
Marine Origin vs. Synthesized Compounds: In Silico Screening for a Potential Drug Against SARS-CoV-2

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
Optibrium—ADME Properties Prediction
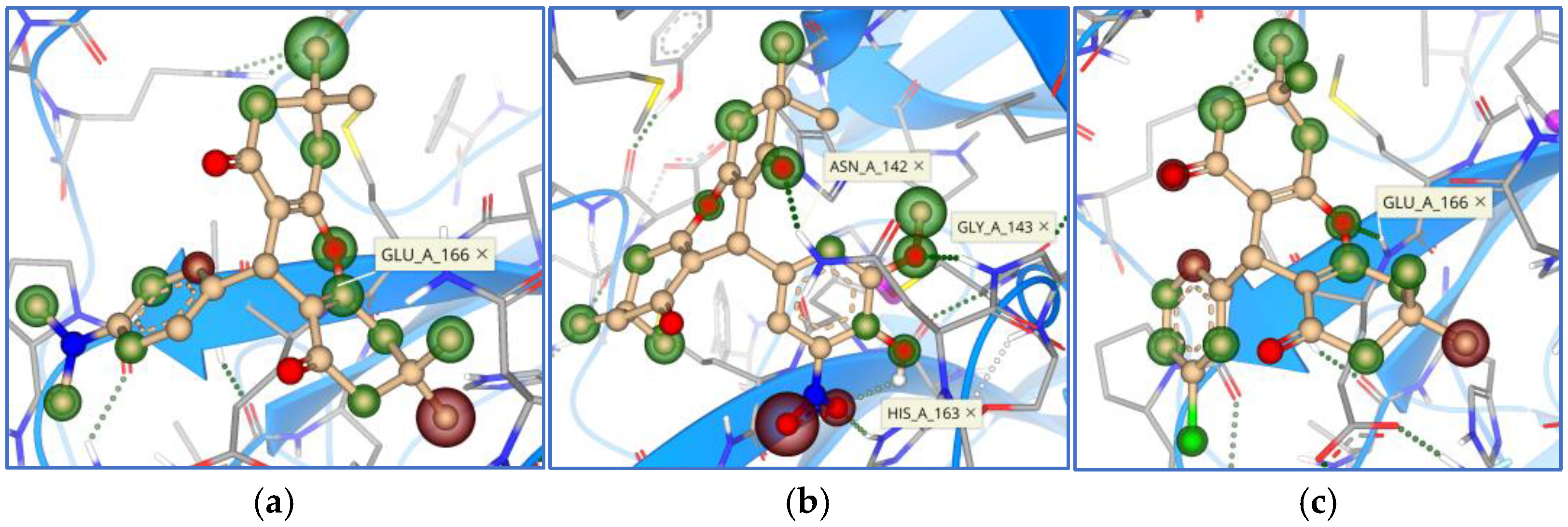
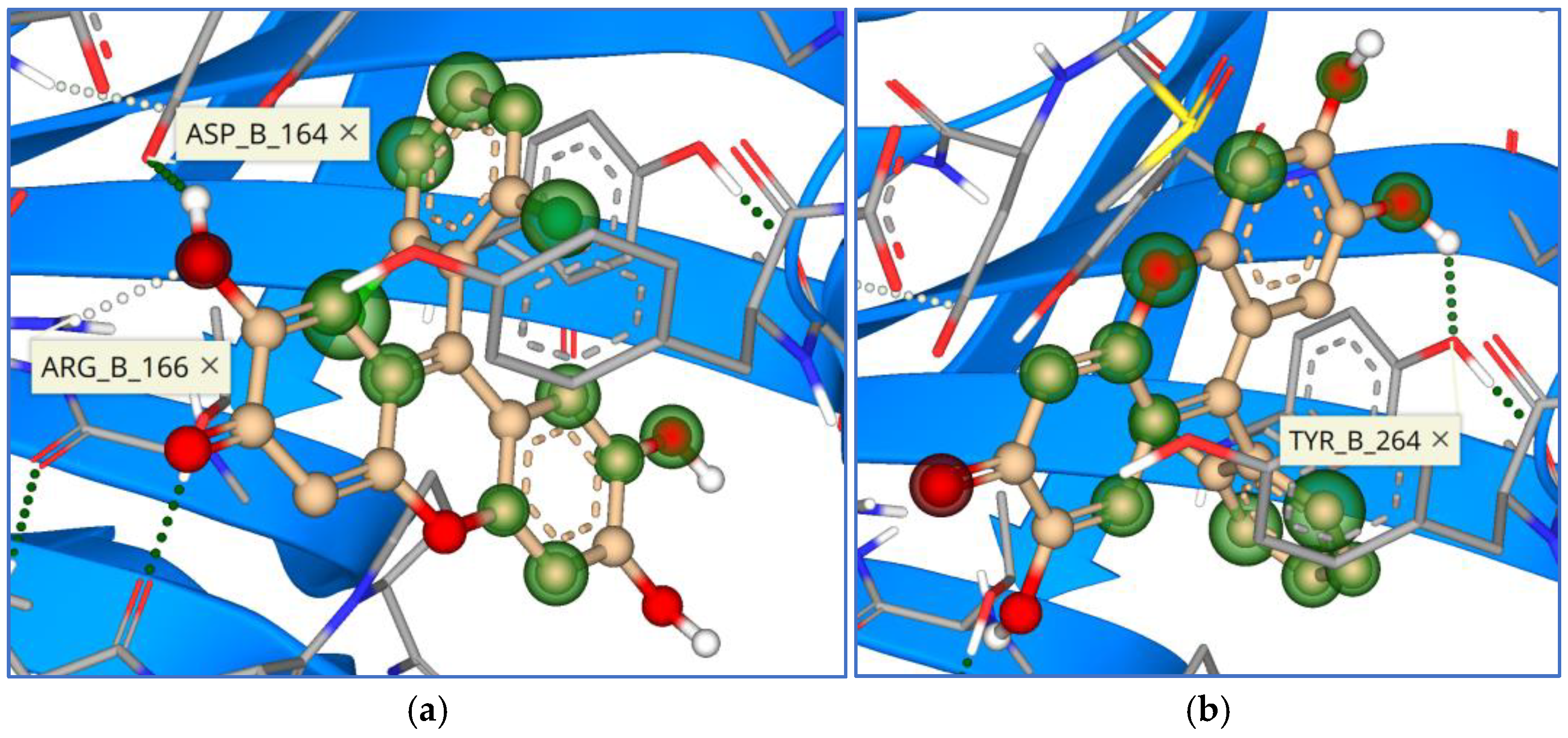
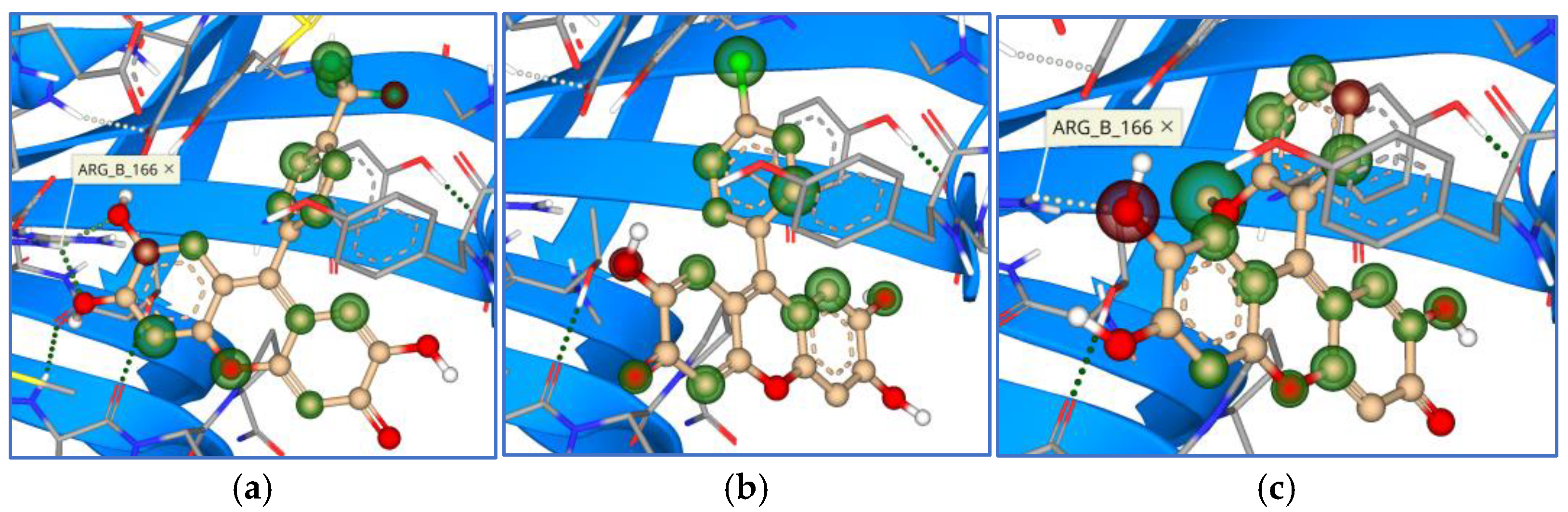
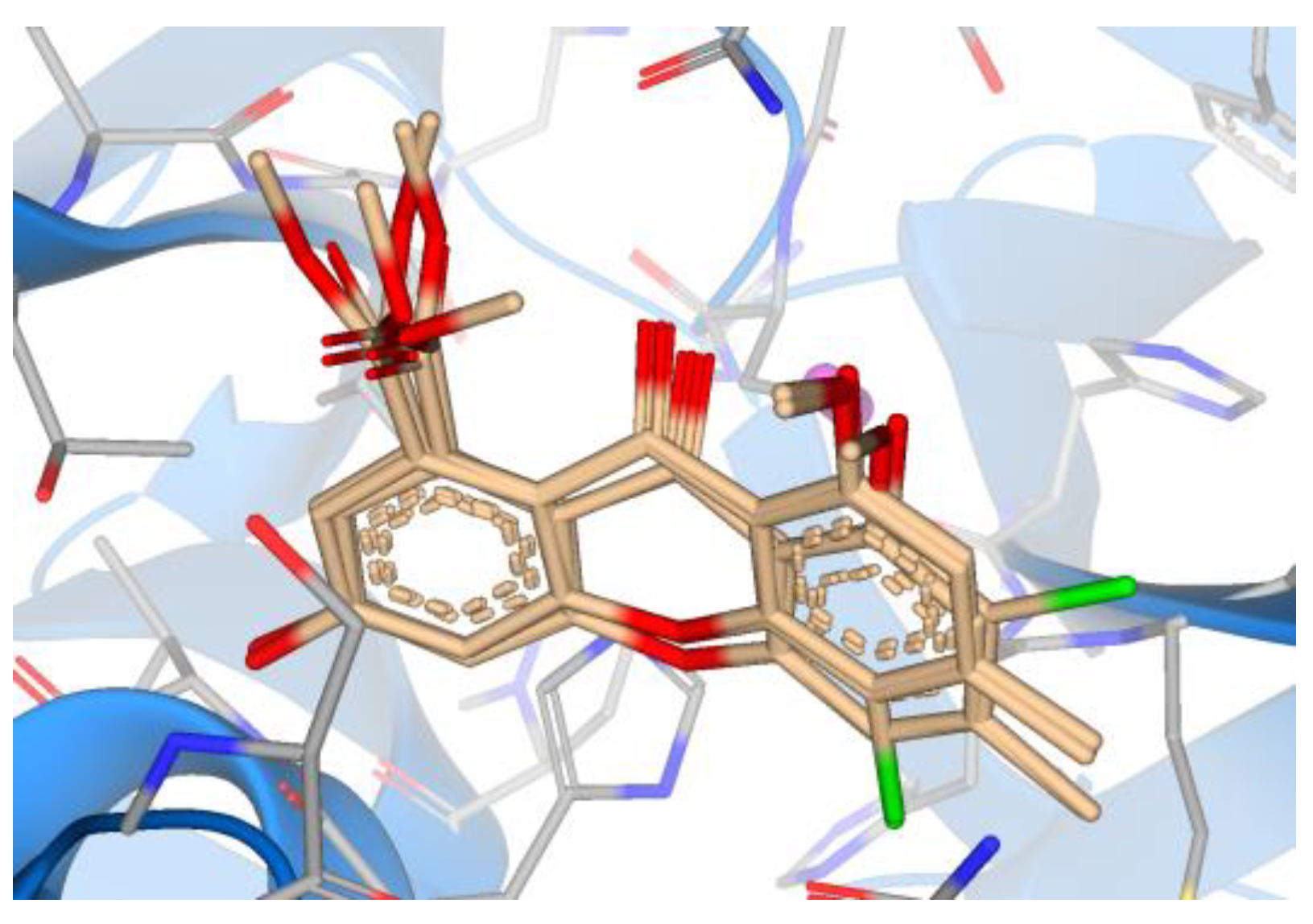
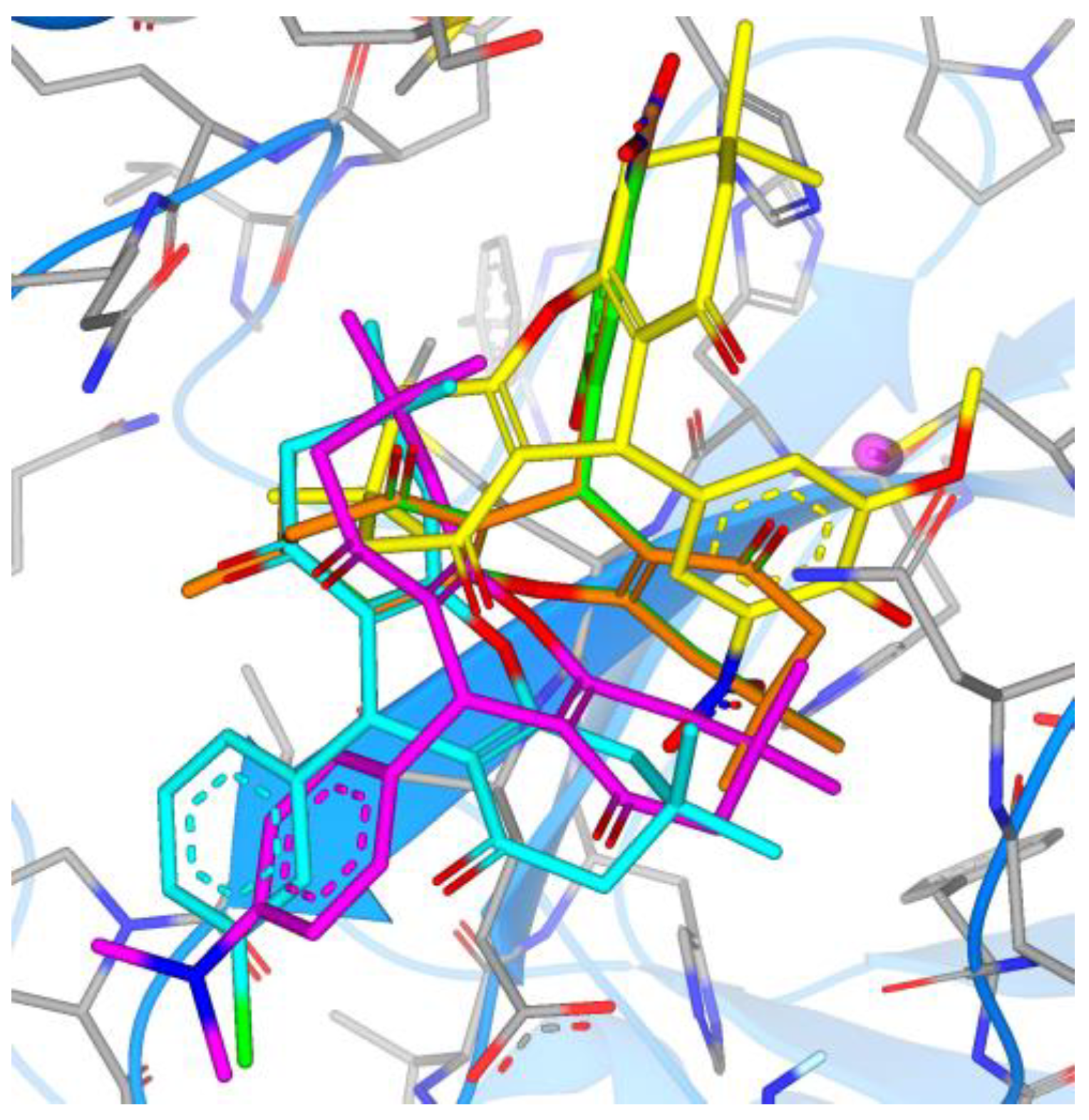
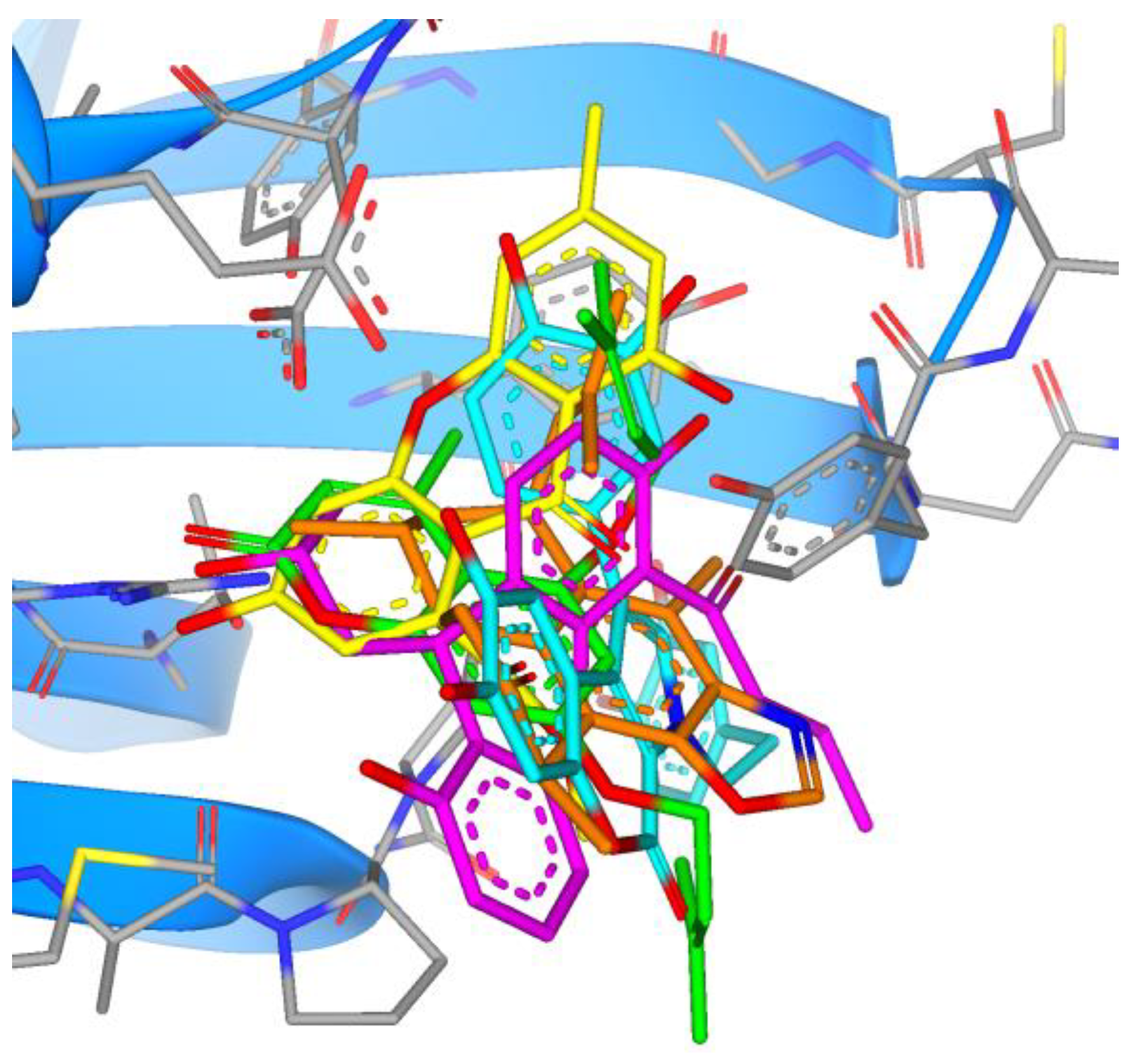
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fan, H.; Qin, S.; Cui, Y. Emergence and characterization of the SARS-CoV-2 JN. 1 variant: Global prevalence and implications for public health. Zoonoses 2024, 4, 994. [Google Scholar] [CrossRef]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.; Lau, E.H.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Number of COVID-19 Cases Reported to WHO (Cumulative Total). Available online: https://data.who.int/dashboards/covid19/cases?n=o (accessed on 5 December 2024).
