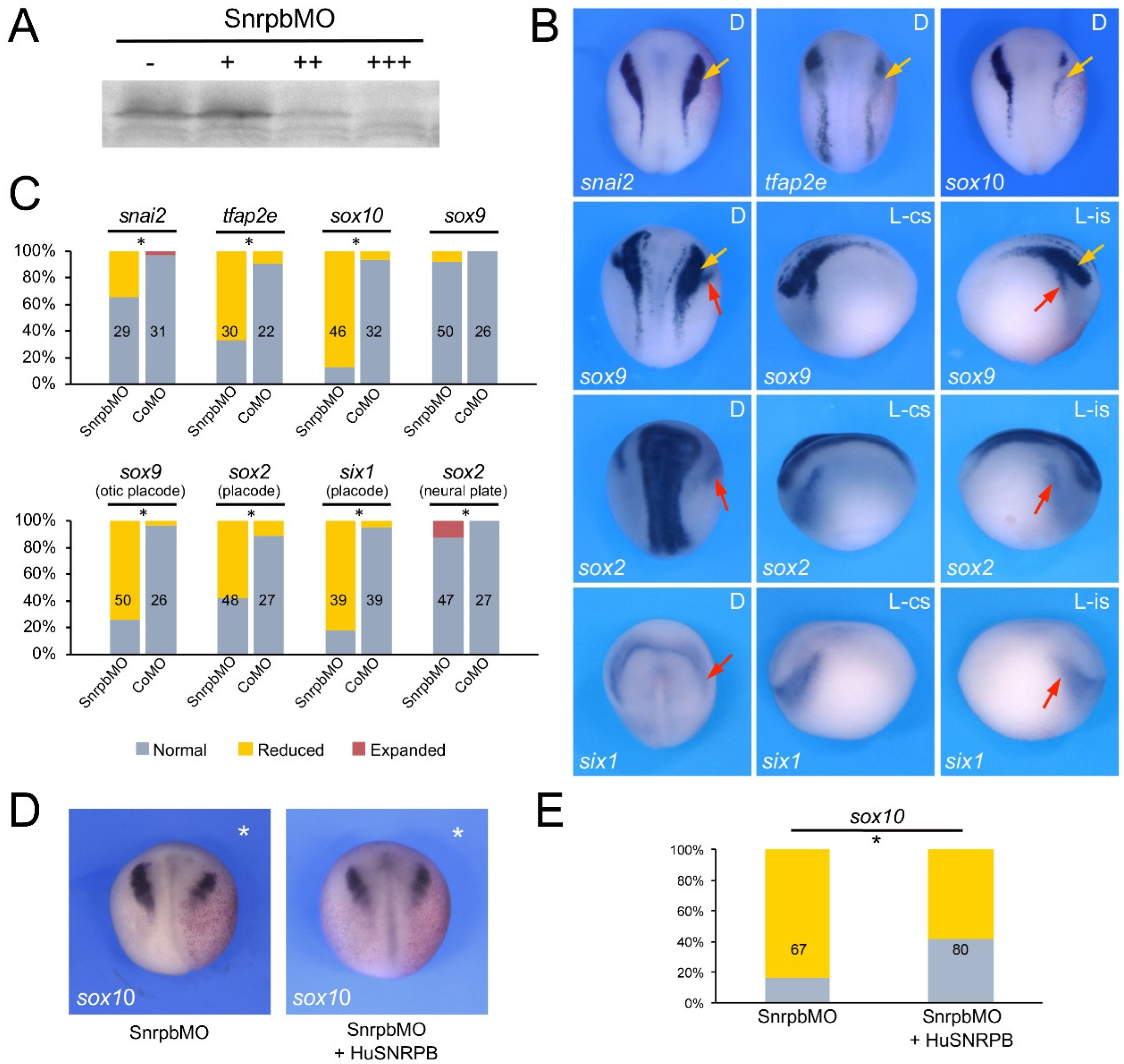
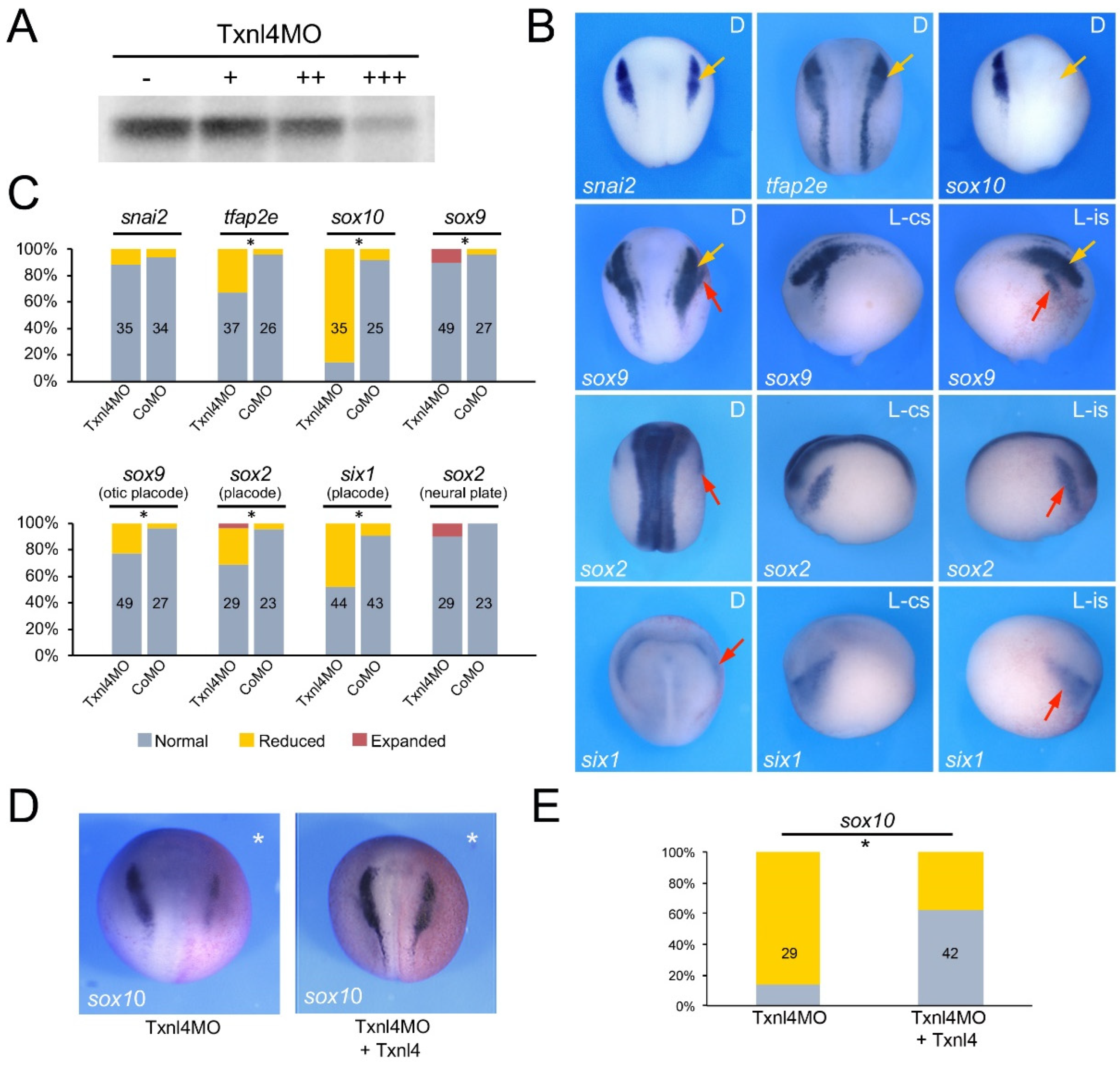
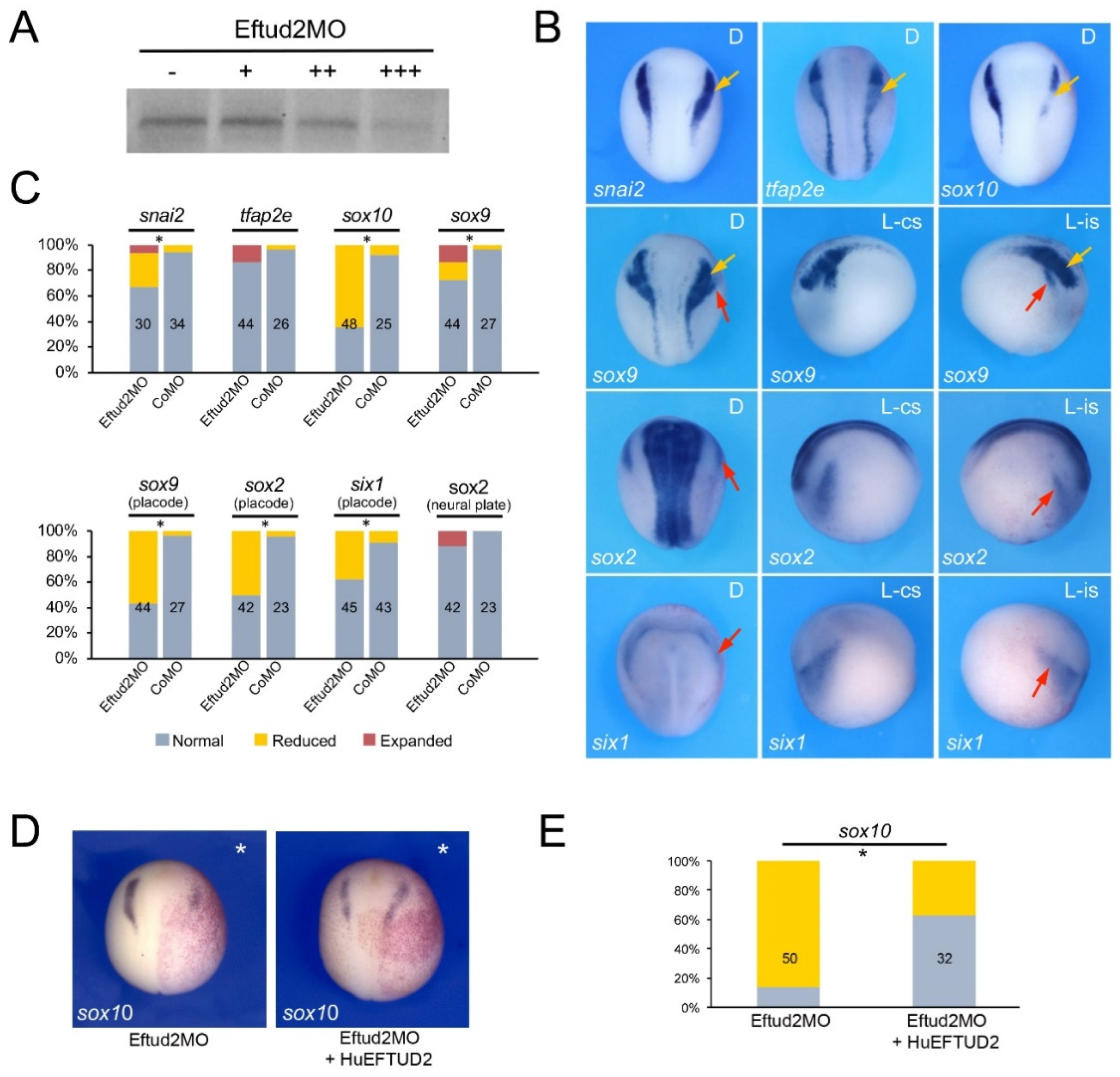
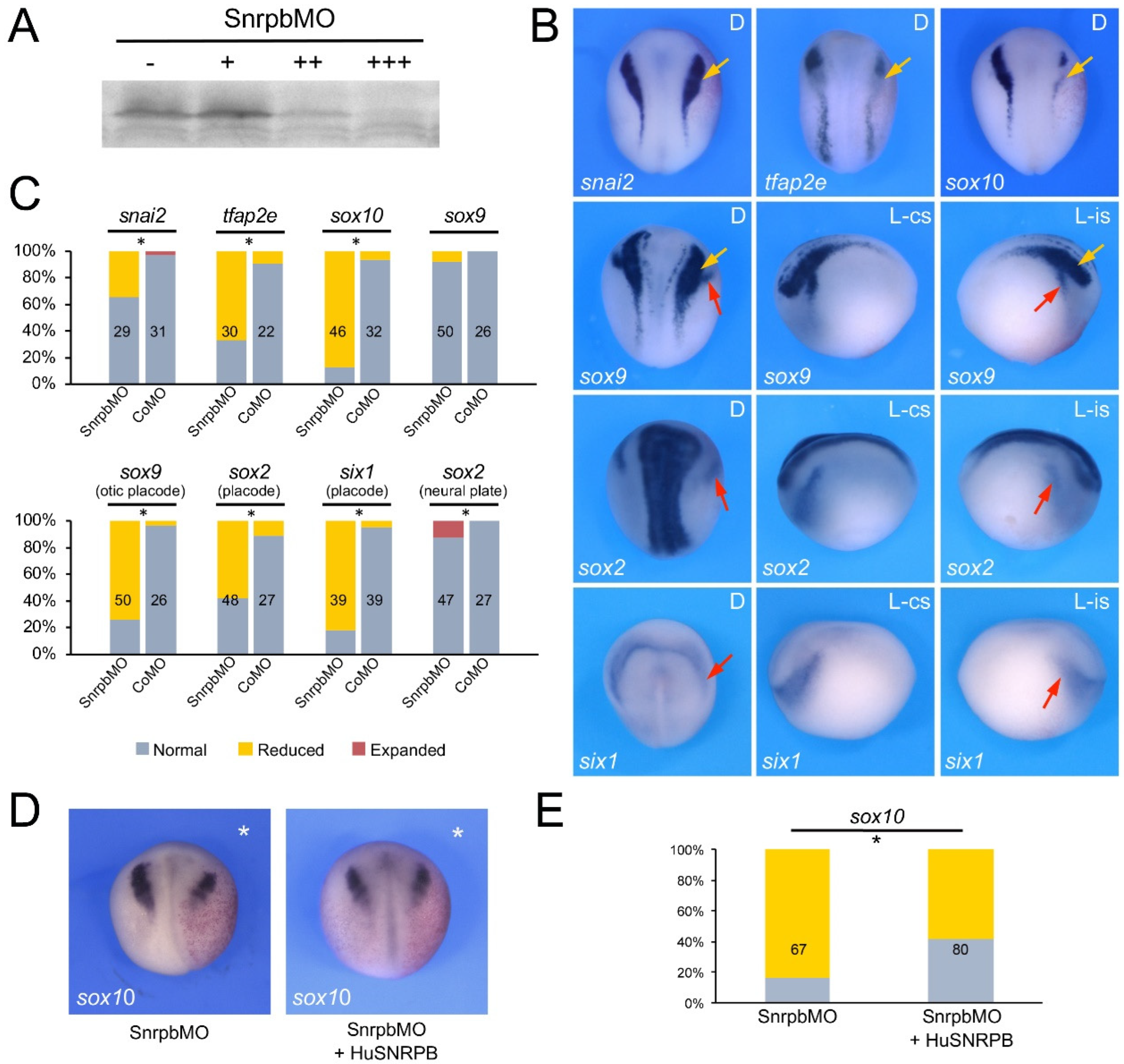
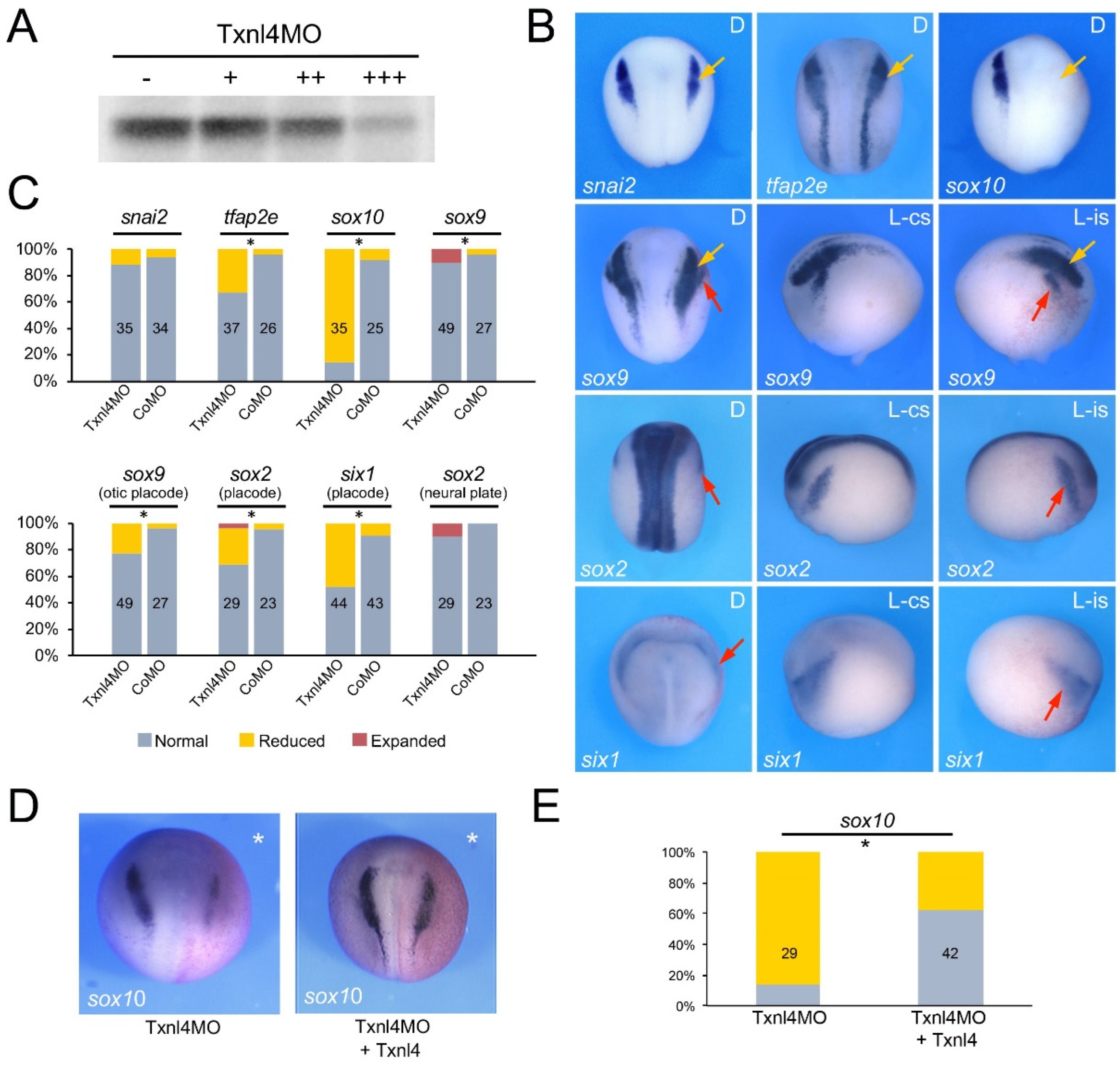
3.2. Eftud2, Snrpb and Txnl4a Knockdown Affect Neural Crest Formation
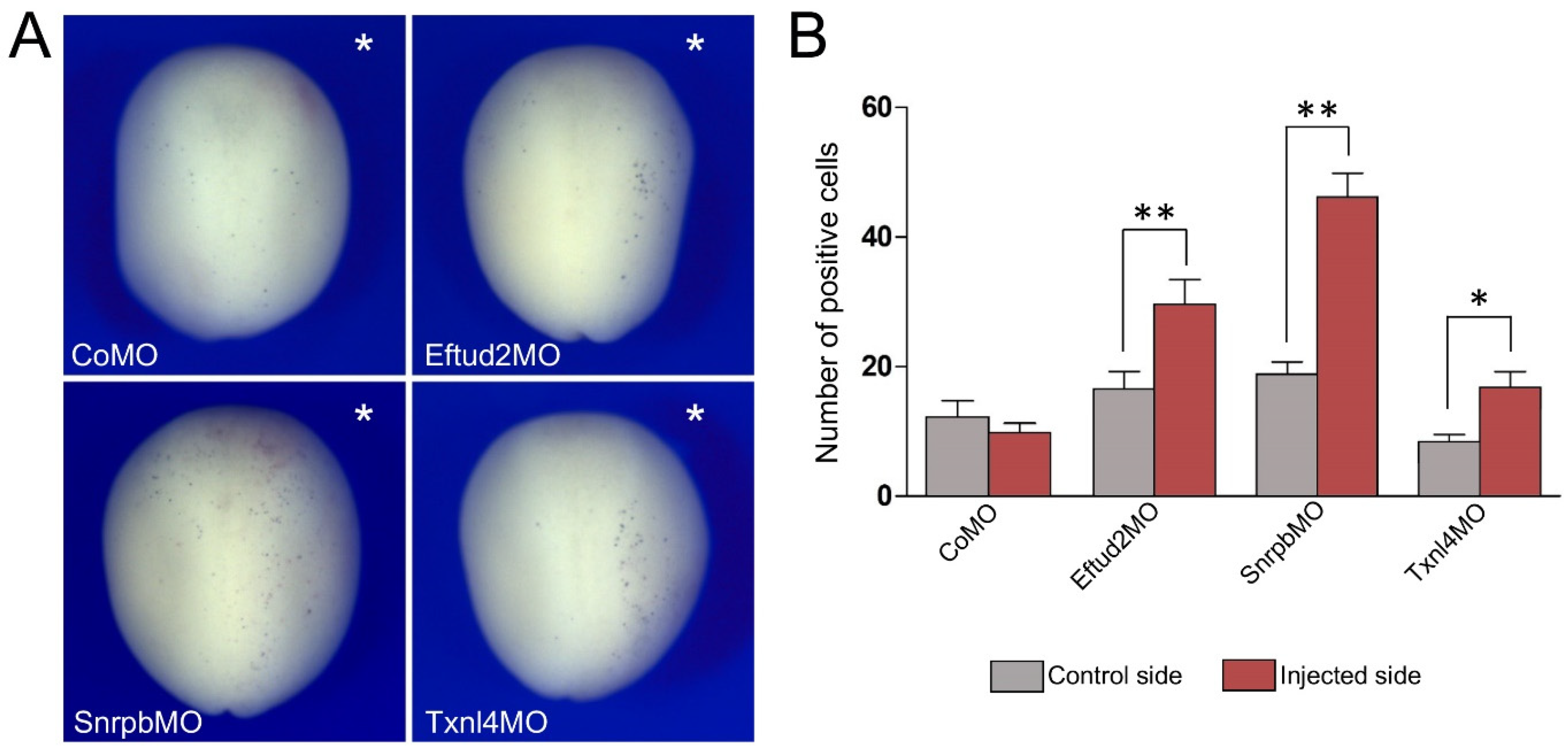
3.3. Increased TUNEL Positivity in Eftud2, Snrpb and Txnl4a Morphant Embryos
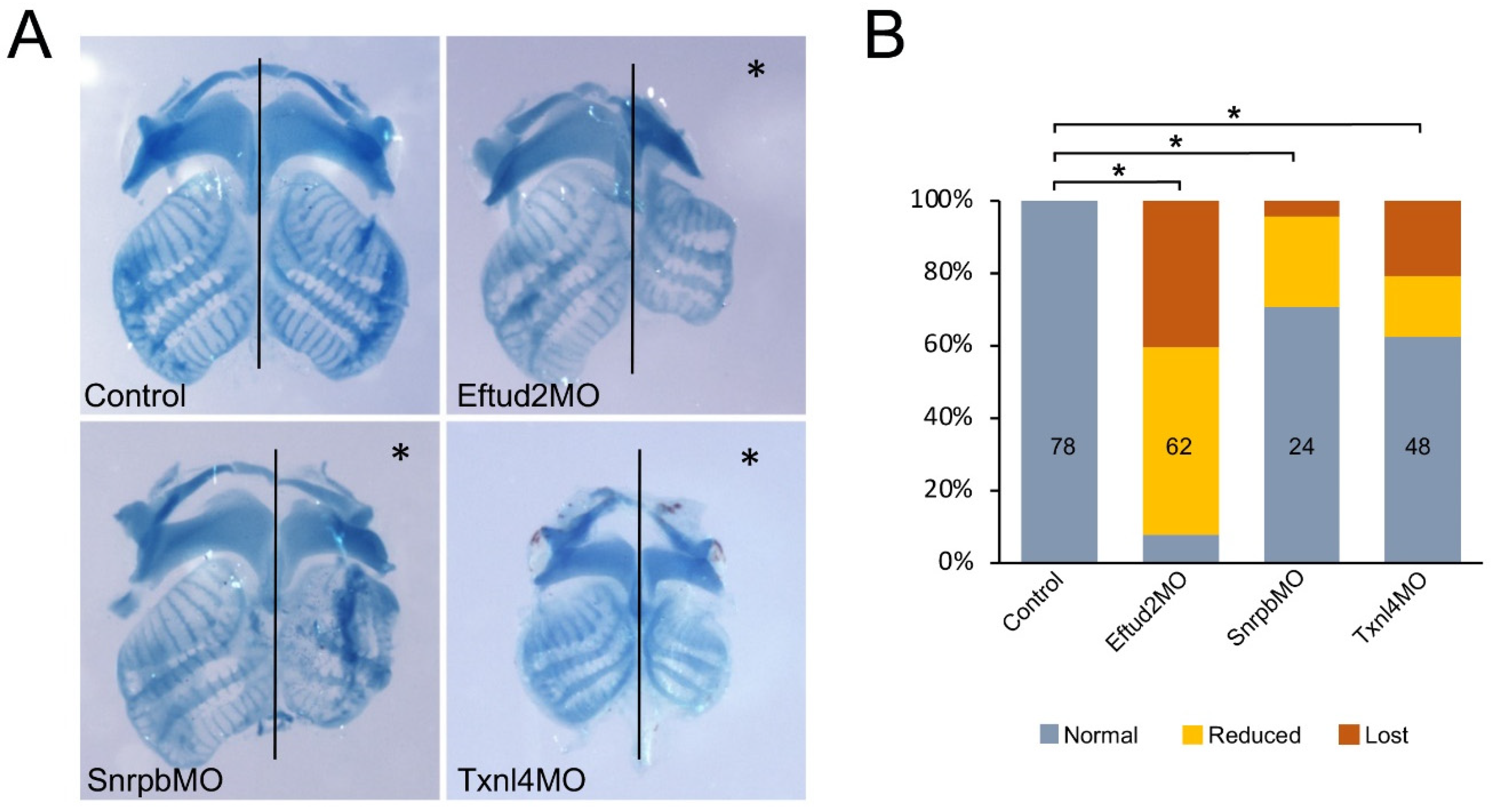
3.4. Eftud2, Snrpb and Txnl4a Knockdown Affect Craniofacial Development
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trainor, P.A.; Andrews, B.T. Facial dysostoses: Etiology, pathogenesis and management. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2013, 163, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Passos-Bueno, M.R.; Ornelas, C.C.; Fanganiello, R.D. Syndromes of the first and second pharyngeal arches: A Review. Am. J. Med. Genet. Part A 2009, 149, 1853–1859. [Google Scholar] [CrossRef]
- Minoux, M.; Rijli, F.M. Molecular mechanisms of cranial NC cell migration and patterning in craniofacial development. Development 2010, 137, 2605–2621. [Google Scholar] [CrossRef] [Green Version]
- Wieczorek, D. Human facial dysostoses. Clin. Genet. 2013, 83, 499–510. [Google Scholar] [CrossRef]
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Lehalle, D.; Wieczorek, D.; Zechi-Ceide, R.M.; Passos-Bueno, M.R.; Lyonnet, S.; Amiel, J.; Gordon, C.T. A review of craniofacial disorders caused by spliceosomal defects. Clin. Genet. 2015, 88, 405–415. [Google Scholar] [CrossRef]
