
Application of Lactose Co-Processed Excipients as an Alternative for Bridging Pharmaceutical Unit Operations: Manufacturing an Omeprazole Tablet Prototype via Direct Compression

 , , ,
, , ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Reagents and Chemicals
2.1.2. Standards, Samples, and Excipients
2.2. Methods
2.2.1. Powder Physical Characterization
Particle Size Distribution
Microscopy
Powder Rheology
Specific Surface Area—Brunauer–Emmett–Teller (BET)
2.2.2. Tablet Production
Blending
Tableting
Coating and Final Packaging
2.2.3. Tablet Physical Characterization (Hardness, Friability, Disintegration, and Thickness)
2.2.4. Analytical Tablet Characterization (Content Uniformity, Assay, Impurities, and Dissolution)
2.2.5. Statistical Analysis
3. Results and Discussion
3.1. Formulation Definition
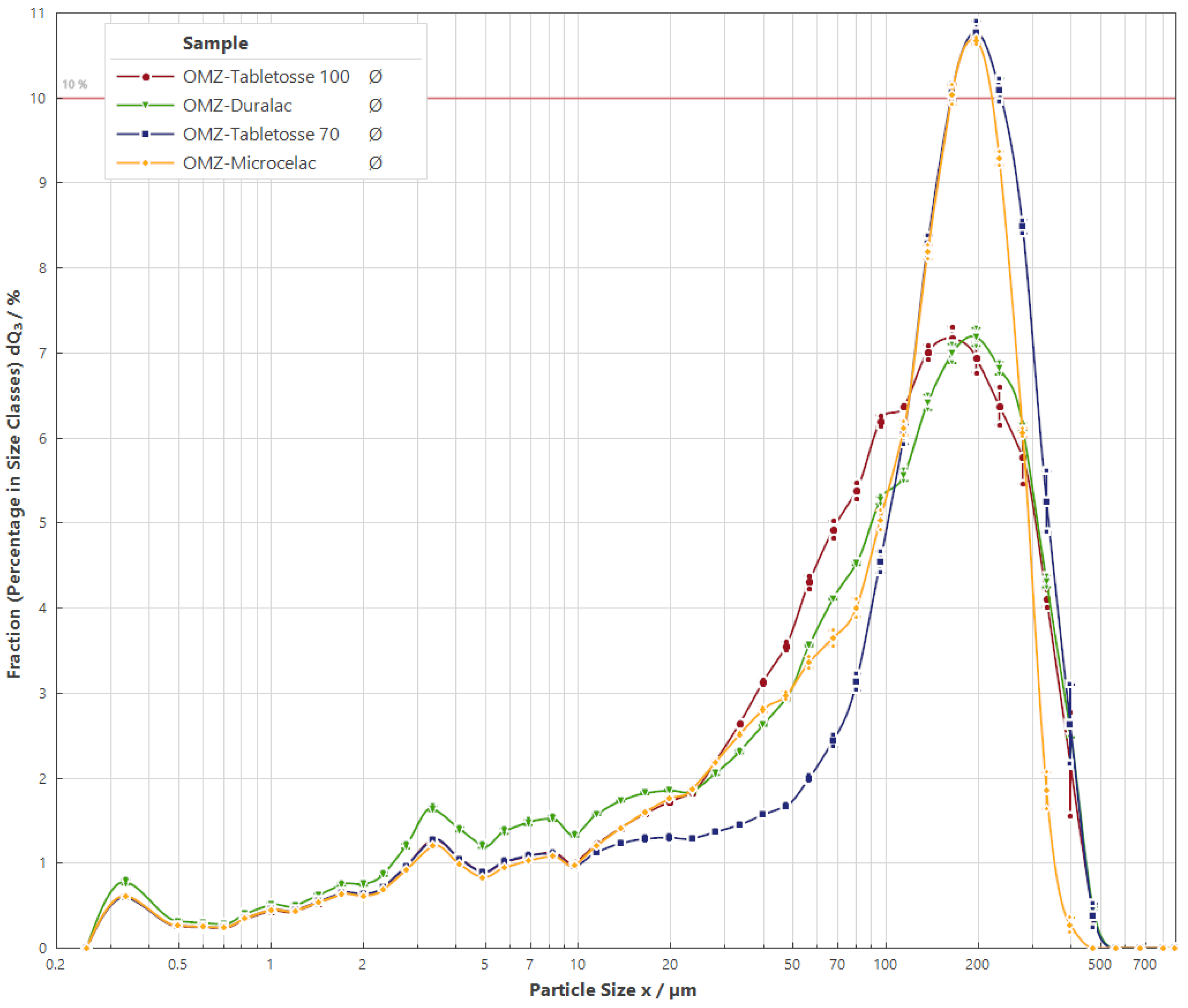
3.2. Particle Size Distribution
3.3. Microscope Picture Analysis
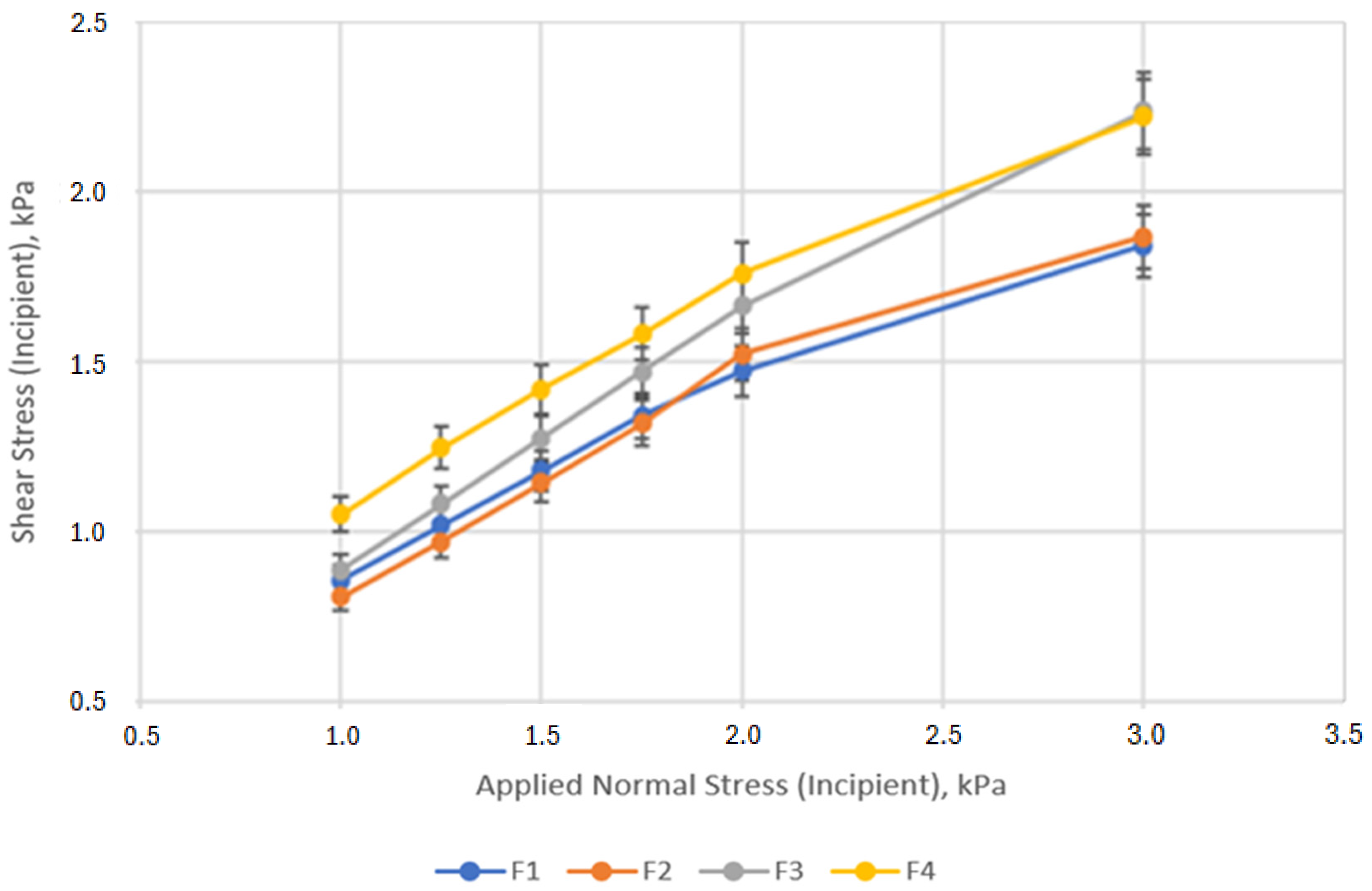
3.4. Powder Rheology
3.5. Specific Surface Area (BET)
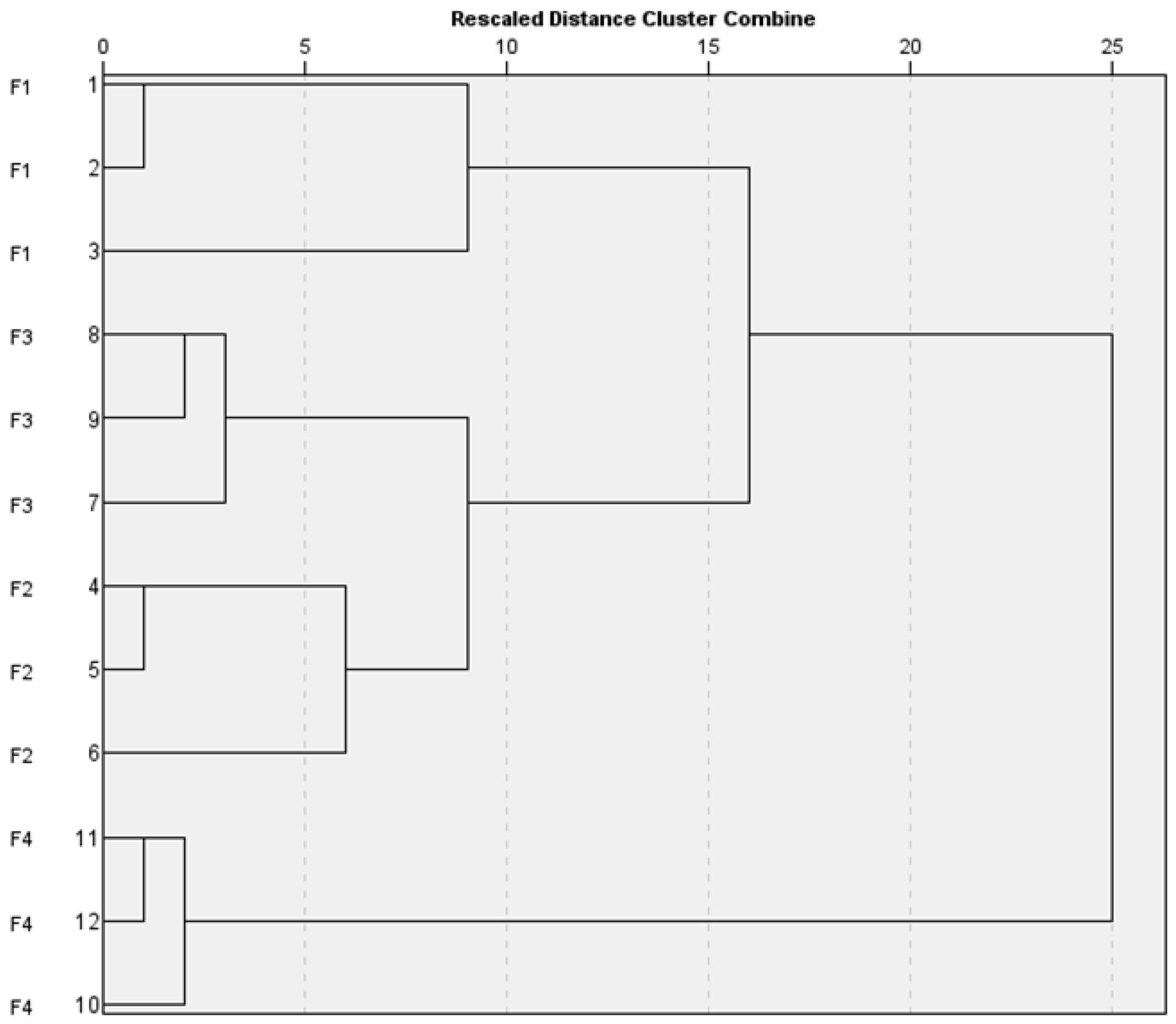
3.6. Statistical Evaluation
3.7. Tablet Manufacturing
3.8. Analytical Characterization
3.8.1. Preliminary Analytical Assessment
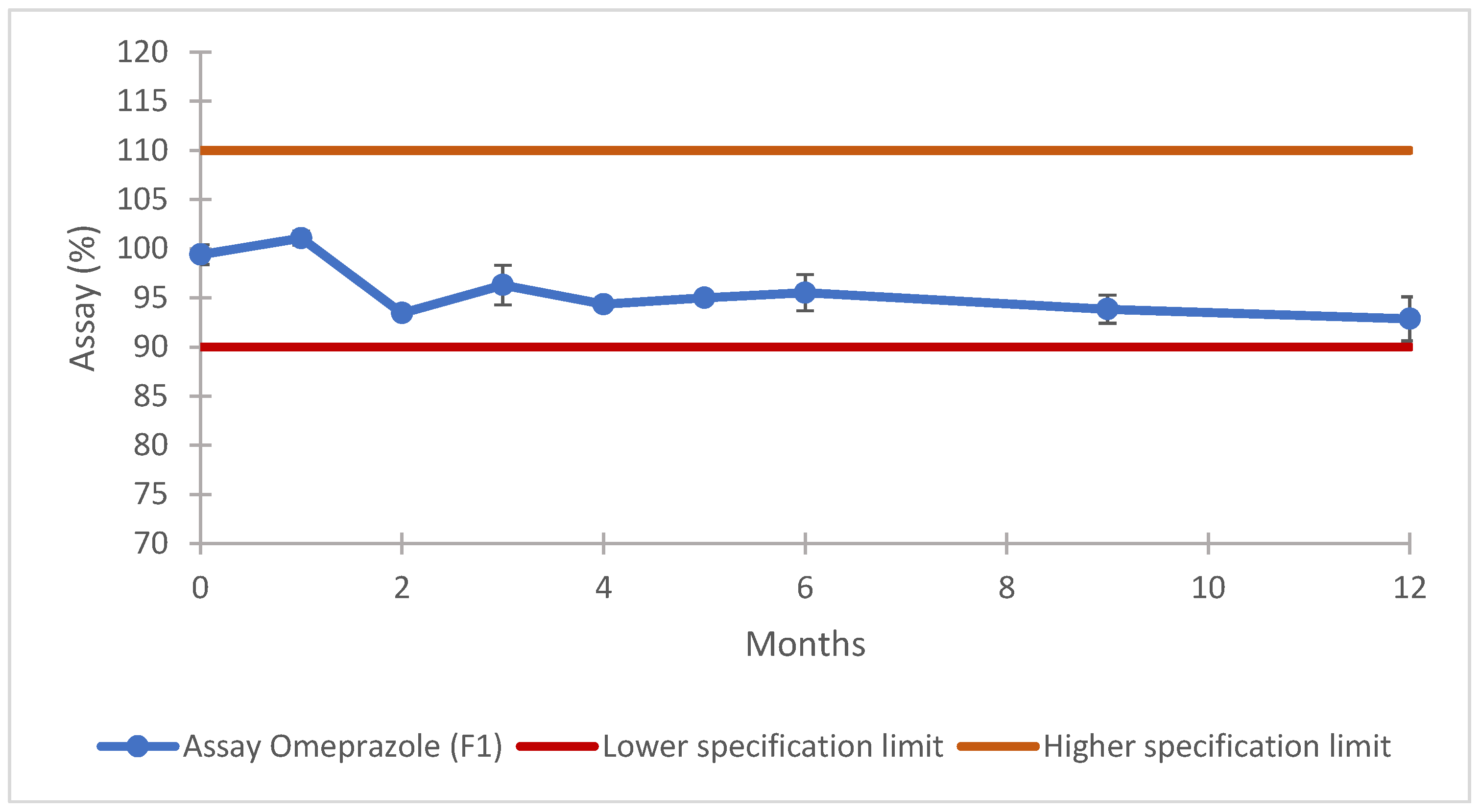
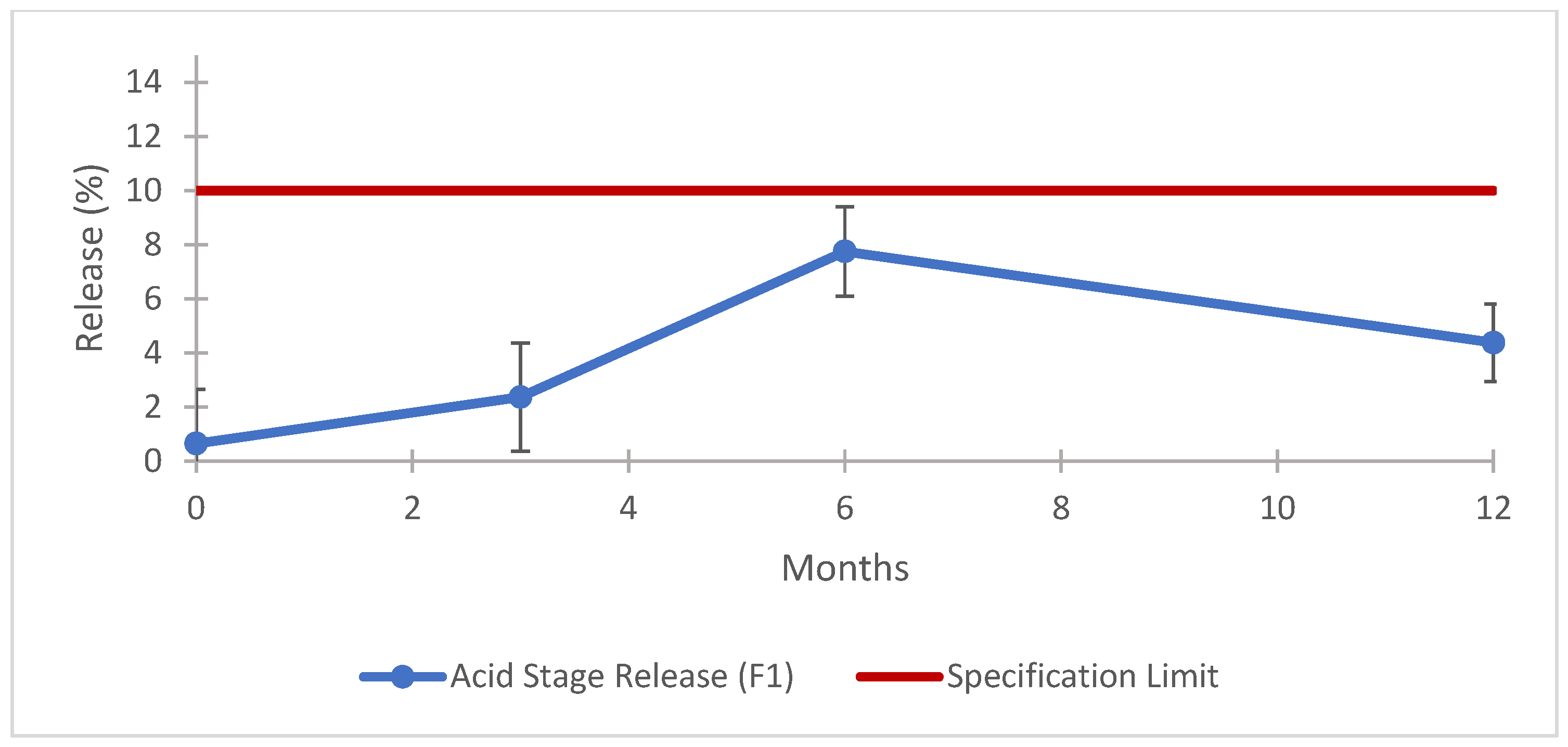
3.8.2. Long-Term Stability Results
3.9. Regulatory Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| °C | Degree Celsius |
| µg | Microgram |
| µm | Micrometre |
| ACS | Acetonitrile |
| AIF | Angle of internal friction |
| AIFe | Effective angle of internal friction |
| API | Active pharmaceutical ingredients |
| AV | Acceptance value |
| BET | Brunauer–Emmett–Teller |
| BJH | Barrett–Joyner–Halenda |
| C | Cohesion |