- Number of COVID-19 Deaths Reported to WHO (Cumulative Total). Available online: https://data.who.int/dashboards/covid19/deaths?n=o (accessed on 5 December 2024).
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Peacock, S.J.; Barclay, W.S.; De Silva, T.I.; Towers, G.J.; Robertson, D.L. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Javed, S.; Farhan, B.A.; Shabbir, M.; Tahseen, A.; Ahmedah, H.T.; Moga, M. Morphology, Pathogenesis, Genome Organization, and Replication of Coronavirus (COVID-19). In The COVID-19 Pandemic, 1st ed.; Apple Academic Press: Palm Bay, FL, USA, 2022; pp. 3–43. [Google Scholar] [CrossRef]
- Zmasek, C.M.; Lefkowitz, E.J.; Niewiadomska, A.; Scheuermann, R.H. Genomic evolution of the coronaviridae family. Virology 2022, 570, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Hu, B.; Wang, L.F. Zoonotic Origin and Evolution of SARS Coronavirus. In Genetics and Evolution of Infectious Diseases, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2024; pp. 841–861. [Google Scholar] [CrossRef]
- Scheller, C.; Krebs, F.; Minkner, R.; Astner, I.; Gil-Moles, M.; Wätzig, H. Physicochemical properties of SARS-CoV-2 for drug targeting, virus inactivation and attenuation, vaccine formulation and quality control. Electrophoresis 2020, 41, 1137–1151. [Google Scholar] [CrossRef]
- Kumar, R.; Srivastava, Y.; Muthuramalingam, P.; Singh, S.K.; Verma, G.; Tiwari, S.; Tandel, N.; Beura, S.K.; Panigrahi, A.R.; Maji, S.; et al. Understanding mutations in human SARS-CoV-2 spike glycoprotein: A systematic review & meta-analysis. Viruses 2023, 15, 856. [Google Scholar] [CrossRef]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Singh, D.; Yi, S.V. On the origin and evolution of SARS-CoV-2. Exp. Mol. Med. 2021, 53, 537–547. [Google Scholar] [CrossRef]
- Chakraborty, C.; Bhattacharya, M.; Dhama, K. SARS-CoV-2 vaccines, vaccine development technologies, and significant efforts in vaccine development during the pandemic: The lessons learned might help to fight against the next pandemic. Vaccines 2023, 11, 682. [Google Scholar] [CrossRef]
- Jadhav, P.; Huang, B.; Osipiuk, J.; Zhang, X.; Tan, H.; Tesar, C.; Endres, M.; Jedrzejczak, R.; Tan, B.; Deng, X.; et al. Structure-based design of SARS-CoV-2 papain-like protease inhibitors. Eur. J. Med. Chem. 2024, 264, 116011. [Google Scholar] [CrossRef]
- Kakavandi, S.; Zare, I.; VaezJalali, M.; Dadashi, M.; Azarian, M.; Akbari, A.; Ramezani Farani, M.; Zalpoor, H.; Hajikhani, B. Structural and non-structural proteins in SARS-CoV-2: Potential aspects to COVID-19 treatment or prevention of progression of related diseases. Cell Commun. Signal. 2023, 21, 110. [Google Scholar] [CrossRef] [PubMed]
- Onyango, O.H. In Silico Models for Anti-COVID-19 Drug Discovery: A Systematic Review. Adv. Pharmacol. Pharm. Sci. 2023, 2023, 4562974. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Shahabi, D.; Imhoff, M.E.; Kovela, S.; Sharma, A.; Hattori, S.I.; Higashi-Kuwata, N.; Mitsuya, H.; Mesecar, A.D. SARS-CoV-2 papain-like protease (PLpro) inhibitory and antiviral activity of small molecule derivatives for drug leads. Bioorg. Med. Chem. Lett. 2023, 96, 129489. [Google Scholar] [CrossRef]
- Sanders, B.C.; Pokhrel, S.; Labbe, A.D.; Mathews, I.I.; Cooper, C.J.; Davidson, R.B.; Phillips, G.; Weiss, K.L.; Zhang, Q.; O’Neill, H.; et al. Potent and selective covalent inhibition of the papain-like protease from SARS-CoV-2. Nat. Commun. 2023, 14, 1733. [Google Scholar] [CrossRef]