- Beauchamp, M.-C.; Alam, S.S.; Kumar, S.; Jerome-Majewska, L.A. Spliceosomopathies and neurocristopathies: Two sides of the same coin? Dev. Dyn. 2020, 249, 924–945. [Google Scholar] [CrossRef]
- Griffin, C.; Saint-Jeannet, J.-P. Spliceosomopathies: Diseases and mechanisms. Dev. Dyn. 2020, 249, 1038–1046. [Google Scholar] [CrossRef]
- Wood, K.A.; Eadsforth, M.A.; Newman, N.W.; O’Keefe, R.T. The role of theU5 snRNP in genetic disorders and cancer. Front. Genet. 2021, 2, 636620. [Google Scholar] [CrossRef]
- Dixon, J.; Jones, N.C.; Sandell, L.L.; Jayasinghe, S.M.; Crane, J.; Rey, J.-P.; Dixon, M.; Trainor, P.A. Tcof1/Treacle is required for NC cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl. Acad. Sci. USA 2006, 103, 13403–13408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devotta, A.; Juraver-Geslin, H.; Gonzalez, J.A.; Hong, C.-S.; Saint-Jeannet, J.-P. Sf3b4-depleted Xenopus embryos: A model to study the pathogenesis of craniofacial defects in Nager syndrome. Dev. Biol. 2016, 415, 371–382. [Google Scholar] [CrossRef]
- Lei, L.; Yan, S.Y.; Yang, R.; Chen, J.Y.; Li, Y.; Bu, Y.; Chang, N.; Zhou, Q.; Zhu, X.; Li, C.Y.; et al. Spliceosomal protein eftud2 mutation leads to p53-dependent apoptosis in zebrafish neural progenitors. Nucleic Acids Res. 2017, 45, 3422–3436. [Google Scholar] [CrossRef] [Green Version]
- Beauchamp, M.-C.; Djedid, A.; Bareke, E.; Mekuri, F.; Aber, R.; Tam, A.S.; Lines, M.A.; Boycott, K.M.; Sterling, P.C.; Fish, J.L.; et al. Mutation in Eftud2 causes craniofacial defects in mice via mis-splicing of Mdm2 and increased P53. Hum. Mol. Genet. 2021, 30, 739–757. [Google Scholar] [CrossRef]
