
Binding of GS-461203 and Its Halogen Derivatives to HCV Genotype 2a RNA Polymerase Drug Resistance Mutants

 ,
, 
Abstract
:1. Introduction
2. Results
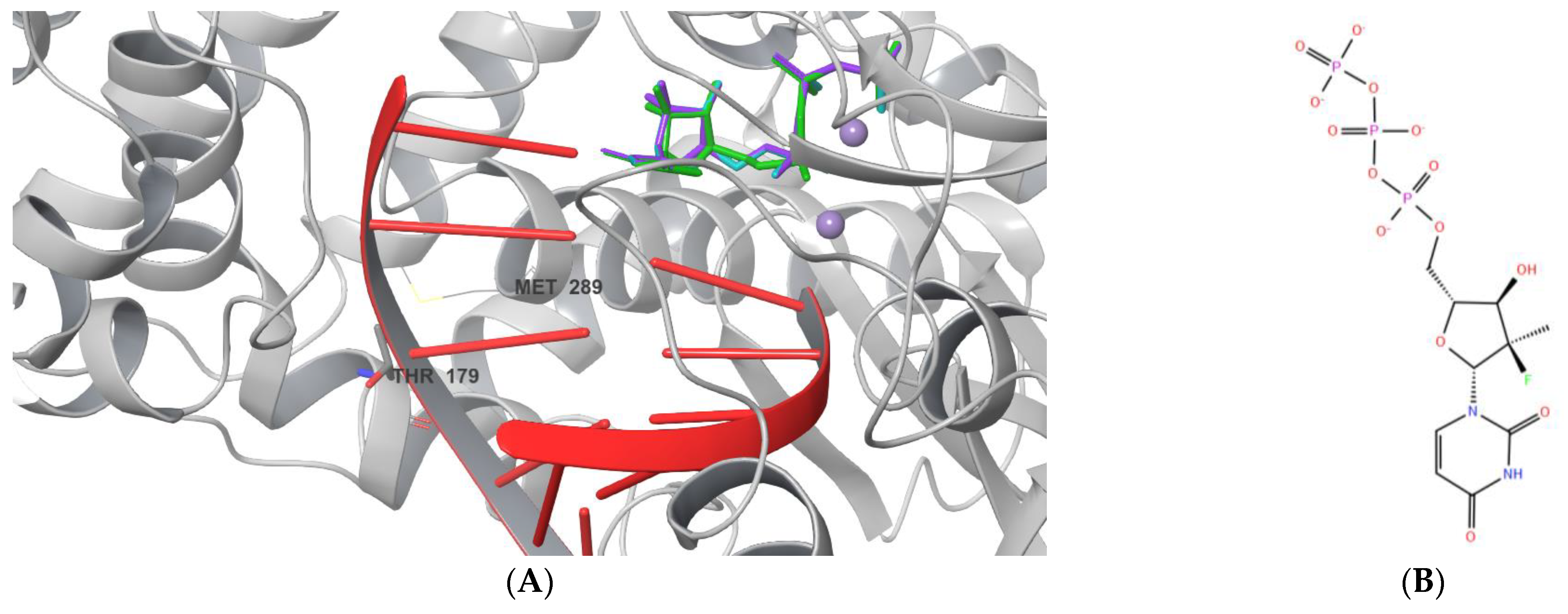
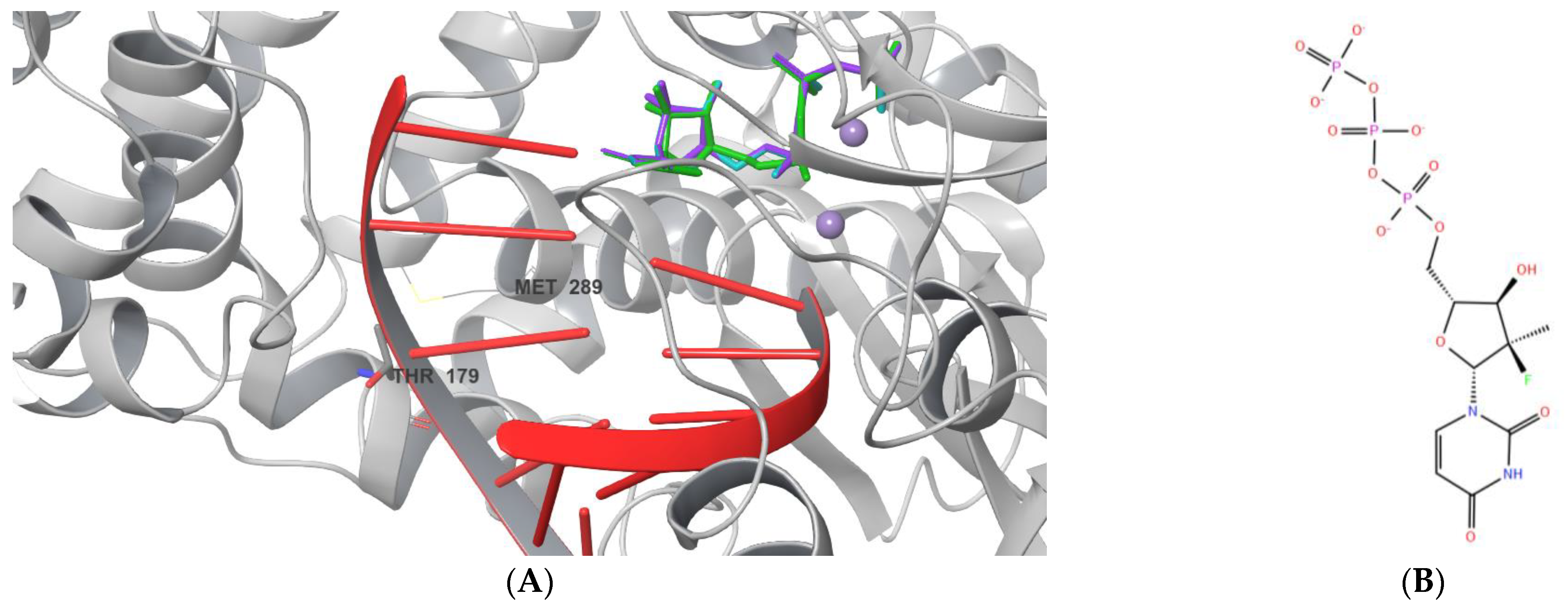
2.1. Ligand Docking
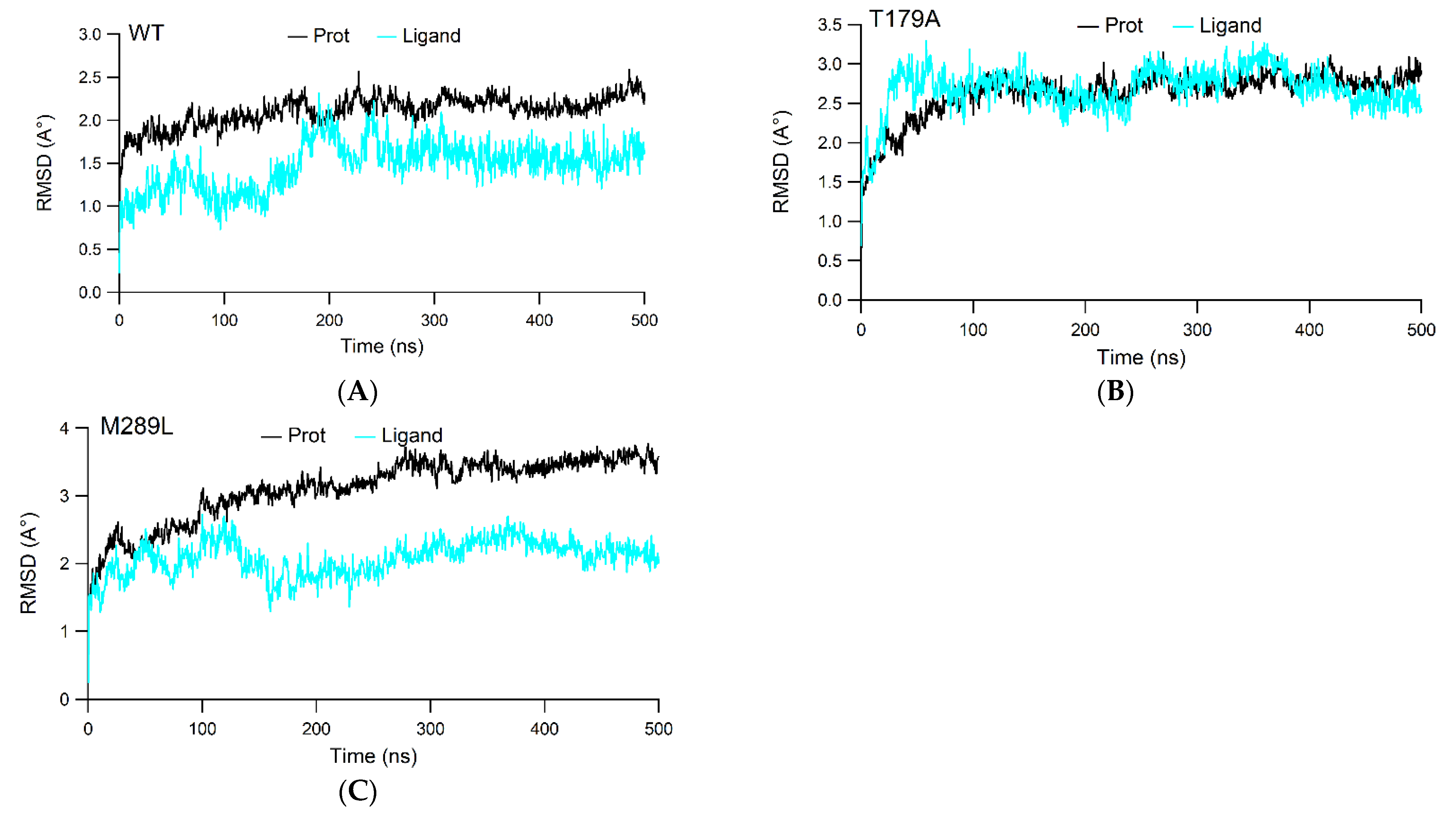
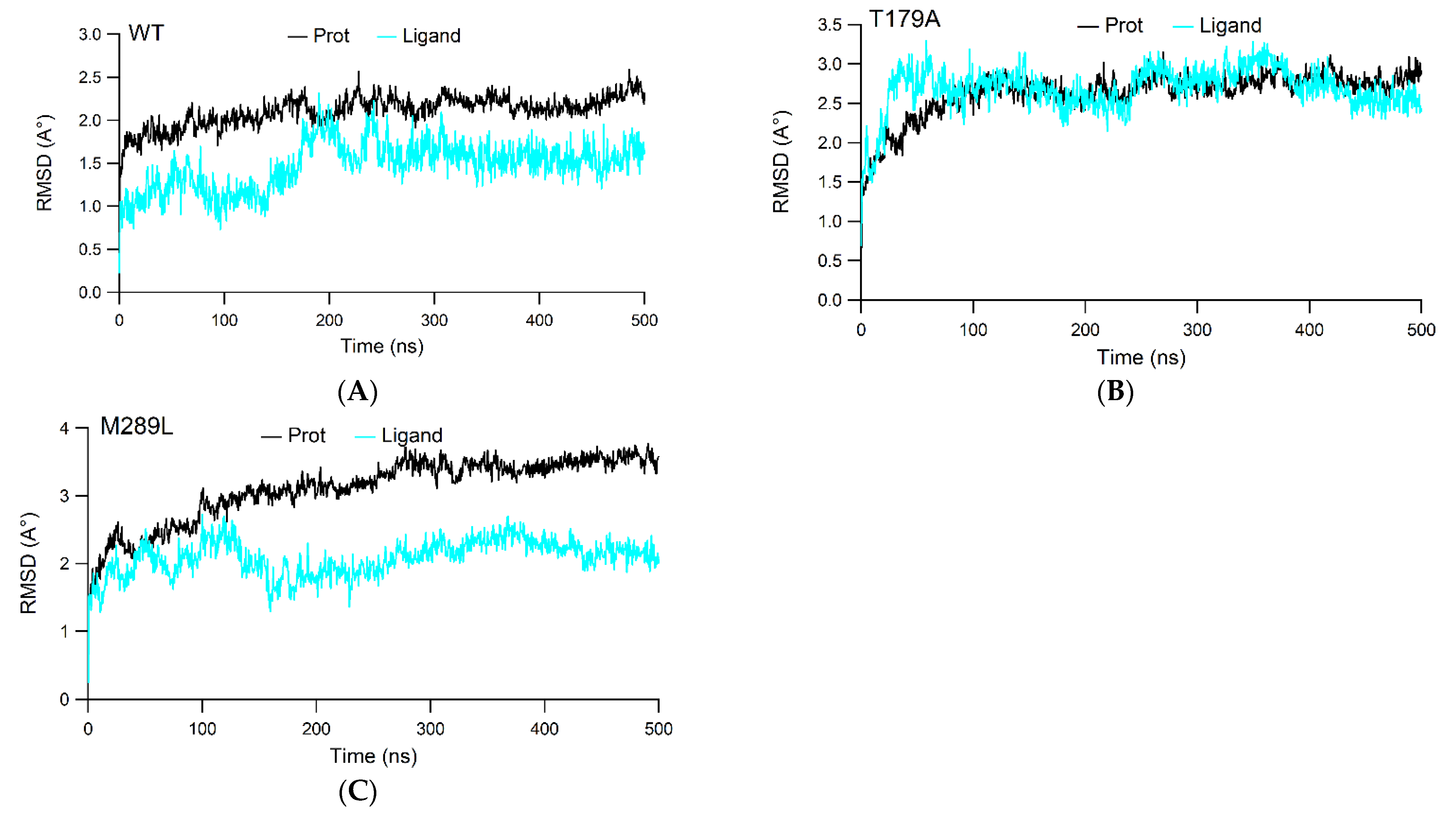