- Funk, L.M.; Poschmann, G.; Rabe von Pappenheim, F.; Chari, A.; Stegmann, K.M.; Dickmanns, A.; Wensien, M.; Eulig, N.; Paknia, E.; Heyne, G.; et al. Multiple redox switches of the SARS-CoV-2 main protease in vitro provide opportunities for drug design. Nat. Commun. 2024, 15, 411. [Google Scholar] [CrossRef] [PubMed]
- Melano, I.; Lo, Y.; Su, W. Characterization of host substrates of SARS-CoV-2 main protease. Front. Microbiol. 2023, 14, 1251705. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.H.; Brewitz, L.; Lukacik, P.; Strain-Damerell, C.; Walsh, M.A.; Schofield, C.J.; Duarte, F. Studies on the selectivity of the SARS-CoV-2 papain-like protease reveal the importance of the P2′ proline of the viral polyprotein. RSC Chem. Biol. 2024, 5, 117–130. [Google Scholar] [CrossRef]
- Citarella, A.; Dimasi, A.; Moi, D.; Passarella, D.; Scala, A.; Piperno, A.; Micale, N. Recent advances in SARS-CoV-2 main protease inhibitors: From nirmatrelvir to future perspectives. Biomolecules 2023, 13, 1339. [Google Scholar] [CrossRef]
- Waqas, M.; Ullah, S.; Halim, S.A.; Rehman, N.U.; Ali, A.; Jan, A.; Muhsinah, A.B.; Khan, A.; Al-Harrasi, A. Targeting papain-like protease by natural products as novel therapeutic potential SARS-CoV-2. Int. J. Biol. Macromol. 2024, 258, 128812. [Google Scholar] [CrossRef]
- Banerjee, P.; Mandhare, A.; Bagalkote, V. Marine natural products as a source of new drugs: An updated patent review. Expert. Opin. Ther. Pat. 2021, 32, 317–363. [Google Scholar] [CrossRef]
- Chhetri, B.K.; Tedbury, P.R.; Sweeney-Jones, A.M.; Mani, L.; Soapi, K.; Manfredi, C.; Sorscher, E.; Sarafianos, S.G.; Kubanek, J. Marine natural products as leads against SARS-CoV-2 infection. J. Nat. Prod. 2022, 85, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Zaporozhets, T.S.; Besednova, N.N. Biologically active compounds from marine organisms in the strategies for combating coronaviruses. AIMS Microbiol. 2020, 6, 470. [Google Scholar] [CrossRef]
- Abdallah, H.M.; El-Halawany, A.M.; Sirwi, A.; El-Araby, A.M.; Mohamed, G.A.; Ibrahim, S.R.; Koshak, A.E.; Asfour, H.Z.; Awan, Z.A.; A. Elfaky, M. Repurposing of some natural product isolates as SARS-COV-2 main protease inhibitors via in vitro cell free and cell-based antiviral assessments and molecular modeling approaches. Pharmaceuticals 2021, 14, 213. [Google Scholar] [CrossRef]
- Wang, S.C.; Chen, Y.; Wang, Y.C.; Wang, W.J.; Yang, C.S.; Tsai, C.L.; Hou, M.H.; Chen, H.F.; Shen, Y.C.; Hung, M.C. Tannic acid suppresses SARS-CoV-2 as a dual inhibitor of the viral main protease and the cellular TMPRSS2 protease. Am. J. Cancer Res. 2020, 10, 4538. [Google Scholar] [CrossRef] [PubMed]
- White, K.M.; Rosales, R.; Yildiz, S.; Kehrer, T.; Miorin, L.; Moreno, E.; Jangra, S.; Uccellini, M.B.; Rathnasinghe, R.; Coughlan, L.; et al. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 2021, 371, 926–931. [Google Scholar] [CrossRef]
- Minagawa, K.; Kouzuki, S.; Yoshimoto, J.U.N.; Kawamura, Y.; Tani, H.; Iwata, T.; Terui, Y.; Nakai, H.; Yagi, S.; Hattori, N.; et al. Stachyflin and acetylstachyflin, novel anti-influenza A virus substances, produced by Stachybotrys sp. RF-7260 I. Isolation, structure elucidation and biological activities. J. Antibiot. 2002, 55, 155–164. [Google Scholar] [CrossRef]