- Nieuwkoop, P.D.; Faber, J. Normal Table of Xenopus Laevis (Daudin); North-Holland Publishing Co., Ltd.: Amesterdam, The Netherlands, 1956. [Google Scholar]
- Slack, J.M.; Forman, D. An interaction between dorsal and ventral regions of the marginal zone in early amphibian embryos. J. Embryol. Exp. Morphol. 1980, 56, 283–299. [Google Scholar] [CrossRef]
- Mayor, R.; Morgan, R.; Sargent, M.G. Induction of the prospective neural crest of Xenopus. Development 1995, 121, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-S.; Devotta, A.; Lee, Y.-H.; Park, B.-Y.; Saint-Jeannet, J.-P. Transcription factor AP2 epsilon (Tfap2e) regulates neural crest specification in Xenopus. Dev. Neurobiol. 2014, 74, 894–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spokony, R.F.; Aoki, Y.; Saint-Germain, N.; Magner-Fink, E.K.; Saint-Jeannet, J.-P. The transcription factor Sox9 is required for cranial neural crest development in Xenopus. Development 2002, 129, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Saint-Germain, N.; Gyda, M.; Magner-Fink, E.K.; Lee, Y.-H.; Credidio, C.; Saint-Jeannet, J.-P. Sox10 regulates the development of neural crest-derived melanocytes in Xenopus. Dev. Biol. 2003, 259, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Mizuseki, K.; Kishi, M.; Matsui, M.; Nakanishi, S.; Sasai, Y. Xenopus Zic-related-1 and Sox-2, two factors induced by chordin, have distinct activities in the initiation of neural induction. Development 1998, 125, 579–587. [Google Scholar] [CrossRef]