2.2. The RMSD Values
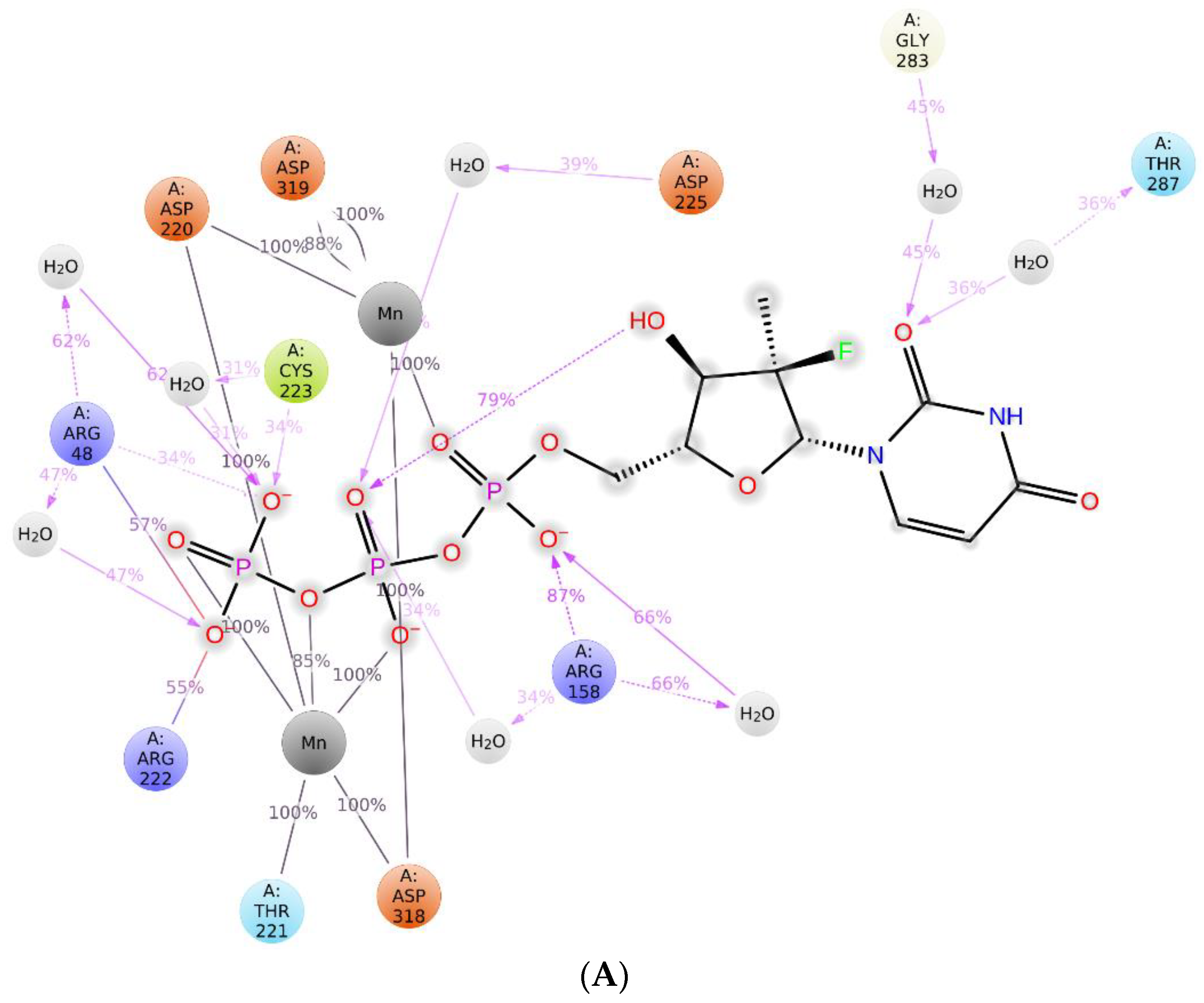
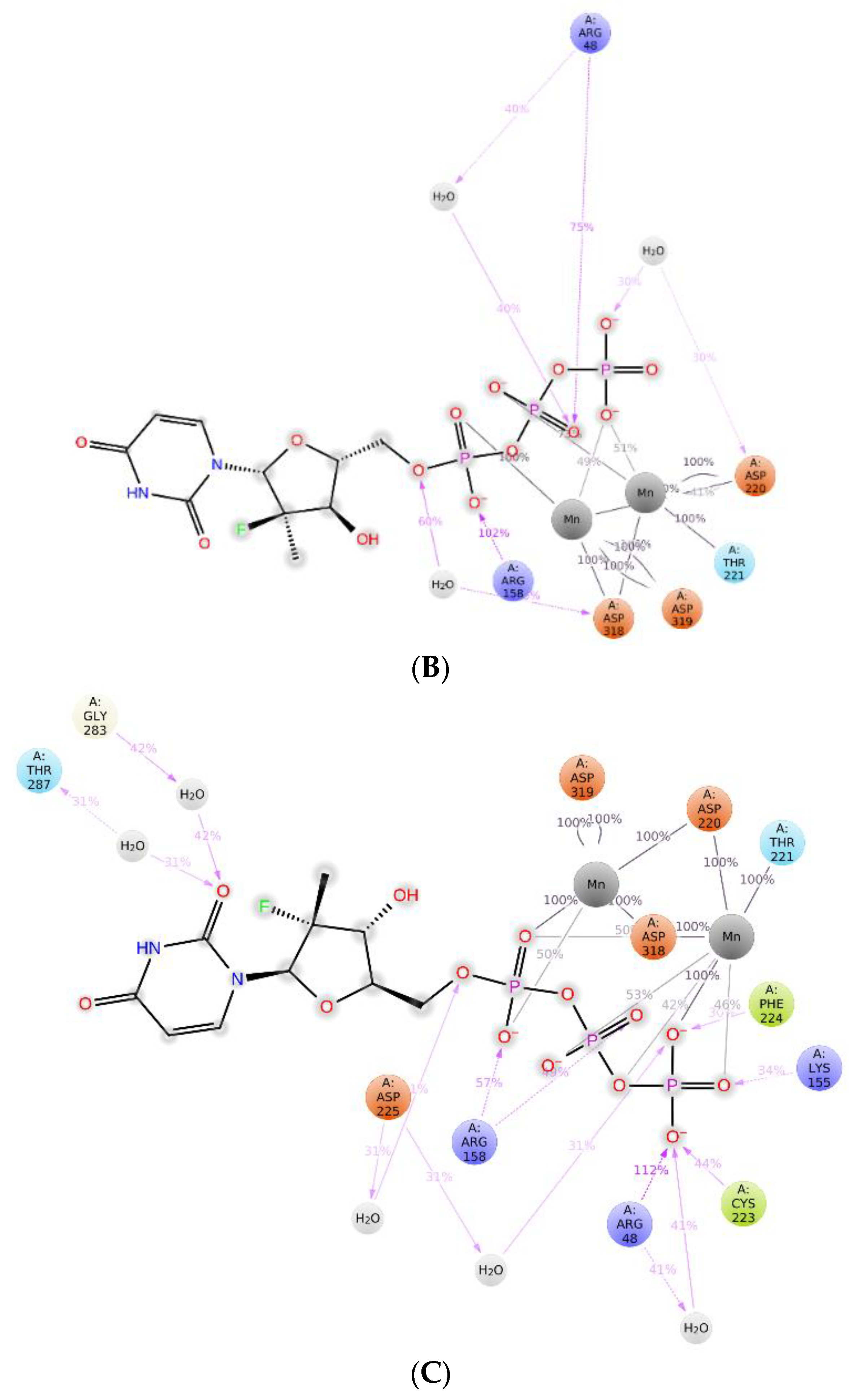
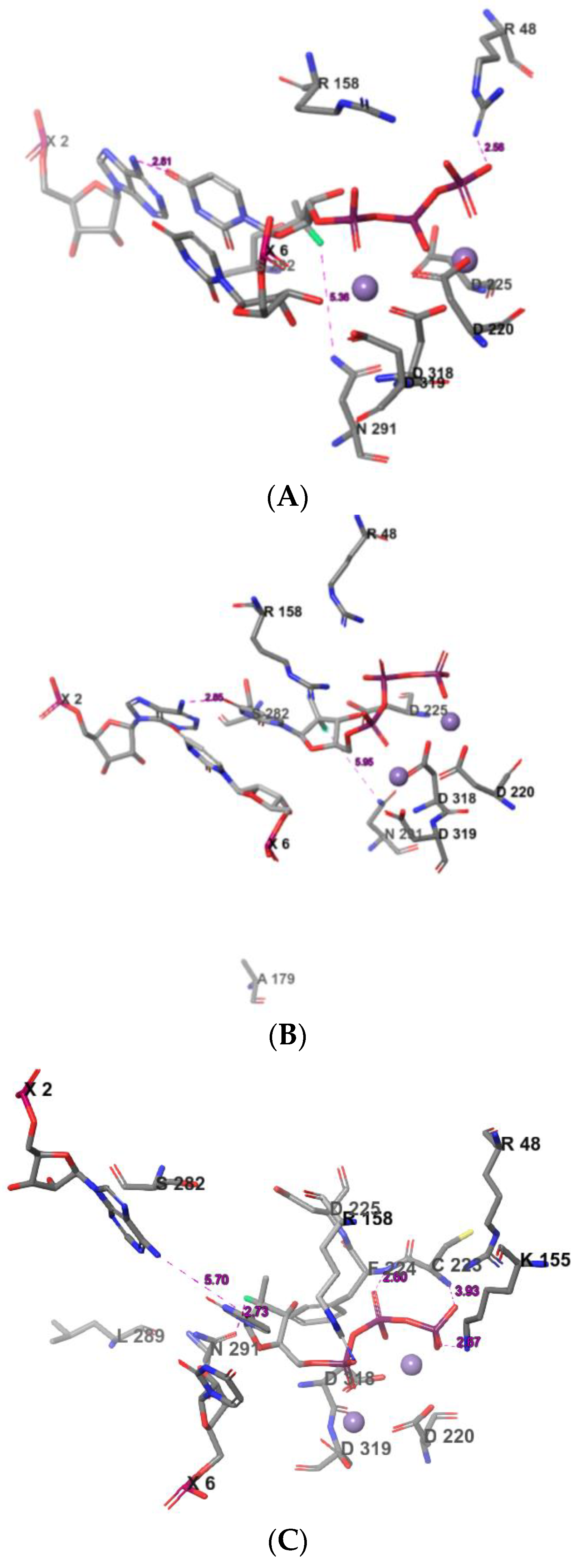