| Cm | Centimetre |
| CPE | Co-processed excipients |
| F | Formulation |
| FDA | Food and Drug Administration |
| Ffc | Flow function coefficient |
| G | Gram |
| GmbH | Gesellschaft mit beschränkter Haftung |
| HCl | Hydrochloric acid |
| HPLC | High-performance liquid chromatography |
| HPLC-RP | High-performance liquid chromatography—reverse phase |
| IBM | International Business Machines Corporation |
| ICH | International Council for Harmonisation |
| kPa | Kilo pascal |
| LTD | Limited company |
| MCC | Microcrystalline cellulose |
| mg | Milligram |
| Min | Minute |
| mL | Milliliter |
| MUPS | Multiple-unit pellet system |
| N | Normality |
| Nm | Nanometre |
| OMZ | Omeprazole |
| OTC | Over-the-counter medication |
| Ph. Eur | European Pharmacopoeia |
| Ph | Potential of hydrogen |
| PSD | Particle size distribution |
| PVA | Polyvinyl alcohol |
| Q | Quality guideline |
| QTPP | Quality Target Product Profile |
| RH | Relative humidity |
| RLD | Reference Listed drug |
| SD | Standard deviation |
| SSA | Specific surface area |
| UK | United Kingdom |
| UPLC | Ultra-high-performance liquid chromatography |
| USA | United States of America |
| USP | United States Pharmacopoeia |
| UYS | Unconfined yield strength |
| WHO | World Health Organization |
References
- Suzuki, Y.; Sugiyama, H.; Kano, M.; Shimono, R.; Shimada, G.; Furukawa, R.; Mano, E.; Motoyama, K.; Koide, T.; Matsui, Y.; et al. Control strategy and methods for continuous direct compression processes. Asian J. Pharm. Sci. 2021, 16, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Shangraw, R.F. Compressed Tablets by Direct Compression; The University of Maryland School of Pharmacy: Baltimore, MD, USA, 1989; ISBN 2013206534. [Google Scholar]