- Pandur, P.D.; Moody, S.A. Xenopus Six1 gene is expressed in neurogenic cranial placodes and maintained in the differentiating lateral lines. Mech. Dev. 2000, 96, 253–257. [Google Scholar] [CrossRef]
- Harland, R.M. In situ hybridization: An improved whole-mount method for Xenopus embryos. Methods Cell Biol. 1991, 36, 685–695. [Google Scholar]
- Saint-Jeannet, J.-P. Whole-Mount In Situ Hybridization of Xenopus Embryos. Cold Spring Harb. Protoc. 2017, 2017, 097287. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.S.; Saint-Jeannet, J.P. The activity of Pax3 and Zic1 regulates three distinct cell fates at the neural plate border. Mol. Biol. Cell 2007, 18, 2192–2202. [Google Scholar]
- Hensey, C.; Gautier, J. Programmed cell death during Xenopus development: A spatio-temporal analysis. Dev. Biol. 1998, 203, 36–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharana, S.K.; Saint-Jeannet, J.-P. Molecular mechanisms of hearing loss in Nager syndrome. Dev. Biol. Dev. Biol. 2021, 476, 200–208. [Google Scholar] [CrossRef]
- Lines, M.; Huang, L.; Schwartzentruber, J.; Douglas, S.L.; Lynch, D.C.; Beaulieu, C.; Guion-Almeida, M.L.; Zechi-Ceide, R.M.; Gener, B.; Gillessen-Kaesbach, G.; et al. Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am. J. Hum. Genet. 2012, 90, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Gordon, C.T.; Petit, F.; Oufadem, M.; Decaestecker, C.; Jourdain, A.-S.; Andrieux, J.; Malan, V.; Alessandri, J.-L.; Baujat, G.; Baumann, C.; et al. EFTUD2 haploinsufficiency leads to syndromic oesophageal atresia. J. Med. Genet. 2012, 49, 737–746. [Google Scholar] [CrossRef]