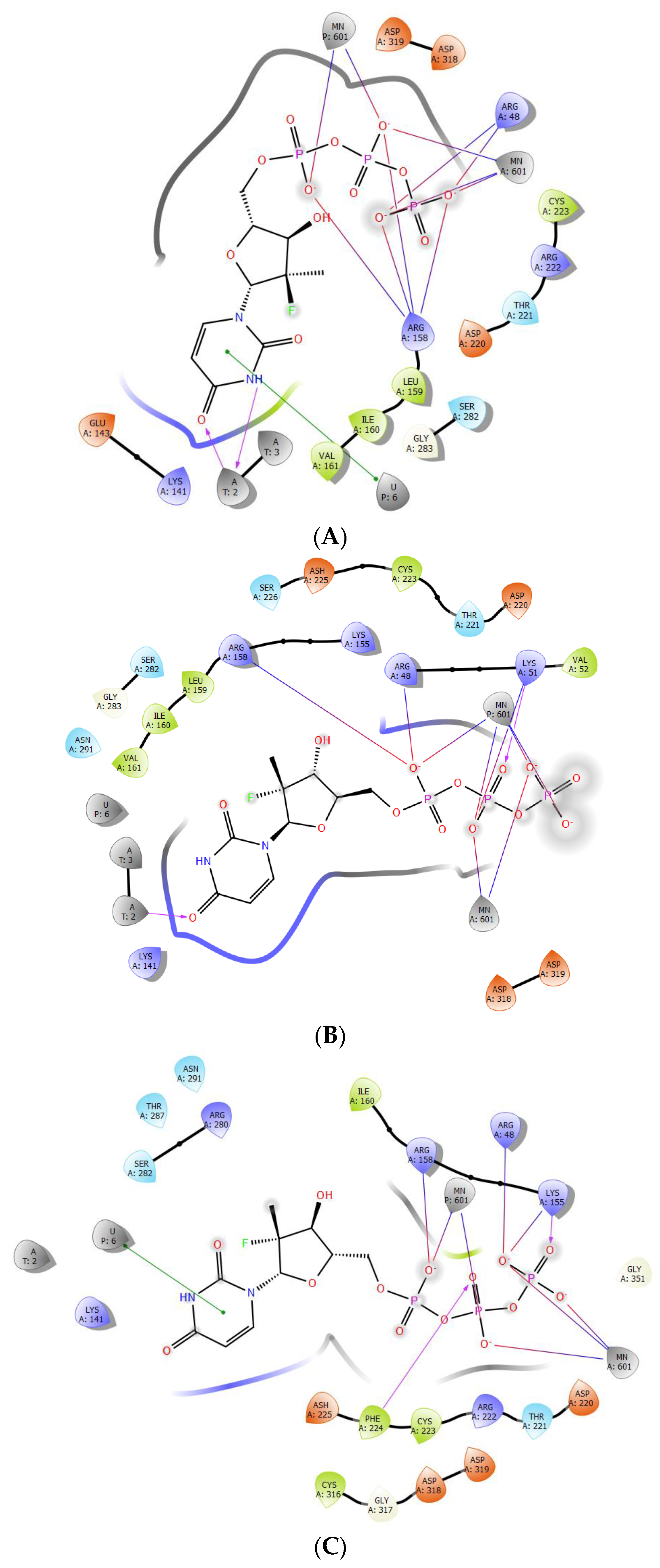
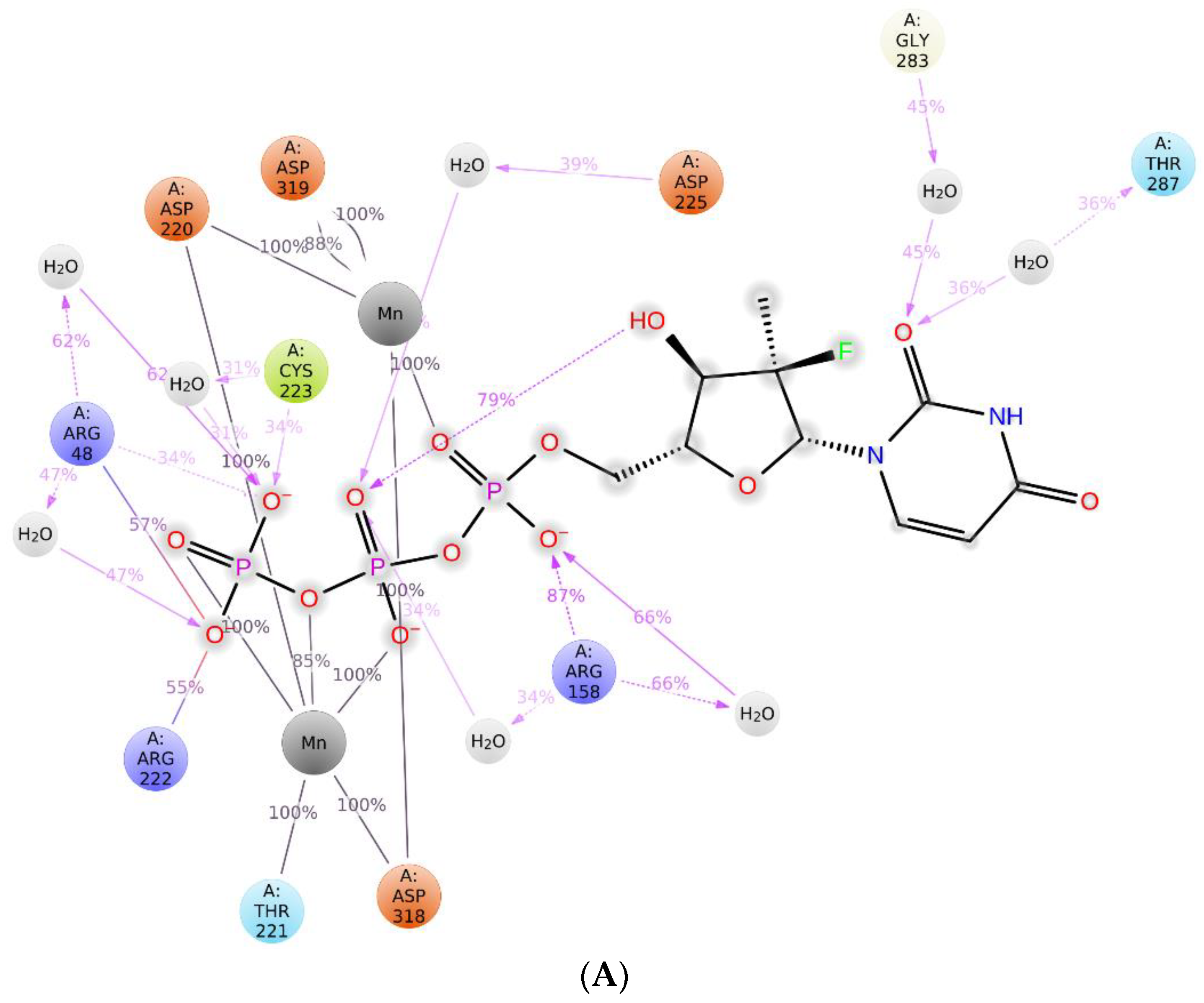
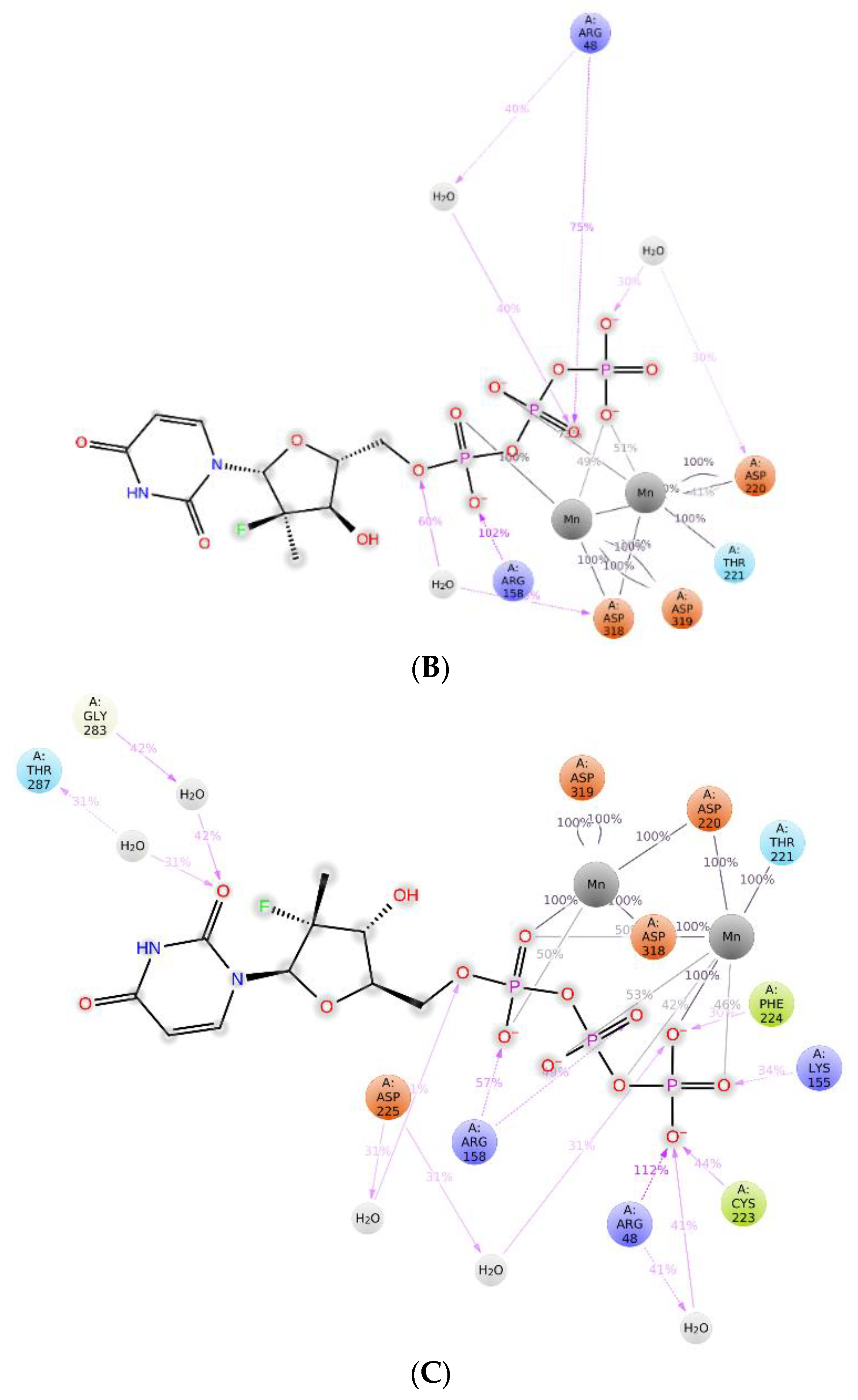
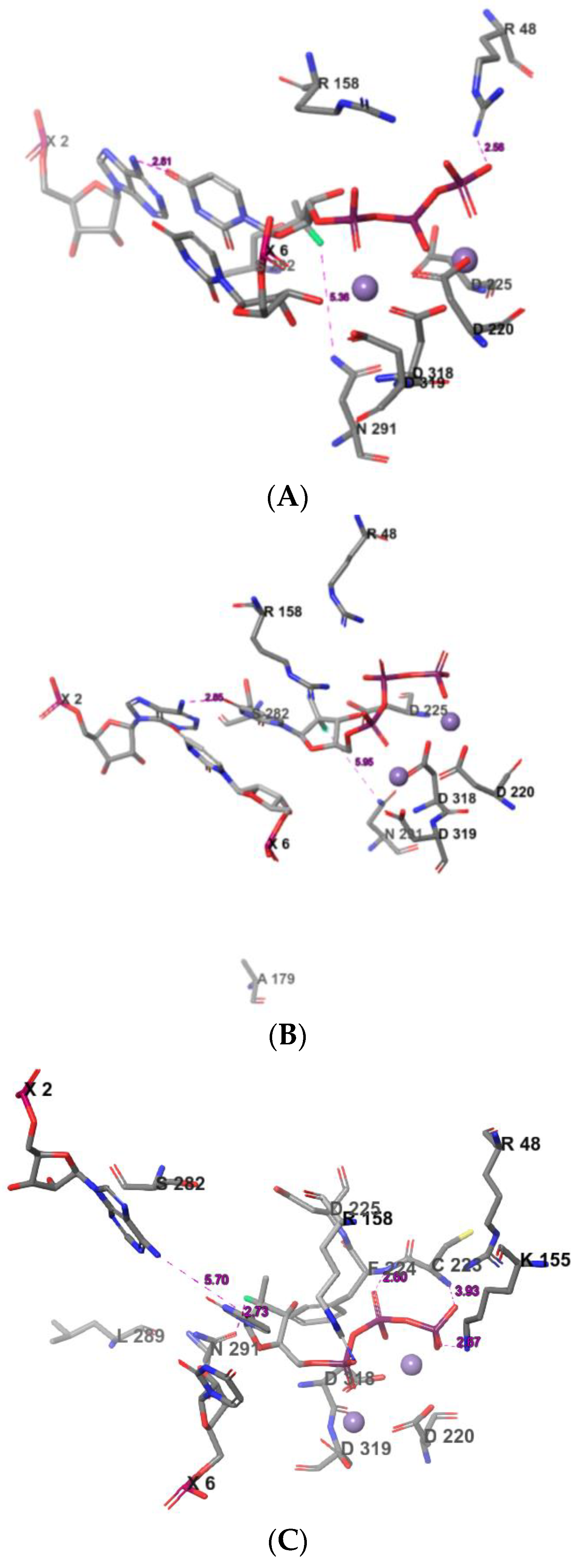