- Chowdary, K.P.R. A comparative evaluation of direct compression and wet granulation methods for formulation of stavudine tabletsTS. J. Glob. Trends Pharm. Sci. 2014, 5, 2000–2003. [Google Scholar]
- Dominik, M.; Vraníková, B.; Svačinová, P.; Elbl, J.; Pavloková, S.; Prudilová, B.B.; Šklubalová, Z.; Franc, A. Comparison of Flow and Compression Properties of Four Lactose-Based Co-Processed Excipients: Cellactose® 80, CombiLac®, MicroceLac® 100, and StarLac®. Pharmaceutics 2021, 13, 1486. [Google Scholar] [CrossRef]
- Kása, P.; Bajdik, J.; Zsigmond, Z.; Pintye-Hódi, K. Study of the compaction behaviour and compressibility of binary mixtures of some pharmaceutical excipients during direct compression. Chem. Eng. Process. Process. Intensif. 2009, 48, 859–863. [Google Scholar] [CrossRef]
- Onofre, F.; O’Donnell, K. Leveraging Direct Compression Technology to Improve Tableting Efficiency|American Pharmaceutical Review—The Review of American Pharmaceutical Business & Technology. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/581695-Leveraging-Direct-Compression-Technology-to-Improve-Tableting-Efficiency/ (accessed on 22 June 2023).
- Rojas, J.; Buckner, I.; Kumar, V. Co-proccessed excipients with enhanced direct compression functionality for improved tableting performance. Drug Dev. Ind. Pharm. 2012, 38, 1159–1170. [Google Scholar] [CrossRef]
- Saha, S.; Shahiwala, A.F. Multifunctional coprocessed excipients for improved tabletting performance. Expert Opin. Drug Deliv. 2009, 6, 197–208. [Google Scholar] [CrossRef]
- Bhatia, V.; Dhingra, A.; Chopra, B.; Guarve, K. Co-processed excipients: Recent advances and future perspective. J. Drug Deliv. Sci. Technol. 2022, 71, 103316. [Google Scholar] [CrossRef]