- Tsujii, S.; Rinehart, K.L.; Gunasekera, S.P.; Kashman, Y.; Cross, S.S.; Lui, M.S.; Pomponi, S.A.; Diaz, M.C. Topsentin, bromotopsentin, and dihydrodeoxybromotopsentin: Antiviral and antitumor bis(indolyl)imidazoles from Caribbean deep-sea sponges of the family Halichondriidae. Structural and synthetic studies. J. Org. Chem. 1988, 53, 5446–5453. [Google Scholar] [CrossRef]
- Ishida, J.; Wang, H.K.; Oyama, M.; Cosentino, M.L.; Hu, C.Q.; Lee, K.H. Anti-AIDS agents. 46. Anti-HIV activity of harman, an anti-HIV principle from Symplocos setchuensis, and its derivatives. J. Nat. Prod. 2001, 64, 958–960. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Min, J.S.; Jang, M.S.; Lee, J.Y.; Shin, Y.S.; Park, C.M.; Song, J.H.; Kim, H.R.; Kim, S.; Jin, Y.H.; et al. Natural bis-benzylisoquinoline alkaloids-tetrandrine, fangchinoline, and cepharanthine, inhibit human coronavirus OC43 infection of MRC-5 human lung cells. Biomolecules 2019, 9, 696. [Google Scholar] [CrossRef]
- Cheng, P.N.L.; Chiang, L.; Lin, C. Antiviral effects of saikosaponins on human coronavirus 229e in vitro. Clin. Exp. Pharmacol. Physiol. 2006, 33, 612–616. [Google Scholar] [CrossRef]
- Hong, S.; Seo, S.H.; Woo, S.J.; Kwon, Y.; Song, M.; Ha, N.C. Epigallocatechin gallate inhibits the uridylate-specific endoribonuclease Nsp15 and efficiently neutralizes the SARS-CoV-2 strain. J. Agric. Food Chem. 2021, 69, 5948–5954. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhu, X.; Hao, L.; Zhao, M.; Hua, Q.; An, F. Bioactive indolyl diketopiperazines from the marine derived endophytic Aspergillus versicolor DY180635. Mar. Drugs 2020, 18, 338. [Google Scholar] [CrossRef] [PubMed]
- Cheohen, C.F.; Esteves, M.E.; da Fonseca, T.S.; Leal, C.M.; Assis, F.D.; Campos, M.F.; Rebelo, R.S.; Allonso, D.; Leitão, G.G.; da Silva, M.L.; et al. In silico screening of phenylethanoid glycosides, a class of pharmacologically active compounds as natural inhibitors of SARS-CoV-2 proteases. Comput. Struct. Biotechnol. J. 2023, 21, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Vegad, U.G.; Gajjar, N.D.; Nagar, P.R.; Chauhan, S.P.; Pandya, D.J.; Dhameliya, T.M. In silico screening, ADMET analysis and MD simulations of phytochemicals of Onosma bracteata Wall. as SARS CoV-2 inhibitors. 3 Biotech 2023, 13, 221. [Google Scholar] [CrossRef]
- Mohapatra, P.K.; Chopdar, K.S.; Dash, G.C.; Mohanty, A.K.; Raval, M.K. In silico screening and covalent binding of phytochemicals of Ocimum sanctum against SARS-CoV-2 (COVID 19) main protease. J. Biomol. Struct. Dyn. 2023, 41, 435–444. [Google Scholar] [CrossRef]
- Hu, C. Marine natural products and human immunity: Novel biomedical resources for anti-infection of SARS-CoV-2 and related cardiovascular disease. Nat. Prod. Bioprospecting 2024, 14, 12. [Google Scholar] [CrossRef] [PubMed]
- Salihović, M.; Osmanović, A.; Špirtović-Halilović, S.; Roca, S.; Meščić, A.; Vujisić, L.; Trifunović, S.; Završnik, D.; Sofić, E. Synthesis, structural, conformational and DFT studies of N-3 and O-4 alkylated regioisomers of 5-(hydroxypropyl) pyrimidine. J. Mol. Struct. 2015, 1091, 170–176. [Google Scholar] [CrossRef]
- Špirtović-Halilović, S.; Veljović, E.; Salihović, M.; Osmanović, A.; Šapčanin, A.; Softić, D.; Roca, S.; Trifunović, S.; Škrijelj, N.; Škrbo, S.; et al. Synthesis, Microbiological Activity and In Silico Investigation for Some Synthesized Coumarin Derivatives. Croat. Chem. Acta 2020, 93, 23–31. [Google Scholar] [CrossRef]