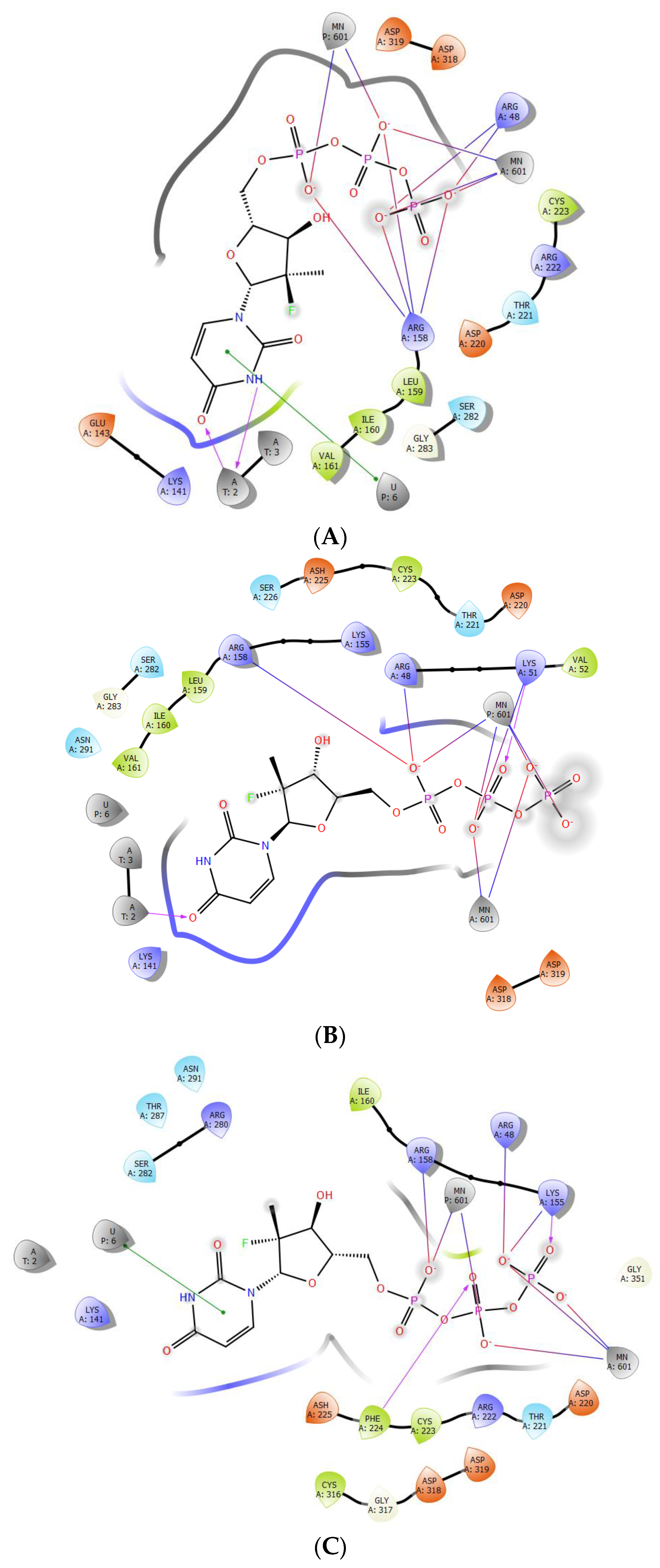
2.3. The Protein–Ligand Interaction Analysis
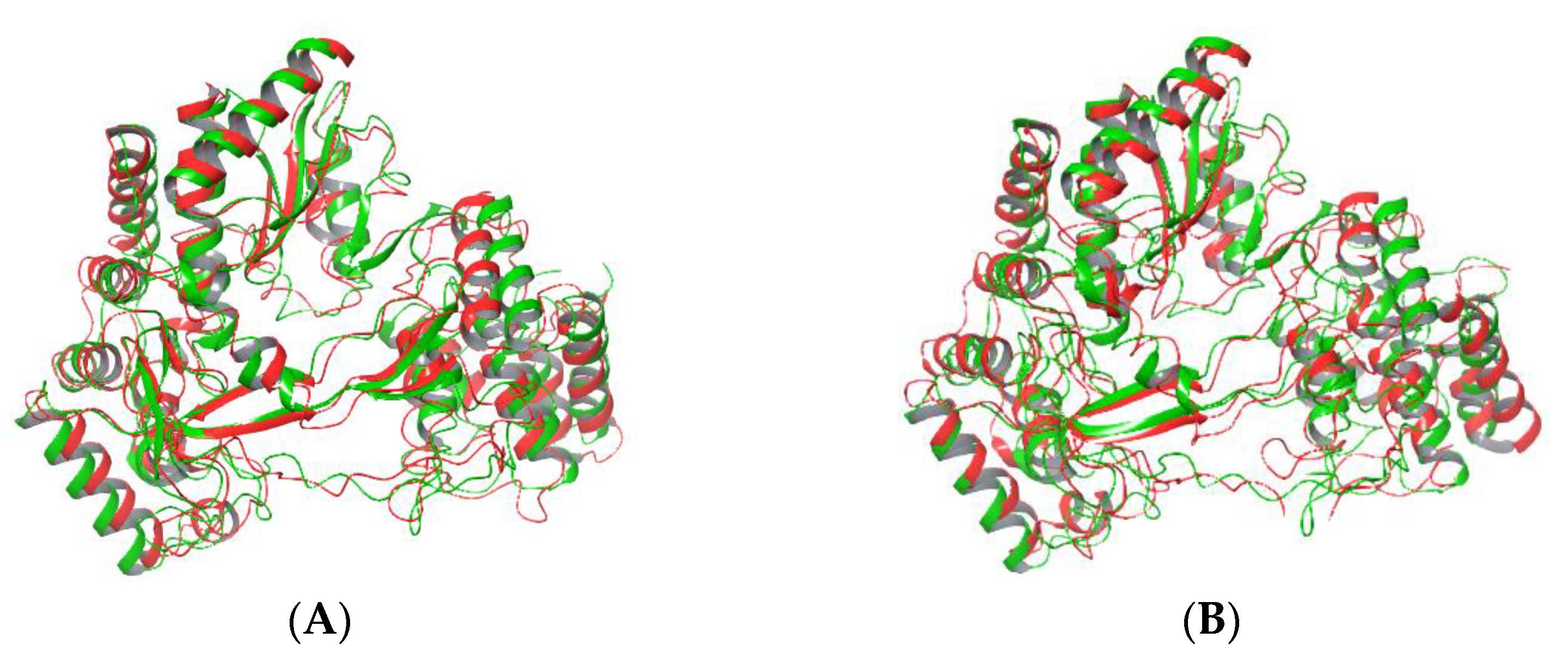
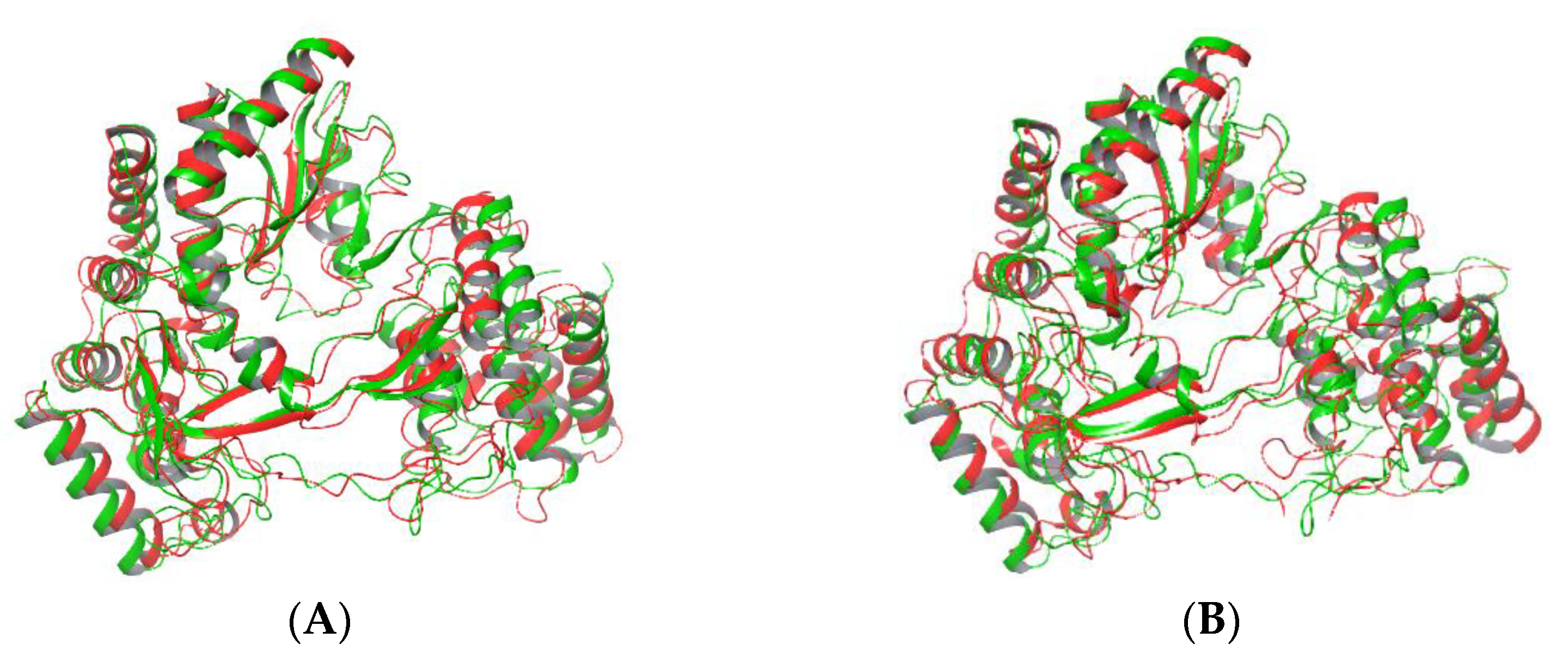