- Rahman, M.S.; Yoshida, N.; Tsuboi, H.; Keila, T.; Sovannarith, T.; Kiet, H.B.; Dararth, E.; Zin, T.; Tanimoto, T.; Kimura, K. Erroneous formulation of delayed-release omeprazole capsules: Alert for importing countries. BMC Pharmacol. Toxicol. 2017, 18, 31. [Google Scholar] [CrossRef]
- Mansuri, N.S.; Parejiya, P.B.; Soniwala, M.M. Research Article Exploring use of quality by design (QbD) principles for development of modified release omeprazole pellets. J. Chem. Pharm. Res. 2015, 7, 630–639. [Google Scholar]
- Mohylyuk, V.; Yerkhova, A.; Katynska, M.; Sirko, V.; Patel, K. Effect of Elevated pH on the Commercial Enteric-Coated Omeprazole Pellets Resistance: Patent Review and Multisource Generics Comparison. AAPS PharmSciTech 2021, 22, 188. [Google Scholar] [CrossRef]
- Srebro, J.; Brniak, W.; Mendyk, A. Formulation of Dosage Forms with Proton Pump Inhibitors: State of the Art, Challenges and Future Perspectives. Pharmaceutics 2022, 14, 2043. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization WHO Model List of Essential Medicines—22nd List; Technical Document; WHO: Geneva, Switzerland, 2021.
- Aubert, J.; Mulder, C.J.; Schrör, K.; Vavricka, S.R. Omeprazole MUPS®: An advanced formulation offering flexibility and predictability for self medication. Selfcare 2011, 2, 1–14. [Google Scholar]
- Chih-Ming Chen, D.; Joseph Chou, M.; Unchalee, K. Omeprazole Formulation. U.S. Patent US20040131684A1, 15 February 2004. [Google Scholar]
- Chih-Ming Chen, D.; Chou, J.C.H.; Weng, T. Omeprazole Formulation. U.S. Patent US6096340A, 1 August 2000. [Google Scholar]