- Bilajac, E.; Osmanović, A.; Glamočlija, U.; Veljović, E.; Imamović, B.; Bečić, E.; Roca, S.; Salihović, M.; Završnik, D.; Špirtović-Halilović, S. Synthesis, In Silico Study and Antitumor Activity of Coumarin Compounds in Lymphoma Cells. Farmacia 2023, 71, 1263–1273. [Google Scholar] [CrossRef]
- Završnik, D.; Špirtović-Halilović, S.; Softić, D. Synthesis, structure and antibacterial activity of 3-substituted derivatives of 4-hydroxycoumarin. Period. Biol. 2011, 113, 93–97. [Google Scholar]
- Veljović, E.; Špirtović-Halilović, S.; Muratović, S.; Valek Žulj, L.; Roca, S.; Trifunović, S.; Osmanović, A.; Završnik, D. 9-Aryl substituted hydroxylated xanthen-3-ones: Synthesis, structure and antioxidant potency evaluation. Croat. Chem. Acta 2015, 88, 121–127. [Google Scholar] [CrossRef]
- Veljović, E.; Špirtović-Halilović, S.; Muratović, S.; Osmanović, A.; Badnjević, A.; Gurbeta, L.; Tatlić, B.; Zorlak, Z.; Imamović, S.; Husić, Đ.; et al. Artificial neural network and docking study in design and synthesis of xanthenes as antimicrobial agents. In CMBEBIH 2017, Proceedings of the International Conference on Medical and Biological Engineering, Sarajevo, Bosnia and Herzegovina, 16–18 March 2017; Badnjević, A., Ed.; Springer: Singapore, 2017; Volume 62, pp. 617–626. [Google Scholar] [CrossRef]
- Zukić, S.; Veljović, E.; Špirtović-Halilović, S.; Muratović, S.; Osmanović, A.; Trifunović, S.; Novaković, I.; Završnik, D. Antioxidant, Antimicrobial and Antiproliferative Activities of Synthesized 2,2,5,5-Tetramethyl-9-aryl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione Derivatives. Croat. Chem. Acta 2018, 91, 1–9. [Google Scholar] [CrossRef]
- Veljović, E.; Špirtović-Halilović, S.; Muratović, S.; Osmanović, A.; Novaković, I.; Trifunović, S.; Završnik, D. Synthesis and biological evaluation of xanthen-1,8-dione derivatives. Bull. Chem. Technol. Bosnia Herzegovina 2018, 51, 13–18. [Google Scholar]
- Glamočlija, U.; Padhye, S.; Špirtović-Halilović, S.; Osmanović, A.; Veljović, E.; Roca, S.; Novaković, I.; Mandić, B.; Turel, I.; Kljun, J.; et al. Synthesis, biological evaluation and docking studies of benzoxazoles derived from thymoquinone. Molecules 2018, 23, 3297. [Google Scholar] [CrossRef]
- Bilajac, E.; Glamočlija, U.; Osmanović, A.; Mahmutović, L.; Sezer, A.; Roca, S.; Špirtović-Halilović, S.; Salihović, M.; Hromić-Jahjefendić, A.; Veljović, E. Analysis of Antitumor Potential of Xanthene Compounds in Lymphoma Cells. Croat. Chem. Acta 2023, 96, 59–68. [Google Scholar] [CrossRef]
- Spirtović-Halilović, S.; Salihović, M.; Osmanović, A.; Veljović, E.; Rahić, O.; Mahmutović, E.; Hadziabdić, J.; Novaković, I.; Roca, S.; Trifunović, S.; et al. In Silico Study of Microbiologically Active Benzoxazole Derivatives. Indian J. Pharm. Sci. 2023, 85, 767–777. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Tan, B.; Zhang, X.; Ansari, A.; Jadhav, P.; Tan, H.; Li, K.; Chopra, A.; Ford, A.; Chi, X.; Ruiz, F.X.; et al. Design of a SARS-CoV-2 papain-like protease inhibitor with antiviral efficacy in a mouse model. Science 2024, 383, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.; Lange, G.; Hindle, S.; Klein, R.; Rarey, M. A consistent description of HYdrogen bond and DEhydration energies in protein–ligand complexes: Methods behind the HYDE scoring function. J. Comput.-Aided Mol. Des. 2013, 27, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Reulecke, I.; Lange, G.; Albrecht, J.; Klein, R.; Rarey, M. Towards an Integrated Description of Hydrogen Bonding and Dehydration: Decreasing False Positives in Virtual Screening with the HYDE Scoring Function. ChemMedChem 2008, 3, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G.A. Fast Flexible Docking Method using an Incremental Construction Algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef]