2.4. Cluster Analysis: The Effect of Mutation on Whole Protein Structure
2.5. MM-GBSA Binding Energy
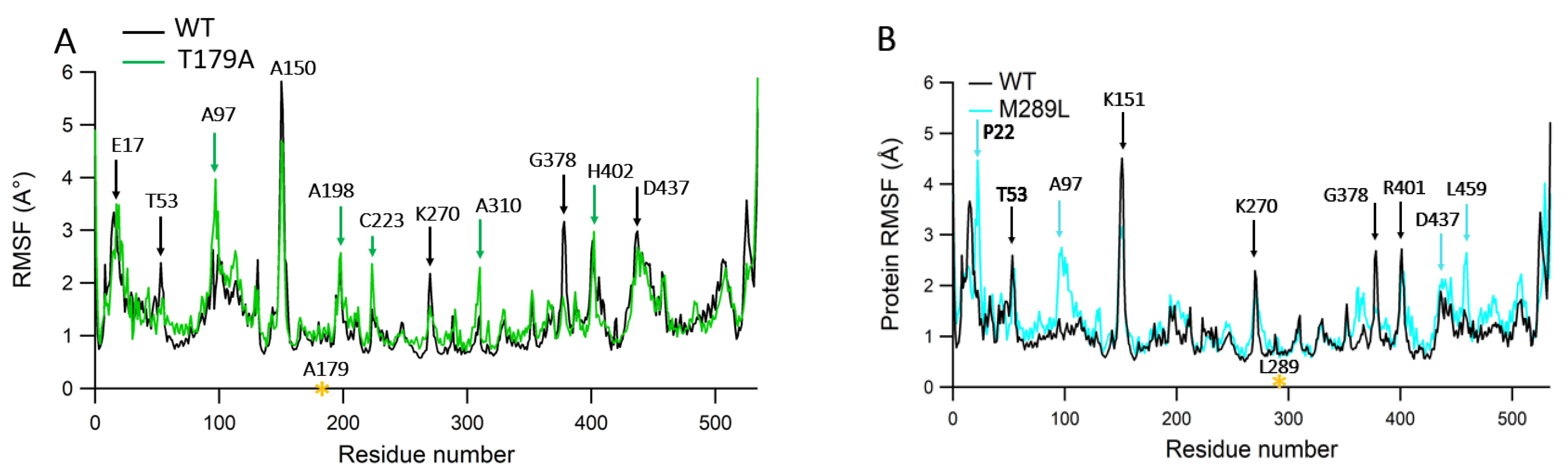