- Palomo Coll, A. Oral Pharmaceutical Preparation Containing Omeprazole. U.S. Patent US5232706A, 3 August 1993. [Google Scholar]
- Bengtsson, I.S.; Lövgren, I. Pharmaceutical Formulation of Omeprazole. U.S. Patent US5690960A, 25 November 1997. [Google Scholar]
- Chauhan, I.; Nutalapati, S.R.K. Multilayer Omeprazole Tablets. U.S. Patent US20090280173A1, 12 November 2009. [Google Scholar]
- Lovgren, K.I.; Pilbrant, A.G.; Yasumura, M.; Morigaki, S.; Oda, M.; Ohishi, N. Coated Omeprazole Tablets. UK Patent GB2189698A, 4 November 1987. [Google Scholar]
- Bergstrand, P.J.A.; Lövgren, K.I. Multiple Unit Tableted Dosage Form of Omeprazole. U.S. Patent US5817338A, 16 October 1998. [Google Scholar]
- Dietrich, R.; Ney, H. Pharmaceutical Preparation in Tablet or Pellet Form for Pantoprazole and Omeprazole. German Patent DE19752843A1, 9 January 2003. [Google Scholar]
- Capua, S.D.; Shterman, N.; Pardo, L.A.; Itach, E. A Stable Pharmaceutical Composition Comprising an Acid Labile. Drug. Patent WO2005092297A2, 6 October 2005. [Google Scholar]
- 〈1216〉 Tablet Friability; USP43-NF38; United States Pharmacopeia: Rockville, MD, USA, 2022. [CrossRef]
- 〈701〉 Disintegration; USP43-NF38; United States Pharmacopeia: Rockville, MD, USA, 2008. [CrossRef]
- Omeprazole Delayed-Release Capsules; USP43-NF38; United States Pharmacopeia: Rockville, MD, USA, 2006. [CrossRef]
- 〈1226〉 Verification of Compendial Procedures; USP43-NF38; United States Pharmacopeia: Rockville, MD, USA, 2019. [CrossRef]
- International Council for Harmonisation Guideline Q2 (R2) Validation of Analytical Procedures. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-q2r2-validation-analytical-procedures-step-2b_en.pdf (accessed on 25 October 2022).