- Gastreich, M.; Lilienthal, M.; Briem, H.; Claussen, H. Ultrafast de novo docking combining pharmacophores and combinatorics. J. Comput.-Aided Mol. Des. 2007, 20, 717–734. [Google Scholar] [CrossRef]
- Duan, Y.; Wang, H.; Yuan, Z.; Yang, H. Structural biology of SARS-CoV-2 Mpro and drug discovery. Curr. Opin. Struct. Biol. 2023, 82, 102667. [Google Scholar] [CrossRef]
- Zagórska, A.; Czopek, A.; Fryc, M.; Jończyk, J. Inhibitors of SARS-CoV-2 main protease (Mpro) as anti-coronavirus agents. Biomolecules 2024, 14, 797. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘Rule of Three’ for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef] [PubMed]
- Yan, A.; Wang, Z.; Cai, Z. Prediction of human intestinal absorption by GA feature selection and support vector machine regression. Int. J. Mol. Sci. 2008, 9, 1961–1976. [Google Scholar] [CrossRef]
- Ghafourian, T.; Amin, Z. QSAR models for the prediction of plasma protein binding. BioImpacts 2013, 3, 21–27. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood–brain barrier. Cold Spring Harbor Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Summerfield, S.G.; Read, K.; Begley, D.J.; Obradovic, T.; Hidalgo, I.J.; Coggon, S.; Lewis, A.V.; Porter, R.A.; Jeffrey, P. Central nervous system drug disposition: The relationship between in situ brain permeability and brain free fraction. J. Pharmacol. Exp. Ther. 2007, 322, 205–213. [Google Scholar] [CrossRef]

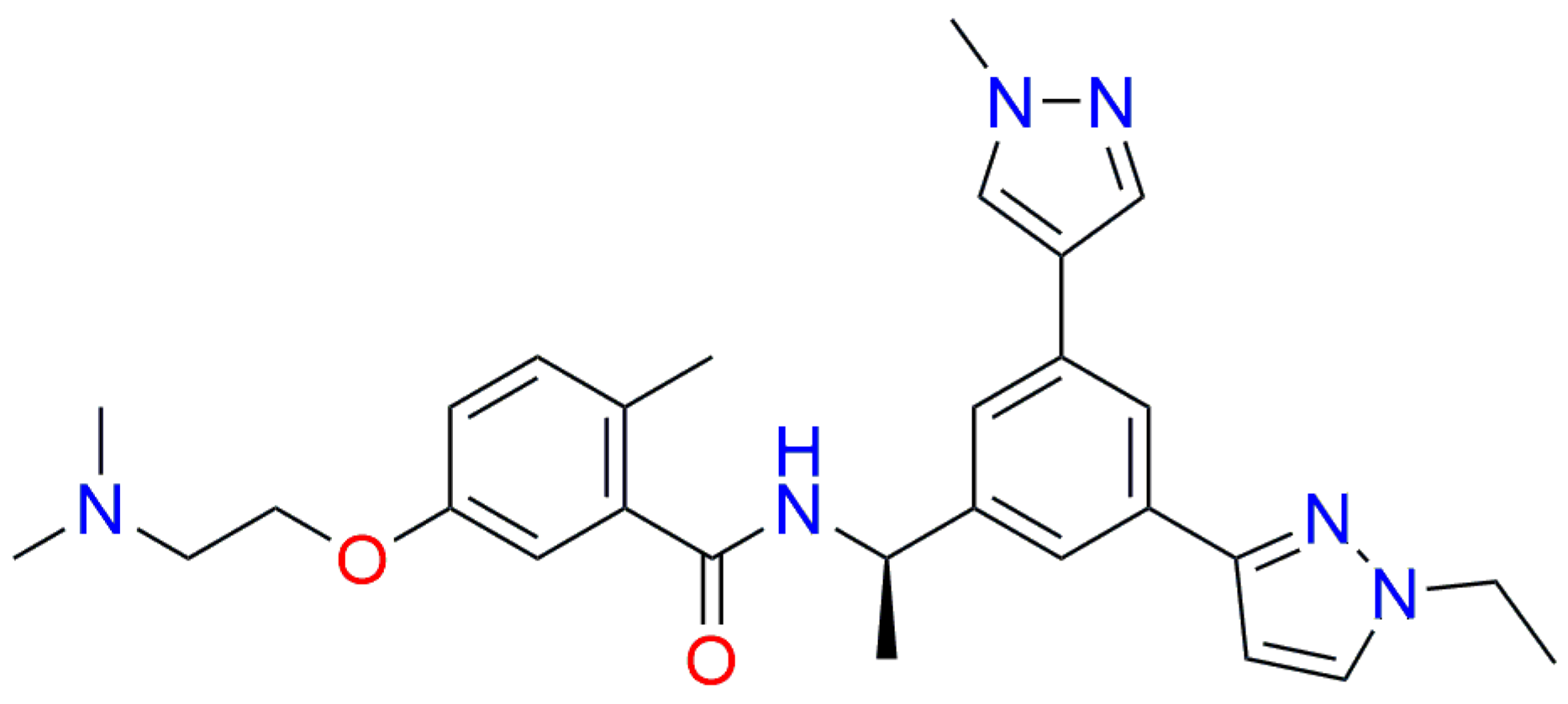
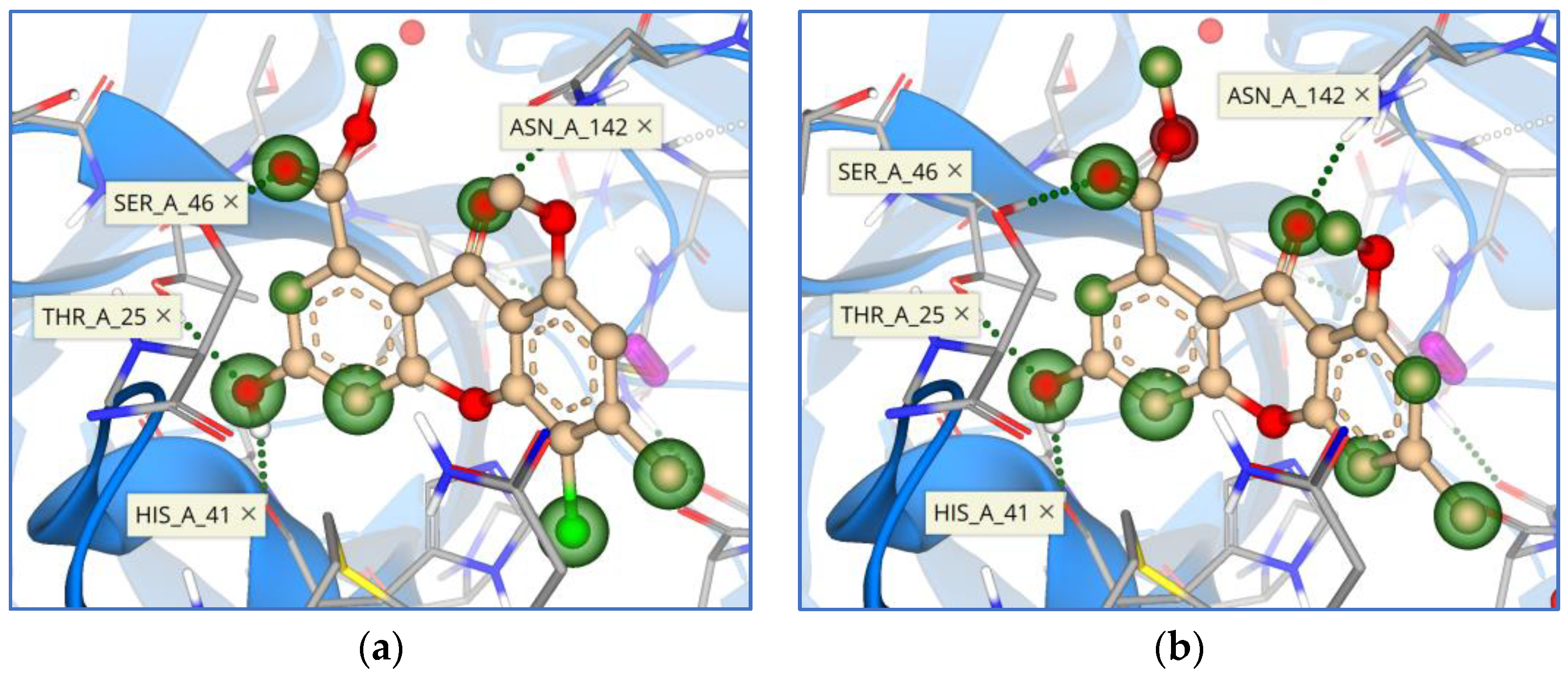
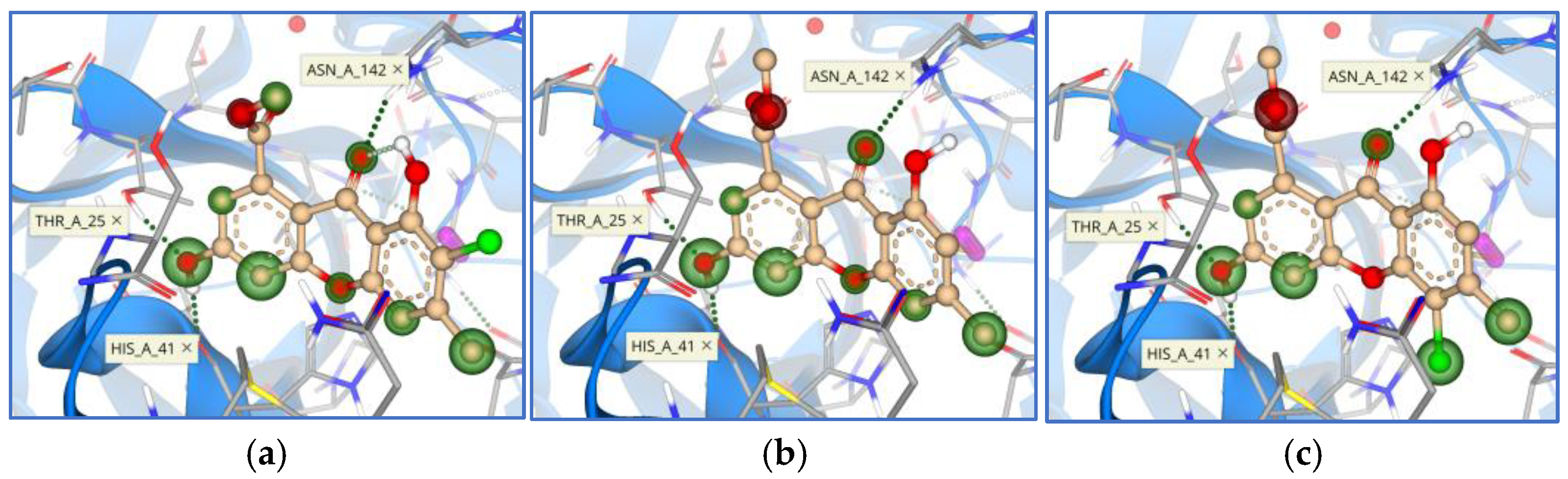
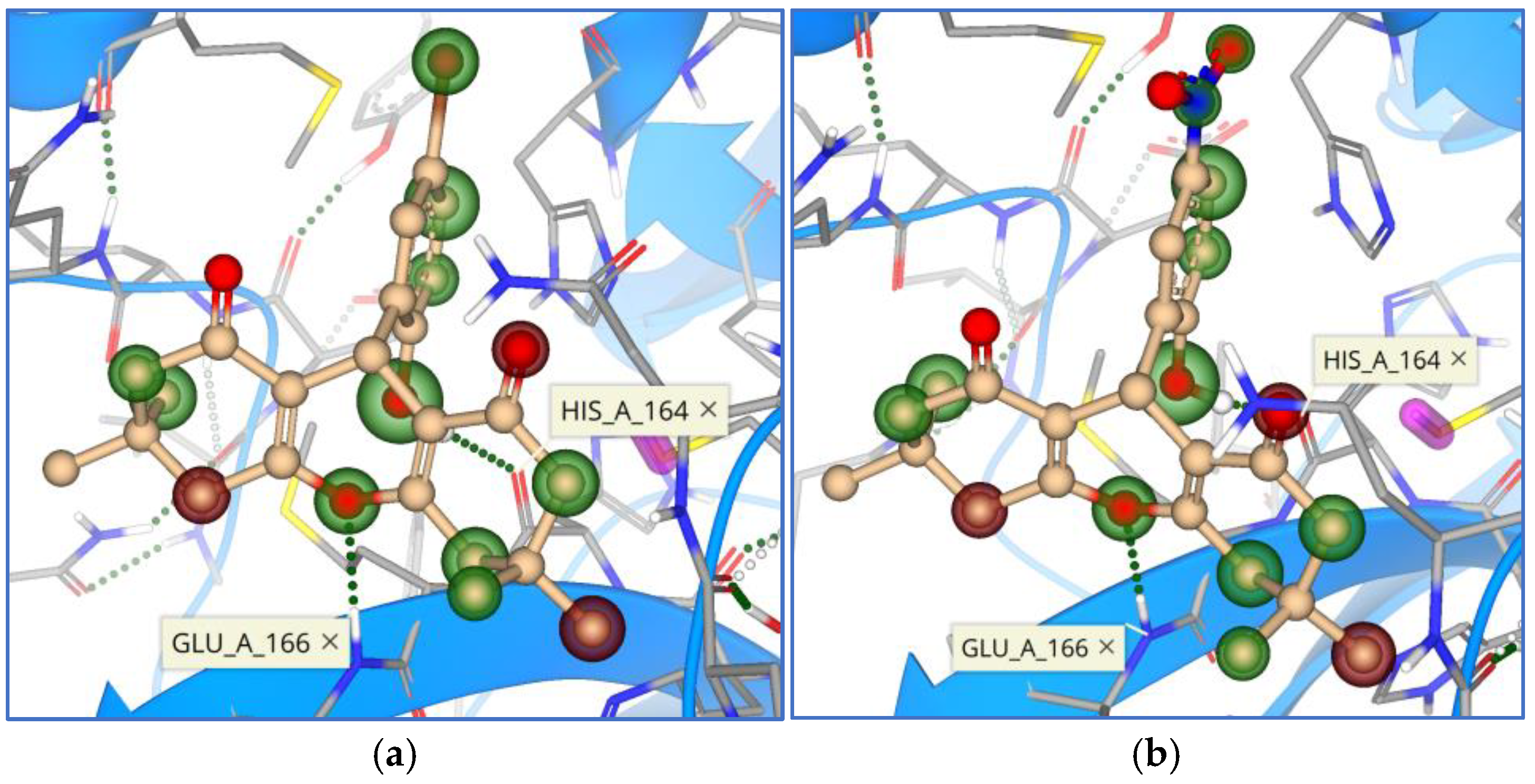




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
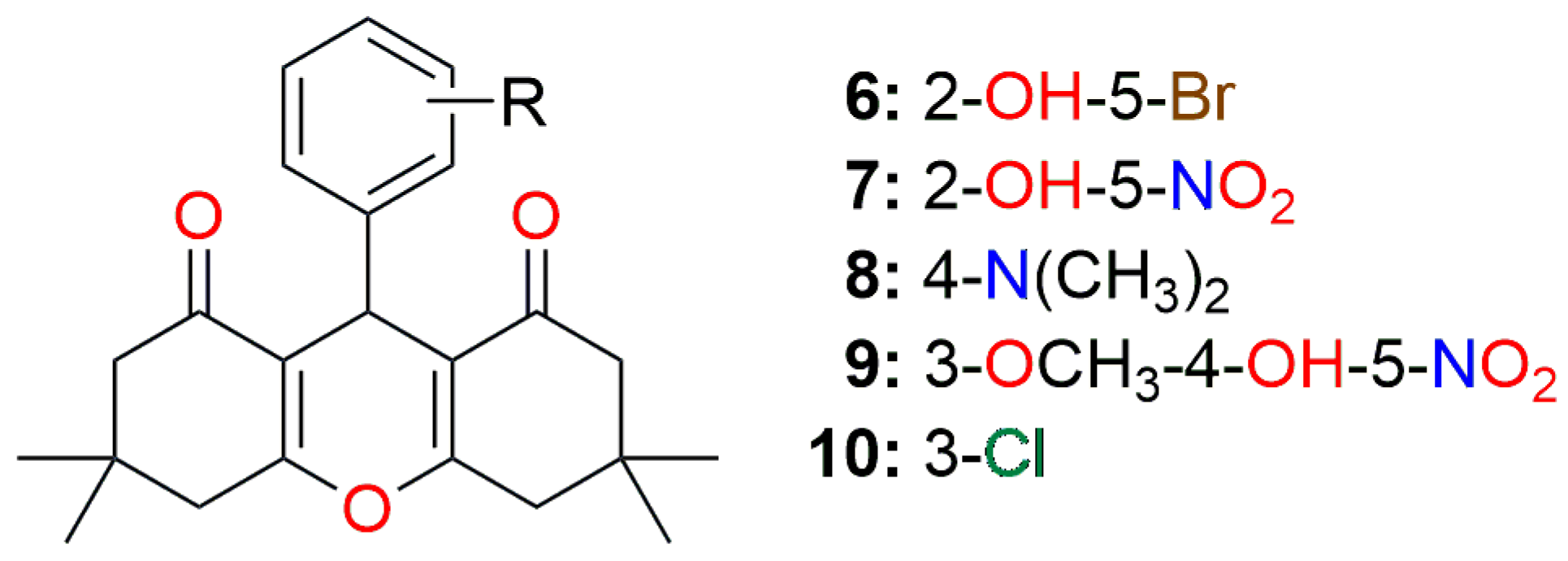
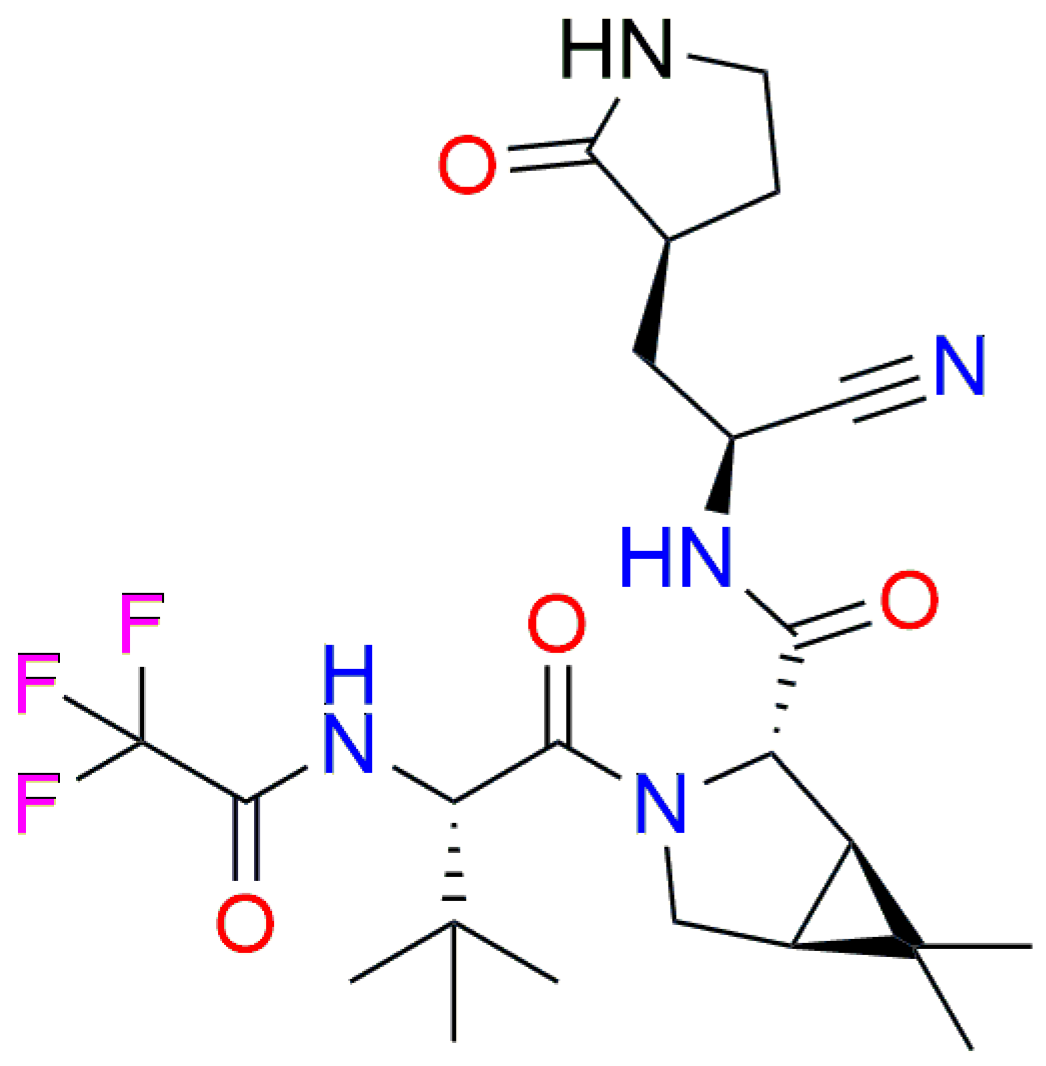
| DDP | Comp. (1) | Comp. (2) | Comp. (3) | Comp. (4) | Comp. (5) |
|---|---|---|---|---|---|
| L/U affinity [mM] | 0.002/0.224 | 0.004/0.442 | 0.035/3.509 | 0.062/6.134 | 0.071/7.024 |
| Mw (g/mol) | 348.7 | 314.3 | 334.7 | 300.3 | 334.7 |
| n H-acc. | 5 | 5 | 5 | 5 | 5 |
| n H-don. | 1 | 1 | 2 | 2 | 2 |
| n rot. bond | 0 | 0 | 0 | 0 | 0 |
| TPSA (Å2) | 82.1 | 82.1 | 93.1 | 93.1 | 93.1 |
| logP | 3.50 | 2.93 | 3.78 | 3.33 | 3.86 |
| logD | 3.50 | 2.93 | 1.56 | 1.01 | 1.50 |
| logS @ pH 7.4 | 1.36 | 2.19 | 3.90 | 3.98 | 3.88 |
| HIA | + | + | + | + | + |
| BBB | - | - | - | - | - |
| PPB90 | High | High | High | High | High |
| 2C9 pKi | 5.66 | 5.46 | 5.48 | 5.28 | 5.44 |
| 2D6 | Medium | Medium | Medium | Medium | Medium |
| P-gp | No | No | No | No | No |
| hERG pIC50 | 5.23 | 5.07 | 4.81 | 4.79 | 4.94 |
| DDP | Comp. * (6) | Comp. (7) | Comp. (8) | Comp. (9) | Comp. (10) |
|---|---|---|---|---|---|
| L/U affinity [mM] | 0.005/0.473 | 0.008/0.747 | 0.012/1.211 | 0.019/1.904 | 0.046/4.594 |
| Mw (g/mol) | 445.4 | 411.5 | 393.5 | 441.5 | 384.9 |
| n H-acc. | 4 | 6 | 3 | 7 | 3 |
| n H-don. | 1 | 1 | 0 | 1 | 0 |
| n rot. bond | 1 | 1 | 1 | 1 | 1 |
| TPSA (Å2) | 63.6 | 109.4 | 46.6 | 118.7 | 43.4 |
| logP | 4.70 | 3.99 | 4.52 | 3.88 | 5.00 |
| logD | 4.70 | 3.99 | 2.58 | 3.88 | 5.00 |
| logS @ pH 7.4 | 0.80 | 0.75 | 2.10 | −0.06 | 0.10 |
| HIA | + | + | + | + | + |