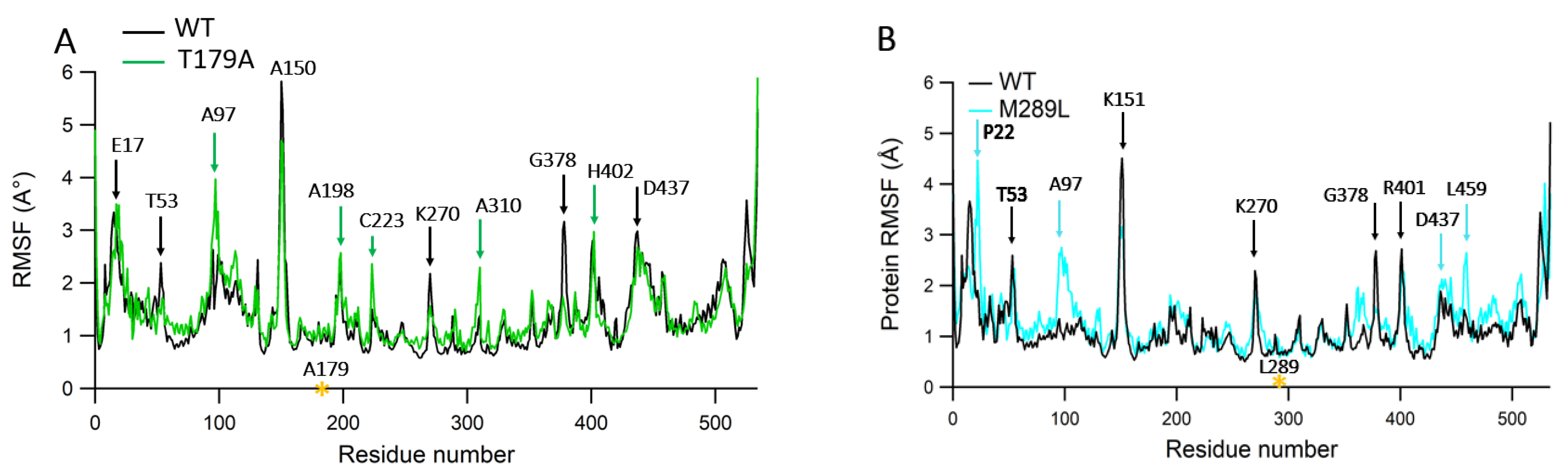
2.6. Protein RMSF Analyses
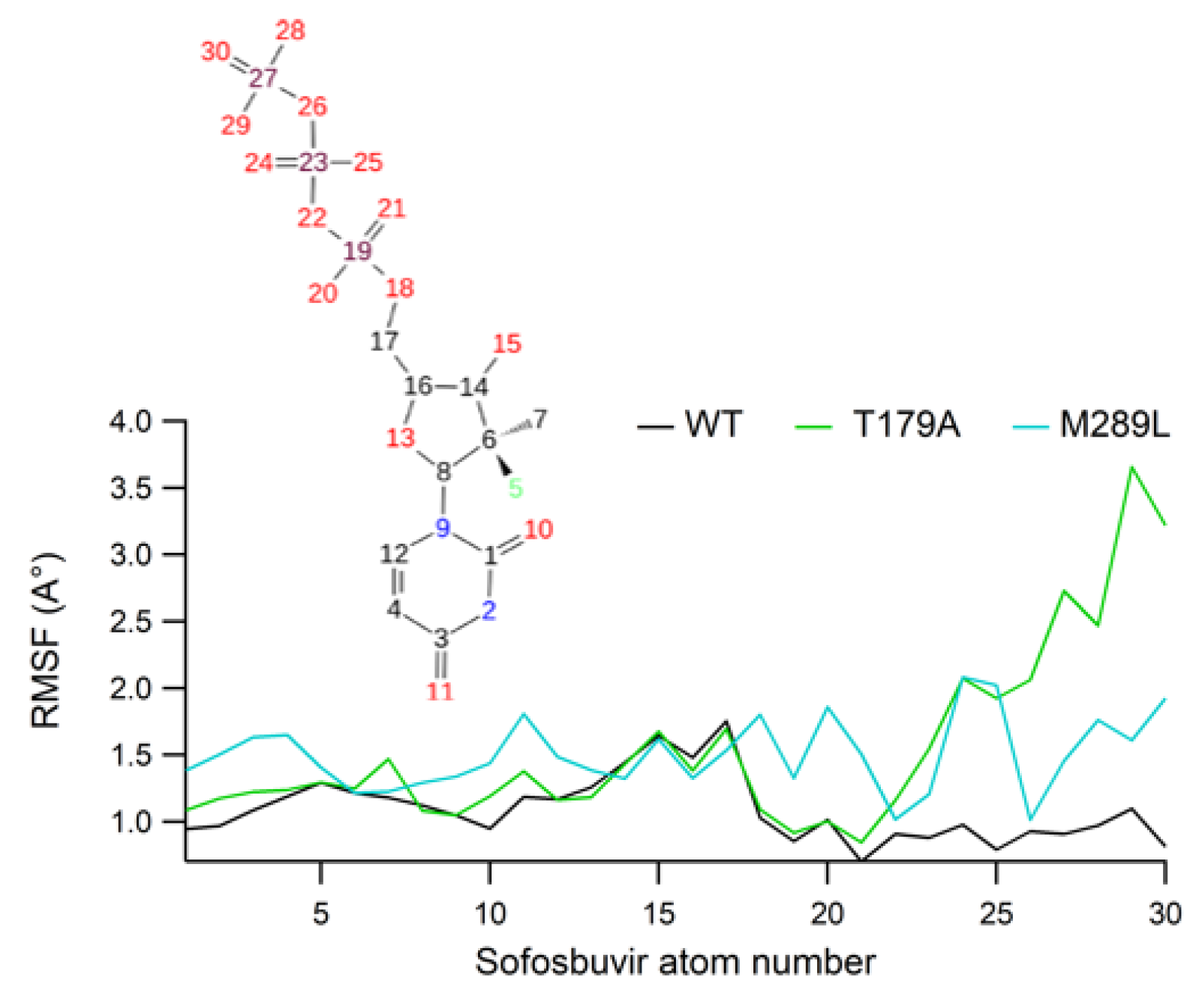
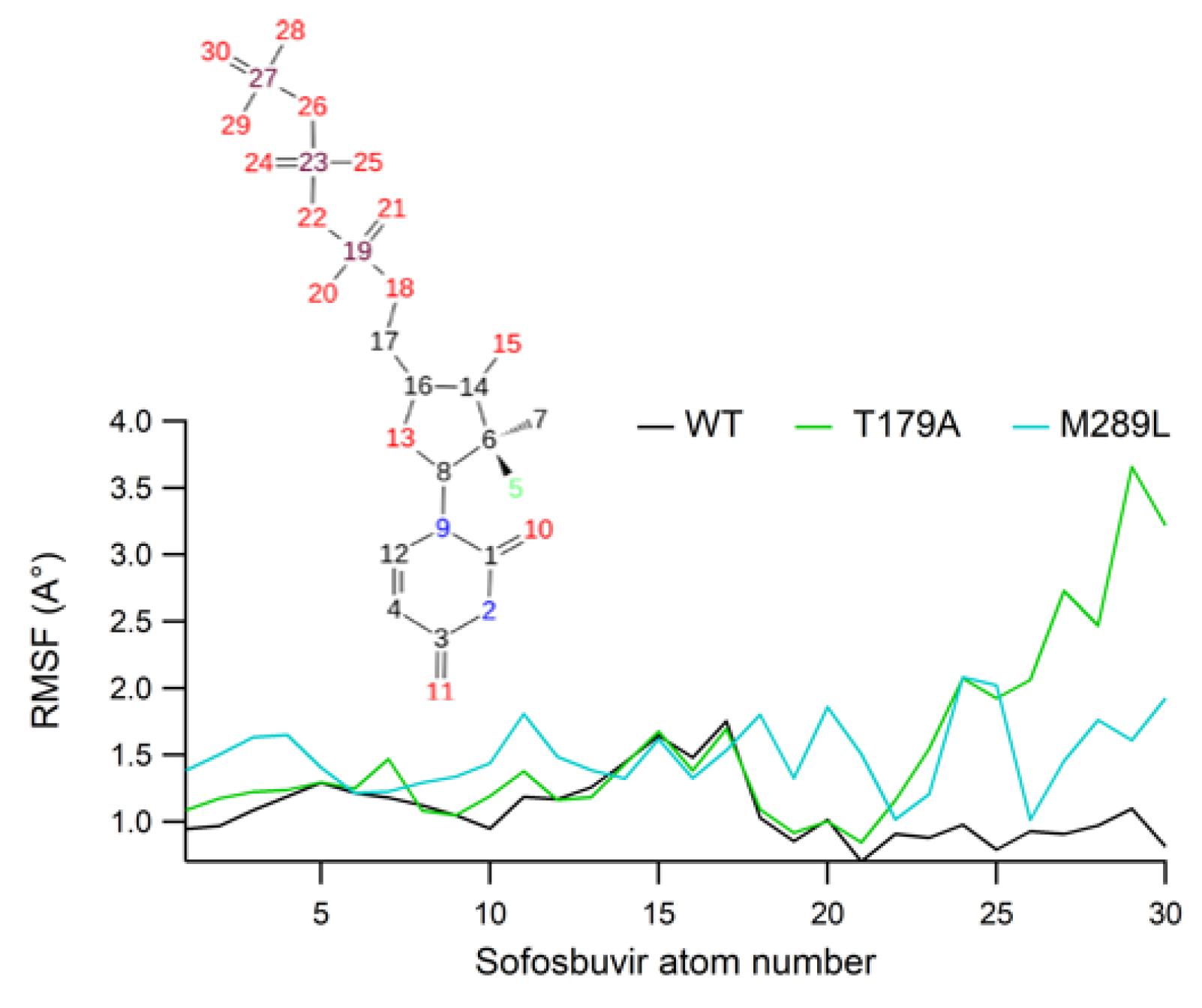
2.7. Ligand RMSF Values