- 〈1225〉 Validation of Compendial Procedures; USP43-NF38; United States Pharmacopeia: Rockville, MD, USA, 2016. [CrossRef]
- FDA Office of Regulatory Affairs Dissolution Methods (Omeprazole). Available online: https://www.accessdata.fda.gov/scripts/cder/dissolution/dsp_SearchResults.cfm (accessed on 26 June 2023).
- International Council of Harmonization Q1A (R2) Stability Testing of New Drug Substances and Products. Available online: https://database.ich.org/sites/default/files/Q1A%28R2%29 Guideline.pdf (accessed on 10 May 2023).
- Procter & Gamble Prilosec OTC Authorized Package Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/021229s006lbl.pdf (accessed on 23 April 2023).
- Niazi, S.K. Handbook of Pharmaceutical Manufacturing Formulations: Compressed Solid Products; CRC Press: Boca Raton, FL, USA, 2016; ISBN 9781420081176. [Google Scholar]
- MEGGLE GmbH & Co. KG. Excipients Brochure; MEGGLE GmbH &, Co. KG: Wasserburg am Inn, Germany, 2023. [Google Scholar]
- Thapa, P.; Choi, D.H.; Kim, M.S.; Jeong, S.H. Effects of granulation process variables on the physical properties of dosage forms by combination of experimental design and principal component analysis. Asian J. Pharm. Sci. 2019, 14, 287–304. [Google Scholar] [CrossRef]
- Kotamarthy, L.; Metta, N.; Ramachandran, R. Understanding the Effect of Granulation and Milling Process Parameters on the Quality Attributes of Milled Granules. Processes 2020, 8, 683. [Google Scholar] [CrossRef]
- Miinea, L.; Mehta, R.; Kallam, M.; Farina, J.; Deorkar, N. Evaluation and Characteristics of a New Direct Compression Performance Excipient. Available online: https://www.pharmtech.com/view/evaluation-and-characteristics-new-direct-compression-performance-excipient (accessed on 25 June 2024).
- MEGGLE Pharma—Excipients & Technology MicroceLac® 100 Product Detail. Available online: https://www.meggle-pharma.com/en/lactose/13-microcelac-100.html (accessed on 24 April 2024).
- Freeman, R. Measuring the flow properties of consolidated, conditioned and aerated powders—A comparative study using a powder rheometer and a rotational shear cell. Powder Technol. 2007, 174, 25–33. [Google Scholar] [CrossRef]
- Busignies, V.; Leclerc, B.; Truchon, S.; Tchoreloff, P. Changes in the specific surface area of tablets composed of pharmaceutical materials with various deformation behaviors. Drug Dev. Ind. Pharm. 2011, 37, 225–233. [Google Scholar] [CrossRef]
- Leung, L.Y.; Mao, C.; Chen, L.P.; Yang, C.-Y. Precision of pharmaceutical powder flow measurement using ring shear tester: High variability is inherent to powders with low cohesion. Powder Technol. 2016, 301, 920–926. [Google Scholar] [CrossRef]
- Manley, L.; Hilden, J.; Valero, P.; Kramer, T. Tablet Compression Force as a Process Analytical Technology (PAT): 100% Inspection and Control of Tablet Weight Uniformity. J. Pharm. Sci. 2019, 108, 485–493. [Google Scholar] [CrossRef]
- Manley, L.; Shi, Z. Characterizing drug product continuous manufacturing residence time distributions of major/minor excipient step changes using near infrared spectroscopy and process parameters. Int. J. Pharm. 2018, 551, 60–66. [Google Scholar] [CrossRef]
- Juban, A.; Nouguier-Lehon, C.; Briancon, S.; Hoc, T.; Puel, F. Predictive model for tensile strength of pharmaceutical tablets based on local hardness measurements. Int. J. Pharm. 2015, 490, 438–445. [Google Scholar] [CrossRef]
- Jarosz, P.J.; Parrott, E.L. Tensile Strengths and Hardness. J. Pharm. Sci. 1982, 71, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Shi, C.; Zhao, L.; Wang, Y.; Shen, L. Influences of different microcrystalline cellulose (MCC) grades on tablet quality and compression behavior of MCC-lactose binary mixtures. J. Drug Deliv. Sci. Technol. 2022, 77, 103893. [Google Scholar] [CrossRef]