| BBB | + | - | + | - | + |
| PPB90 | High | High | High | High | High |
| 2C9 pKi | 5.34 | 5.27 | 5.20 | 5.32 | 5.29 |
| 2D6 | High | High | High | High | High |
| P-gp | Yes | Yes | Yes | Yes | No |
| hERG pIC50 | 4.96 | 4.79 | 5.24 | 4.44 | 4.83 |
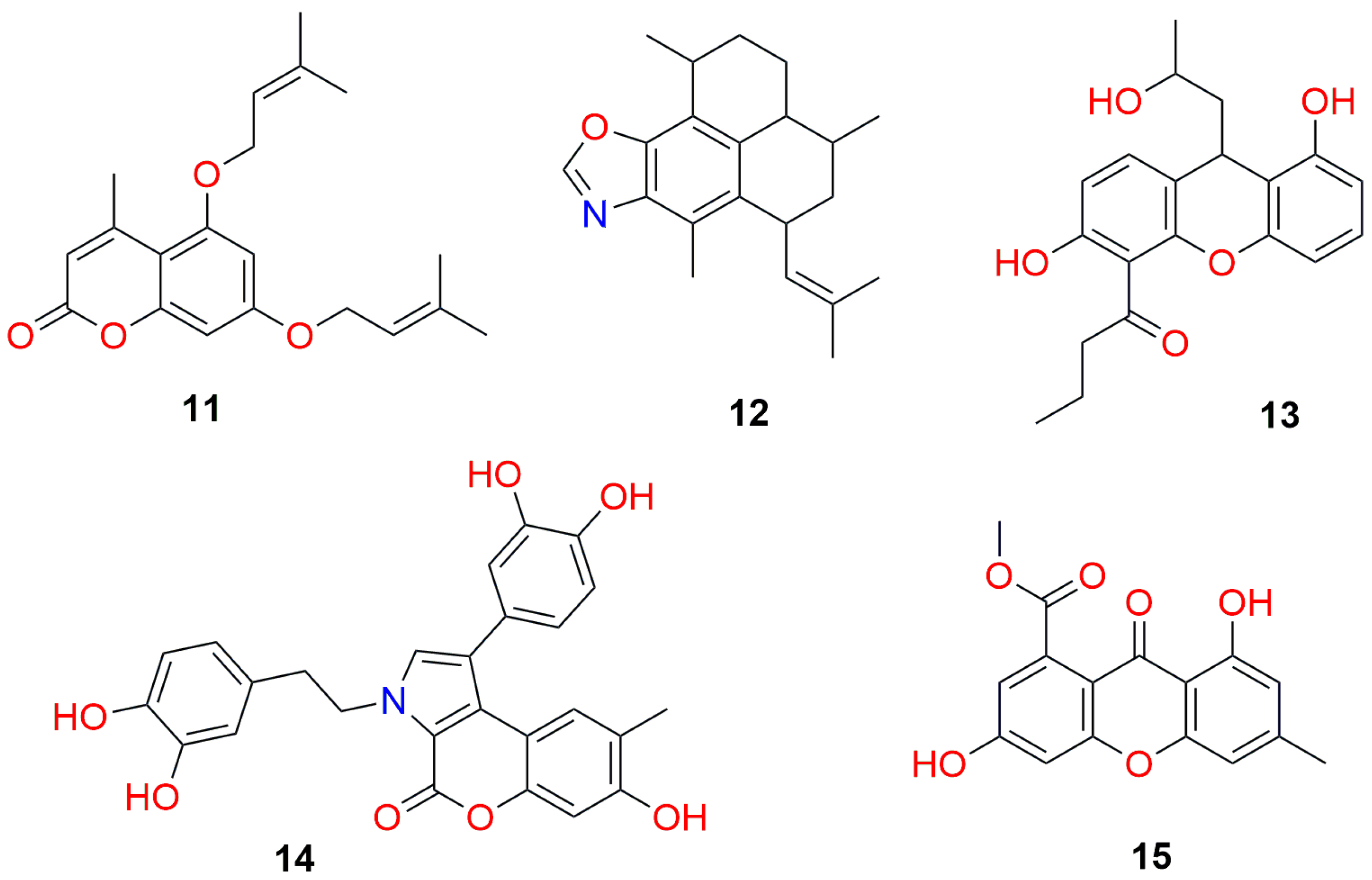
| DDP | Comp (11) | Comp. (12) | Comp. (13) | Comp. (14) | Comp. (15) |
|---|---|---|---|---|---|
| L/U affinity [mM] | 0.0001/0.010 | 0.003/0.299 | 0.003/0.341 | 0.004/0.431 | 0.005/0.483 |
| Mw (g/mol) | 328.4 | 309.4 | 342.4 | 461.4 | 300.3 |
| n H-acc. | 4 | 2 | 4 | 8 | 5 |
| n H-don. | 0 | 0 | 3 | 6 | 2 |
| n rot. bond | 4 | 1 | 4 | 3 | 0 |
| TPSA (Å2) | 44.76 | 26.03 | 86.99 | 152.61 | 93.06 |
| logP | 4.28 | 6.46 | 3.82 | 2.51 | 3.33 |
| logD | 4.28 | 6.46 | 3.82 | 2.51 | 1.01 |
| logS @ pH 7.4 | 1.08 | 0.57 | 1.55 | 2.39 | 3.98 |
| HIA | + | + | + | - | + |
| BBB | + | + | - | - | - |
| PPB90 | High | High | High | Low | High |
| 2C9 pKi | 4.86 | 5.23 | 5.33 | 5.83 | 5.28 |
| 2D6 | Medium | Very high | High | Medium | Medium |
| P-gp | No | Yes | Yes | Yes | No |
| hERG pIC50 | 5.74 | 6.06 | 5.07 | 5.70 | 4.79 |
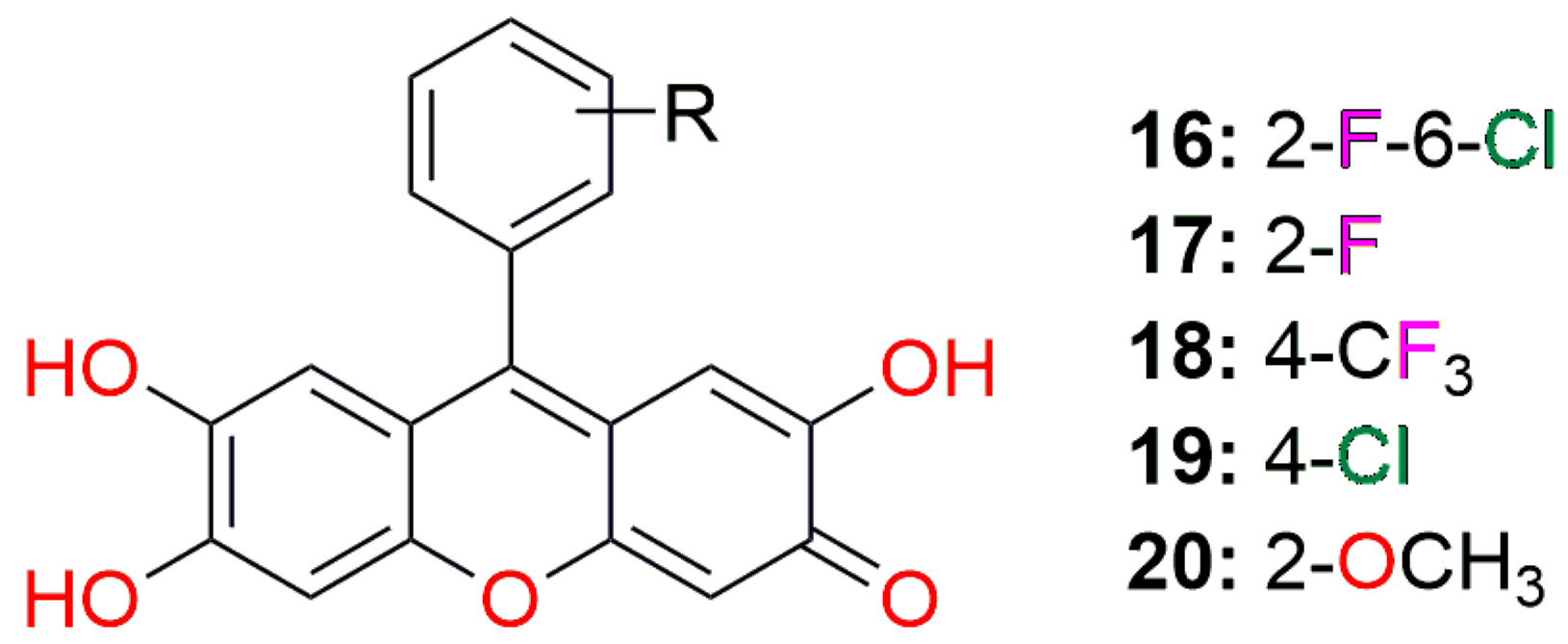
| DDP | Comp. (16) | Comp. (17) | Comp. (18) | Comp. (19) | Comp. (20) |
|---|---|---|---|---|---|
| L/U affinity [mM] | 0.0017/0.171 | 0.0023/0.223 | 0.0024/0.242 | 0.0025/0.246 | 0.0029/0.286 |
| Mw (g/mol) | 372.7 | 338.3 | 388.3 | 354.7 | 350.3 |
| n H-acc. | 2 | 1 | 2 | 0 | 1 |
| n H-don. | 4 | 4 | 4 | 4 | 5 |
| n rot. bond | 3 | 3 | 3 | 3 | 3 |
| TPSA (Å2) | 86.99 | 86.99 | 86.99 | 86.99 | 96.22 |
| logP | 3.90 | 3.46 | 3.83 | 3.95 | 3.23 |
| logD | 3.90 | 3.46 | 3.83 | 3.95 | 3.23 |
| logS @ pH 7.4 | 2.07 | 2.51 | 2.21 | 2.46 | 2.59 |
| HIA | + | + | + | + | + |
| BBB | - | - | - | - | - |
| PPB90 | High | High | High | High | Low |
| 2C9 pKi | 6.04 | 5.96 | 6.14 | 5.99 | 5.92 |
| 2D6 | Low | Low | High | Low | Low |
| P-gp | No | No | No | No | No |
| hERG pIC50 | 5.03 | 5.06 | 5.20 | 4.99 | 4.81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Published by MDPI on behalf of the Österreichische Pharmazeutische Gesellschaft. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osmanović, A.; Salihović, M.; Veljović, E.; Hindija, L.; Pazalja, M.; Malenica, M.; Selmanagić, A.; Špirtović-Halilović, S. Marine Origin vs. Synthesized Compounds: In Silico Screening for a Potential Drug Against SARS-CoV-2. Sci. Pharm. 2025, 93, 2. https://doi.org/10.3390/scipharm93010002
Osmanović A, Salihović M, Veljović E, Hindija L, Pazalja M, Malenica M, Selmanagić A, Špirtović-Halilović S. Marine Origin vs. Synthesized Compounds: In Silico Screening for a Potential Drug Against SARS-CoV-2. Scientia Pharmaceutica. 2025; 93(1):2. https://doi.org/10.3390/scipharm93010002
Chicago/Turabian StyleOsmanović, Amar, Mirsada Salihović, Elma Veljović, Lamija Hindija, Mirha Pazalja, Maja Malenica, Aida Selmanagić, and Selma Špirtović-Halilović. 2025. "Marine Origin vs. Synthesized Compounds: In Silico Screening for a Potential Drug Against SARS-CoV-2" Scientia Pharmaceutica 93, no. 1: 2. https://doi.org/10.3390/scipharm93010002
APA StyleOsmanović, A., Salihović, M., Veljović, E., Hindija, L., Pazalja, M., Malenica, M., Selmanagić, A., & Špirtović-Halilović, S. (2025). Marine Origin vs. Synthesized Compounds: In Silico Screening for a Potential Drug Against SARS-CoV-2. Scientia Pharmaceutica, 93(1), 2. https://doi.org/10.3390/scipharm93010002