- Lynch, D.C.; Revil, T.; Schwartzentruber, J.; Bhoj, E.J.; Innes, A.M.; Lamont, R.E.; Lemire, E.G.; Chodirker, B.N.; Taylor, J.P.; Zackai, E.H.; et al. Disrupted auto-regulation of the spliceosomal complex gene SNRPB causes cerebro-costo-mandibular syndrome. Nat. Commun. 2014, 5, 4483. [Google Scholar] [CrossRef] [Green Version]
- Wieczorek, D.; Newman, W.G.; Wieland, T.; Berulava, T.; Kaffe, M.; Falkenstein, D.; Betz, C.; Graf, E.; Schwartmayr, T.; Douzgou, S.; et al. Compound heterozygosity of low-frequency promoter deletions and rare loss-of-function mutations in TXNL4A causes Burn-McKeown syndrome. Am. J. Hum. Genet. 2014, 95, 698–707. [Google Scholar] [CrossRef] [Green Version]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, apotential mediator of p53 tumor supression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Bernier, F.P.; Caluseriu, O.; Ng, S.; Schwartzentruber, J.; Buckingham, K.J.; Innes, A.M.; Jabs, E.W.; Innis, J.W.; Schuette, J.L.; Gorski, J.L.; et al. Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am. J. Hum. Genet. 2012, 90, 925–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit, F.; Escande, F.; Jourdain, A.S.; Porchet, N.; Amiel, J.; Doray, B.; Delrue, M.; Flori, E.; Kim, C.; Marlin, S.; et al. Nager syndrome: Confirmation of SF3B4haploinsufficiency as the major cause. Clin. Genet. 2013, 86, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Czeschik, J.C.; Voigt, C.; Alanay, Y.; Albrecht, B.; Avci, S.; Fitzpatrick, D.; Goudie, D.R.; Hehr, U.; Hoogeboom, A.J.; Kayserili, H.; et al. Clinical and mutation data in 12 patients with the clinical diagnosis of Nager syndrome. Hum. Genet. 2013, 132, 885–898. [Google Scholar] [CrossRef]