- Janssen, P.H.M.; Berardi, A.; Kok, J.H.; Thornton, A.W.; Dickhoff, B.H.J. The impact of lactose type on disintegration: An integral study on porosity and polymorphism. Eur. J. Pharm. Biopharm. 2022, 180, 251–259. [Google Scholar] [CrossRef]
- van Kamp, H.V.; Bolhuis, G.K.; Kussendrager, K.D.; Lerk, C.F. Studies on tableting properties of lactose. IV. Dissolution and disintegration properties of different types of crystalline lactose. Int. J. Pharm. 1986, 28, 229–238. [Google Scholar] [CrossRef]
- Ziffels, S.; Steckel, H. Influence of amorphous content on compaction behaviour of anhydrous α-lactose. Int. J. Pharm. 2010, 387, 71–78. [Google Scholar] [CrossRef]
- Thio, D.R.; Heng, P.W.S.; Chan, L.W. MUPS Tableting—Comparison between Crospovidone and Microcrystalline Cellulose Core Pellets. Pharmaceutics 2022, 14, 2812. [Google Scholar] [CrossRef]
- 〈905〉 Uniformity of Dosage Units; USP42-NF37; United States Pharmacopeia: Rockville, MD, USA, 2022. [CrossRef]
- 〈2〉 Oral Drug Products—Product Quality Tests; USP43-NF38; United States Pharmacopeia: Rockville, MD, USA, 2024. [CrossRef]
- 〈711〉 Dissolution; USP42-NF37; United States Pharmacopeia: Rockville, MD, USA, 2023. [CrossRef]
- MEGGLE Pharma—Excipients & Technology Tablettose® 100 Product Detail. Available online: https://www.meggle-pharma.com/en/lactose/8-tablettose-100.html (accessed on 24 April 2024).
- MEGGLE Pharma—Excipients & Technology Tablettose® 70 Product Detail. Available online: https://www.meggle-pharma.com/en/lactose/6-tablettose-70.html (accessed on 24 April 2024).
- Bowles, B.J.; Dziemidowicz, K.; Lopez, F.L.; Orlu, M.; Tuleu, C.; Edwards, A.J.; Ernest, T.B. Co-Processed Excipients for Dispersible Tablets–Part 1: Manufacturability. AAPS PharmSciTech 2018, 19, 2598–2609. [Google Scholar] [CrossRef]
- Challener, C.A. Formulating with Coprocessed Excipients: Current Trends. Pharm. Technol. 2023, 47, 18–21. [Google Scholar]
- Guy, T. Challenges Facing Pharmaceutical Excipients|American Pharmaceutical Review—The Review of American Pharmaceutical Business & Technology. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/177716-Challenges-Facing-Pharmaceutical-Excipients/ (accessed on 17 May 2024).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mg/Tablet | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| OMZ | Microcelac® 100 | Tabletosse® 70 | Tabletosse® 100 | Avicel PH102 | DuraLac® H | Explotab® | Pruv® | Total | |
| F1 | 20.0 | 170.0 | - | - | - | - | 8.0 | 2.0 | 200.0 |
| F2 | 20.0 | - | 170.0 | - | - | - | 8.0 | 2.0 | 200.0 |
| F3 | 20.0 | - | - | 170.0 | - | - | 8.0 | 2.0 | 200.0 |
| F4 | 20.0 | - | - | - | 42.5 | 127.5 | 8.0 | 2.0 | 200.0 |
| Chromatographic Feature | Content Uniformity, Assay and Dissolution | Impurities |
|---|---|---|
| Mobile phases | A: 10 mM ammonium bicarbonate buffer pH 8.75 B: Acetonitrile | A: Glycine 3 g/L adjusted to pH 9.0 B: Acetonitrile |
| Gradient program | %A0min = 90, %A3min = 90, %A10min = 40, %A11min = 90 and %A15min = 90 | %A0min = 88, %A20min = 40, %A21min = 88 and %A25min = 88 |
| Flow | 1.9 mL/min | 1.2 mL/min |
| Injection volume | 5 µL (content uniformity, assay) and 40 µL (dissolution) | 10 µL |
| Time per injection | 15 min | 25 min |
| Column temperature | 35 °C | 20 °C |
| Sample temperature | 20 °C | 20 °C |
| Wavelength | 305 nm | 305 nm |
| Stability Test | Timepoint (Months) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Initial | 1 | 2 | 3 | 4 | 5 | 6 | 9 | 12 | |
| Assay | X | X | X | X | X | X | X | X | X |
| Dissolution | X | X | X | X | |||||
| Impurities | X | X | X | X | |||||
| OMZ | F1 | F2 | F3 | F4 | |
|---|---|---|---|---|---|
| D10 [μm] | 0.79 ± 0.02 | 6.26 ± 0.11 | 5.87±0.08 | 5.99 ± 0.10 | 4.31 ± 0.14 |
| D50 [μm] | 3.71 ± 0.02 | 113.23 ± 1.37 | 143.06 ± 1.90 | 94.79 ± 1.10 | 93.30 ± 1.53 |
| D90 [μm] | 13.82 ± 2.93 | 247.26 ± 1.39 | 294.80 ± 5.73 | 276.66 ± 3.94 | 283.35 ± 1.45 |
| Cohesion, kPa | UYS, kPa | FF | AIF, ° | |
|---|---|---|---|---|
| F1 | 0.25 ± 0.02 | 0.89 ± 0.06 | 5.57 ± 0.27 | 31.68 ± 0.51 |
| F2 | 0.09 ± 0.06 | 0.34 ± 0.21 | 18.51 ± 10.71 | 35.30 ± 1.35 |
| F3 | 0.21 ± 0.09 | 0.78 ± 0.31 | 7.35 ± 3.11 | 34.75 ± 2.17 |
| F4 | 0.36 ± 0.02 | 1.38 ± 0.06 | 4.11 ± 0.18 | 35.03 ± 0.89 |
| Formulation | SSA (m2/g) |
|---|---|
| F1 | 0.6708 |
| F2 | 0.5906 |
| F3 | 0.6174 |
| F4 | 0.7023 |
| Formulation | Average Weight Variation (mg) | Average Hardness (N) | Friability (%) | Disintegration (min) |
|---|---|---|---|---|
| F1 | 198.9 ± 1.5 | 78.1 ± 7.4 | 0.1 | 0.8 |
| F2 | 204.9 ± 0.4 | 46.9 ± 7.4 | 0.2 | 0.5 |
| F3 | 200.1 ± 2.8 | 45.8 ± 7.9 | 0.2 | 0.5 |
| F4 | 192.7 ± 7.4 | 43.8 ± 11.4 | 0.1 | 1.0 |
| Prilosec® | 333.5 ± 3.8 | 154.0 ± 14.6 | n.a. | 7.5 |
| Test | Acceptance Criteria | Justification |
|---|---|---|
| Uniformity of dosage units | Acceptance value (AV) < 15.0 | USP <905> [52] |
| Assay | 90.0–110.0% | USP <2> [53] |
| Impurities Omeprazole-related compounds F and G 5-Methoxy-1H-benzimidazole-2-thiol Any other individual impurity Total impurities | 0.5% 0.5% 0.5% 2 | [27] |
| Dissolution (Acid Stage) | NMT 10% after 2 h | [27,54] |
| Dissolution (Buffer Stage) | NLT 75% (Q) at 45 min | [27] |
| Formulation | Uniformity of Dosage Units | Assay (%) | Dissolved at 45 Minutes (%) | ||
|---|---|---|---|---|---|
| Mean of Individuals (%) | AV | Nominal | Normalized per Assay | ||
| F1 | 100.6 ± 1.4 | 3.0 | 99.4 ± 1.0 | 95 ± 2 | 96 ± 2 |
| F2 | 98.9 ± 0.8 | 2.0 | 96.7 ± 1.0 | 90 ± 1 | 93 ± 1 |
| F3 | 96.3 ± 1.0 | 5.0 | 96.2 ± 0.6 | 89 ± 2 | 93 ± 2 |
| F4 | 98.3 ± 3.0 | 7.3 | 96.0 ± 2.8 | 91 ± 1 | 95 ± 3 |
| Prilosec® | 98.2 ± 3.0 | 7.5 | 98.2 ± 2.6 * | 92 ± 4 | 94 ± 5 |
| Formulation | Average Weight Variation (mg) | Average Hardness (N) | Friability (%) | Disintegration (min) |
|---|---|---|---|---|
| F1-Scale up Batch | 200.8 ± 1.5 | 102.1 ± 6.5 | 0.09 | 1.5 |
| Impurity | Acceptance Criteria (%) | Timepoint (Months) | |||
|---|---|---|---|---|---|
| Initial | 6 | 9 | 12 | ||
| Omeprazole related compounds F and G | 0.5 | 0.0 ± 0.0 | 0.3 ± 0.0 | 0.2 ± 0.0 | 0.2 ± 0.0 |
| 5-Methoxy-1H-benzimidazole-2-thiol | 0.5 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Any other individual impurity | 0.5 | 0.2 ± 0.0 | 0.3 ± 0.1 | 0.4 ± 0.0 | 0.4 ± 0.0 |
| Total impurities | 2 | 0.6 ± 0.1 | 0.9 ± 0.1 | 1.2 ± 0.1 | 1.4 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Österreichische Pharmazeutische Gesellschaft. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lara Garcia, R.A.; Afonso Urich, J.A.; Afonso Urich, A.I.; Jeremic, D.; Khinast, J. Application of Lactose Co-Processed Excipients as an Alternative for Bridging Pharmaceutical Unit Operations: Manufacturing an Omeprazole Tablet Prototype via Direct Compression. Sci. Pharm. 2025, 93, 24. https://doi.org/10.3390/scipharm93020024
Lara Garcia RA, Afonso Urich JA, Afonso Urich AI, Jeremic D, Khinast J. Application of Lactose Co-Processed Excipients as an Alternative for Bridging Pharmaceutical Unit Operations: Manufacturing an Omeprazole Tablet Prototype via Direct Compression. Scientia Pharmaceutica. 2025; 93(2):24. https://doi.org/10.3390/scipharm93020024
Chicago/Turabian StyleLara Garcia, Raymar Andreina, Jesús Alberto Afonso Urich, Andreina Isabel Afonso Urich, Dalibor Jeremic, and Johannes Khinast. 2025. "Application of Lactose Co-Processed Excipients as an Alternative for Bridging Pharmaceutical Unit Operations: Manufacturing an Omeprazole Tablet Prototype via Direct Compression" Scientia Pharmaceutica 93, no. 2: 24. https://doi.org/10.3390/scipharm93020024
APA StyleLara Garcia, R. A., Afonso Urich, J. A., Afonso Urich, A. I., Jeremic, D., & Khinast, J. (2025). Application of Lactose Co-Processed Excipients as an Alternative for Bridging Pharmaceutical Unit Operations: Manufacturing an Omeprazole Tablet Prototype via Direct Compression. Scientia Pharmaceutica, 93(2), 24. https://doi.org/10.3390/scipharm93020024