- Drivas, T.G.; Taylor, J.A.; Zackai, E.H. The final demise of Rodriguez lethal acrofacial dysostosis: A case report and review of the literature. Am. J. Med. Genet. 2019, 179A, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Deml, B.; Reis, L.M.; Muheisen, S.; Bick, D.; Semina, E.V. EFTUD2 deficiency in vertebrates: Identification of a novel human mutation and generation of a zebrafish model. Birth Defects Research. Part A Clin. Mol. Teratol. 2015, 103, 630–640. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Yang, Y.; He, Y.; Li, Q.; Wang, X.; Sun, C.; Wang, L.; An, Y.; Luo, F. EFTUD2 gene deficiency disrupts osteoblast maturation and inhibits chondrocyte differentiation via activation of the p53 signaling pathway. Hum. Genom. 2019, 13, 63. [Google Scholar] [CrossRef] [Green Version]
- Wood, K.A.; Rowlands, C.F.; Qureshi, W.M.S.; Thomas, H.B.; Buczek, W.A.; Briggs, T.A.; Hubbard, S.J.; Hentges, K.E.; Newman, W.G.; O’Keefe, R.T. Disease modeling of core pre-mRNA splicing factor haploinsufficiency. Hum. Mol. Genet. 2019, 28, 3704–3723. [Google Scholar] [CrossRef]
- Wood, K.A.; Rowlands, C.F.; Thomas, H.B.; Woods, S.; O’Flaherty, J.; Douzgou, S.; Kimber, S.J.; Newman, W.G.; O’Keefe, R.T. Modelling the developmental spliceosomal craniofacial disorder Burn-McKeown syndrome using induced pluripotent stem cells. PLoS ONE 2020, 15, e0233582. [Google Scholar] [CrossRef]
- Favaro, F.P.; Alvizi, L.; Zechi-Ceide, R.M.; Bertola, D.; Felix, T.M.; de Souza, J.; Raskin, S.; Twigg, S.R.; Weiner, A.M.; Armas, P.; et al. A noncoding expansion in EIF4A3 causes Richieri-Costa-Pereira syndrome, a craniofacial disorder associated with limb defects. Am. J. Human Genet. 2014, 94, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.E.; Kobayashi, G.S.; Musso, C.M.; Allen, M.; Ishiy, F.A.; De Caires, L.C.; Goulart, E.; Griesi-Oliveira, K.; Zechi-Ceide, R.M.; Richieri-Costa, A.; et al. EIF4A3 deficient human iPSCs and mouse models demonstrate neural crest defects that underlie Richieri-Costa-Pereira syndrome. Hum. Mol. Genet. 2017, 26, 2177–2191. [Google Scholar] [CrossRef] [Green Version]
- Timberlake, A.T.; Griffin, C.; Heike, C.L.; Hing, A.V.; Cunningham, M.L.; Chitayat, D.; Davis, M.R.; Doust, S.J.; Drake, A.F.; Duenas-Roque, M.M.; et al. Haploinsufficiency of SF3B2 causes craniofacial microsomia. Nat. Commun. 2021, 12, 4680. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, M.C.; Djedid, A.; Daupin, K.; Clokie, K.; Kumar, S.; Majewski, J.; Jerome-Majewska, L.A. Loss of function mutation of Eftud2, the gene responsible for mandibulofacial dysostosis with microcephaly (MFDM), leads to pre-implantation arrest in mouse. PLoS ONEs 2019, 14, e0219280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, T.; Takechi, M.; Yokoyama, N.; Hiraoka, Y.; Ishikubo, H.; Usami, T.; Furutera, T.; Taga, Y.; Hirate, Y.; Kanai-Azuma, M.; et al. Heterozygous mutation of the splicing factor Sf3b4 affects development of the axial skeleton and forebrain in mouse. Dev. Dyn. 2019, 249, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Gu, B.; Bowen, M.; Aryan, F.; Zalc, A.; Liang, J.; Flynn, R.A.; Swigut, T.; Chang, H.Y.; Attardi, L.D.; et al. Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders. Nature 2018, 554, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, B.-Y.; Tachi-Duprat, M.; Ihewulezi, C.; Devotta, A.; Saint-Jeannet, J.-P. The Core Splicing Factors EFTUD2, SNRPB and TXNL4A Are Essential for Neural Crest and Craniofacial Development. J. Dev. Biol. 2022, 10, 29. https://doi.org/10.3390/jdb10030029
Park B-Y, Tachi-Duprat M, Ihewulezi C, Devotta A, Saint-Jeannet J-P. The Core Splicing Factors EFTUD2, SNRPB and TXNL4A Are Essential for Neural Crest and Craniofacial Development. Journal of Developmental Biology. 2022; 10(3):29. https://doi.org/10.3390/jdb10030029
Chicago/Turabian StylePark, Byung-Yong, Melanie Tachi-Duprat, Chibuike Ihewulezi, Arun Devotta, and Jean-Pierre Saint-Jeannet. 2022. "The Core Splicing Factors EFTUD2, SNRPB and TXNL4A Are Essential for Neural Crest and Craniofacial Development" Journal of Developmental Biology 10, no. 3: 29. https://doi.org/10.3390/jdb10030029
APA StylePark, B.-Y., Tachi-Duprat, M., Ihewulezi, C., Devotta, A., & Saint-Jeannet, J.-P. (2022). The Core Splicing Factors EFTUD2, SNRPB and TXNL4A Are Essential for Neural Crest and Craniofacial Development. Journal of Developmental Biology, 10(3), 29. https://doi.org/10.3390/jdb10030029