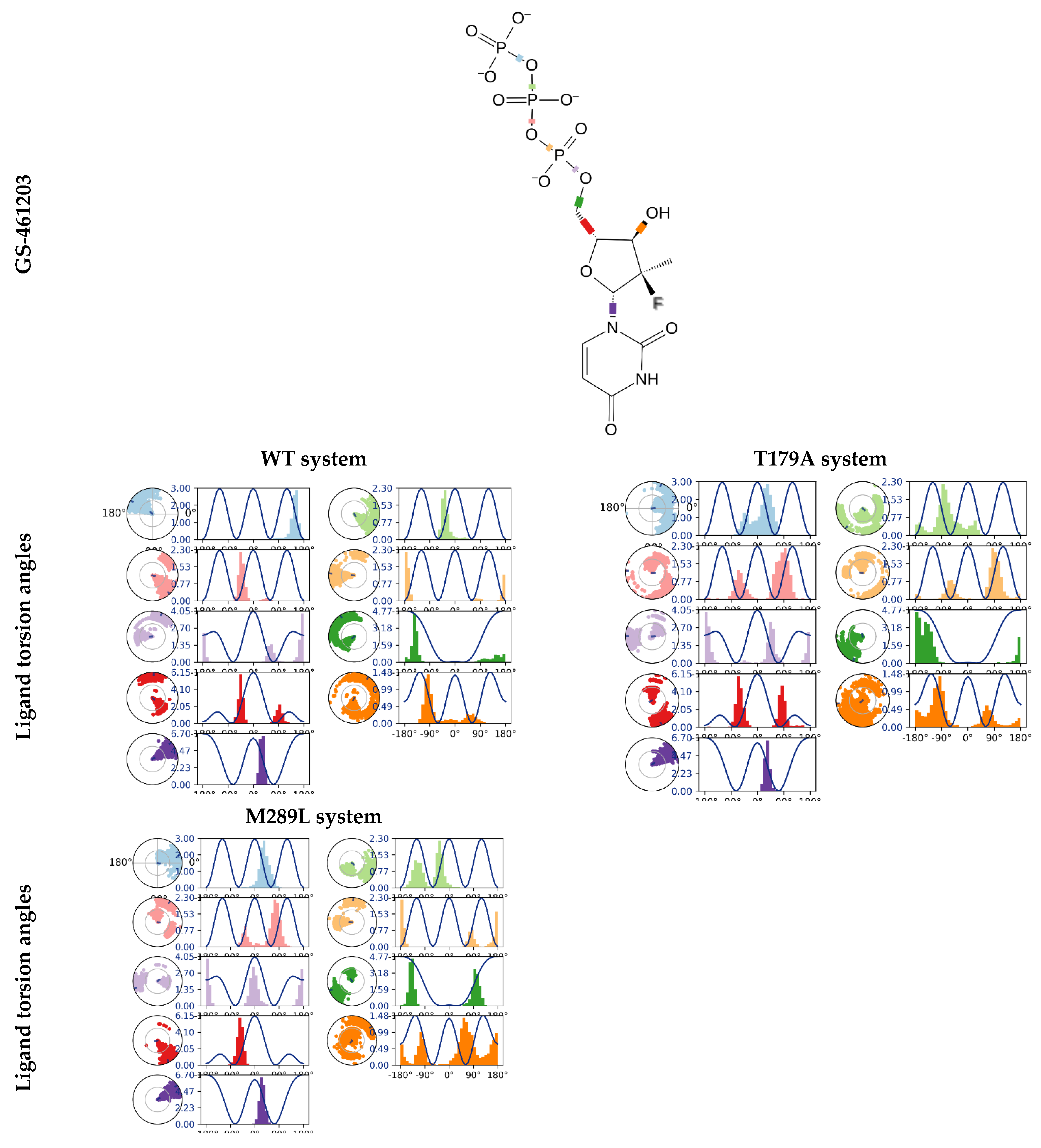
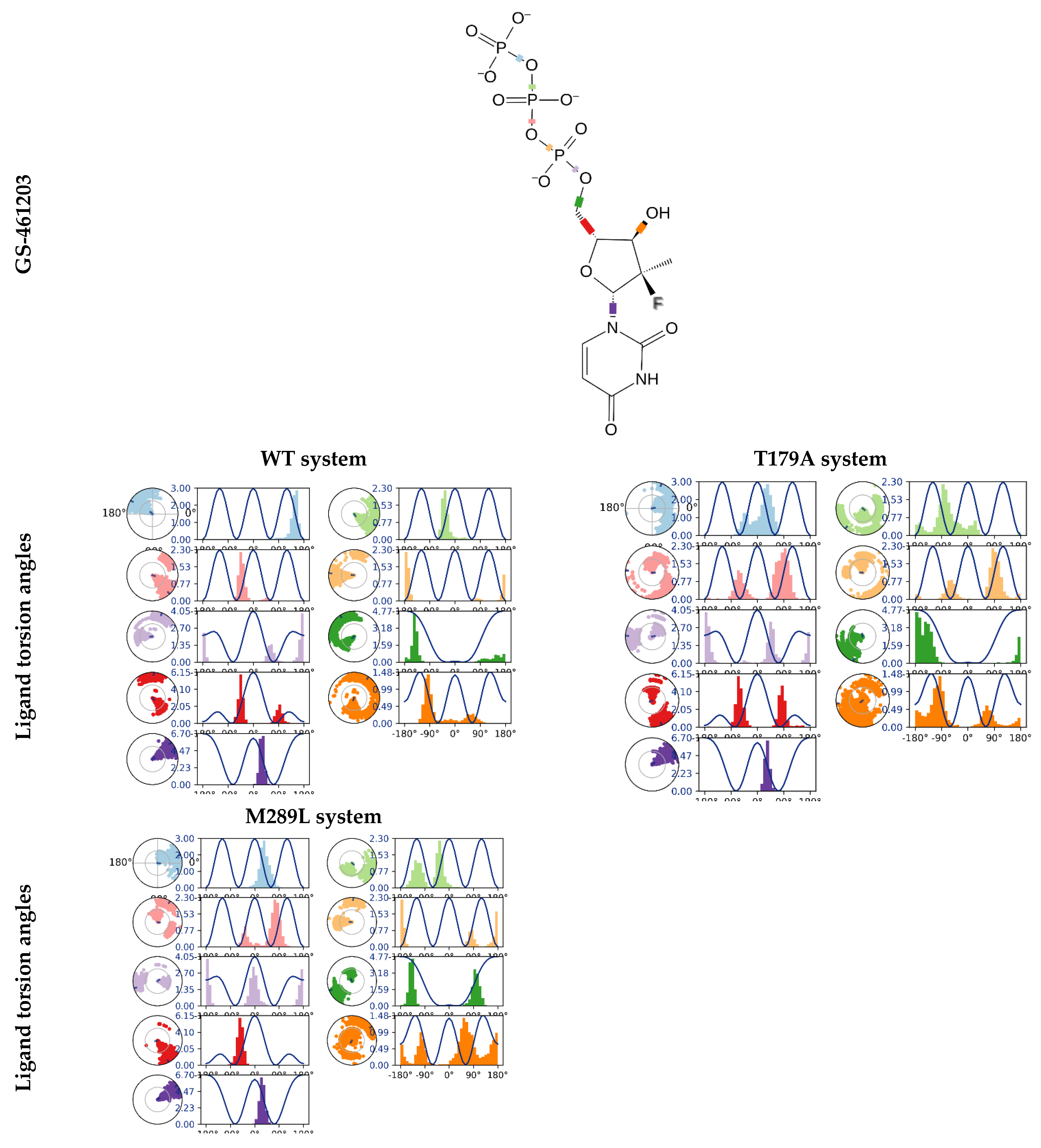
2.8. The GS-461203 Dihedral Angle Profiles
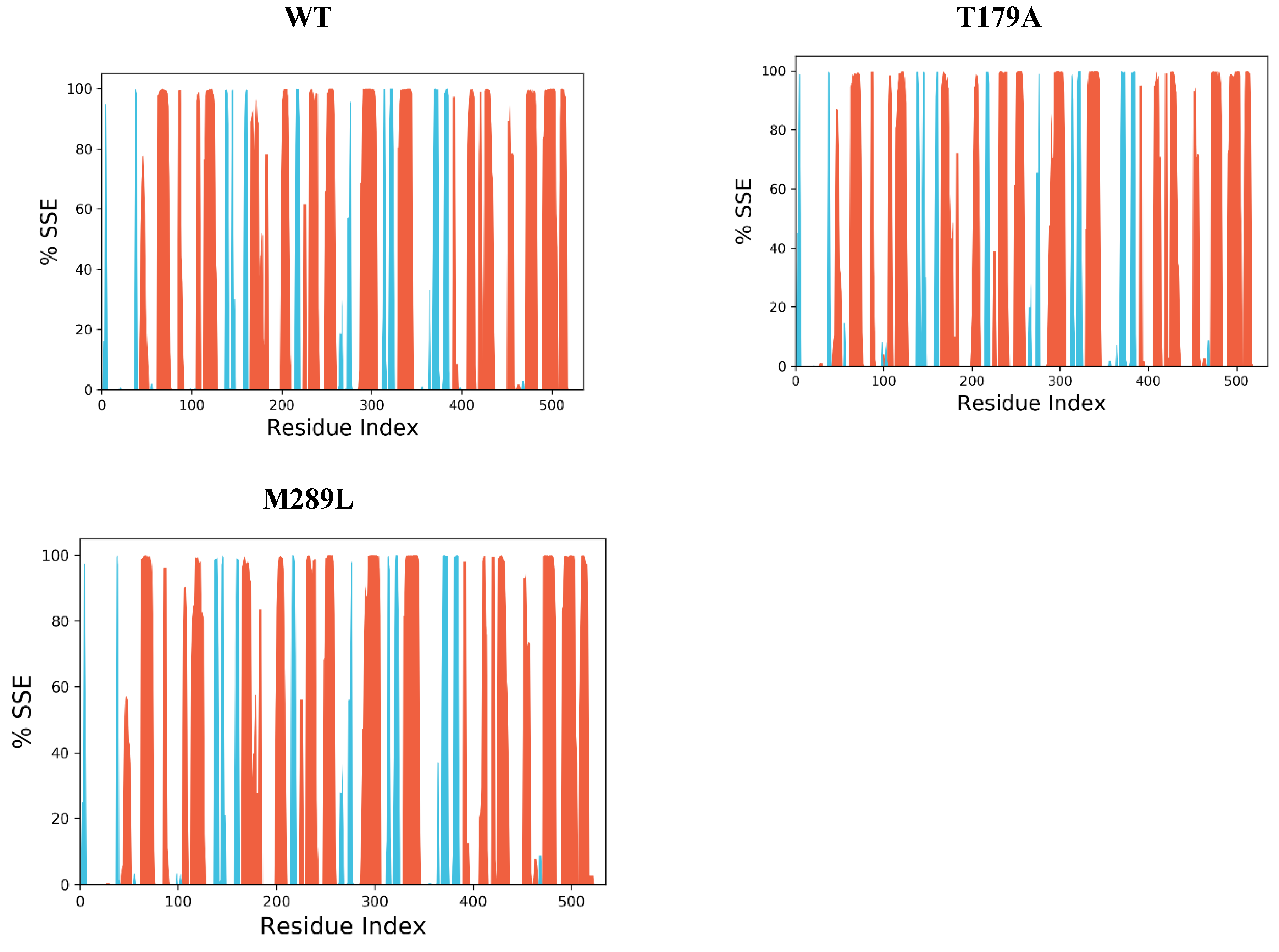
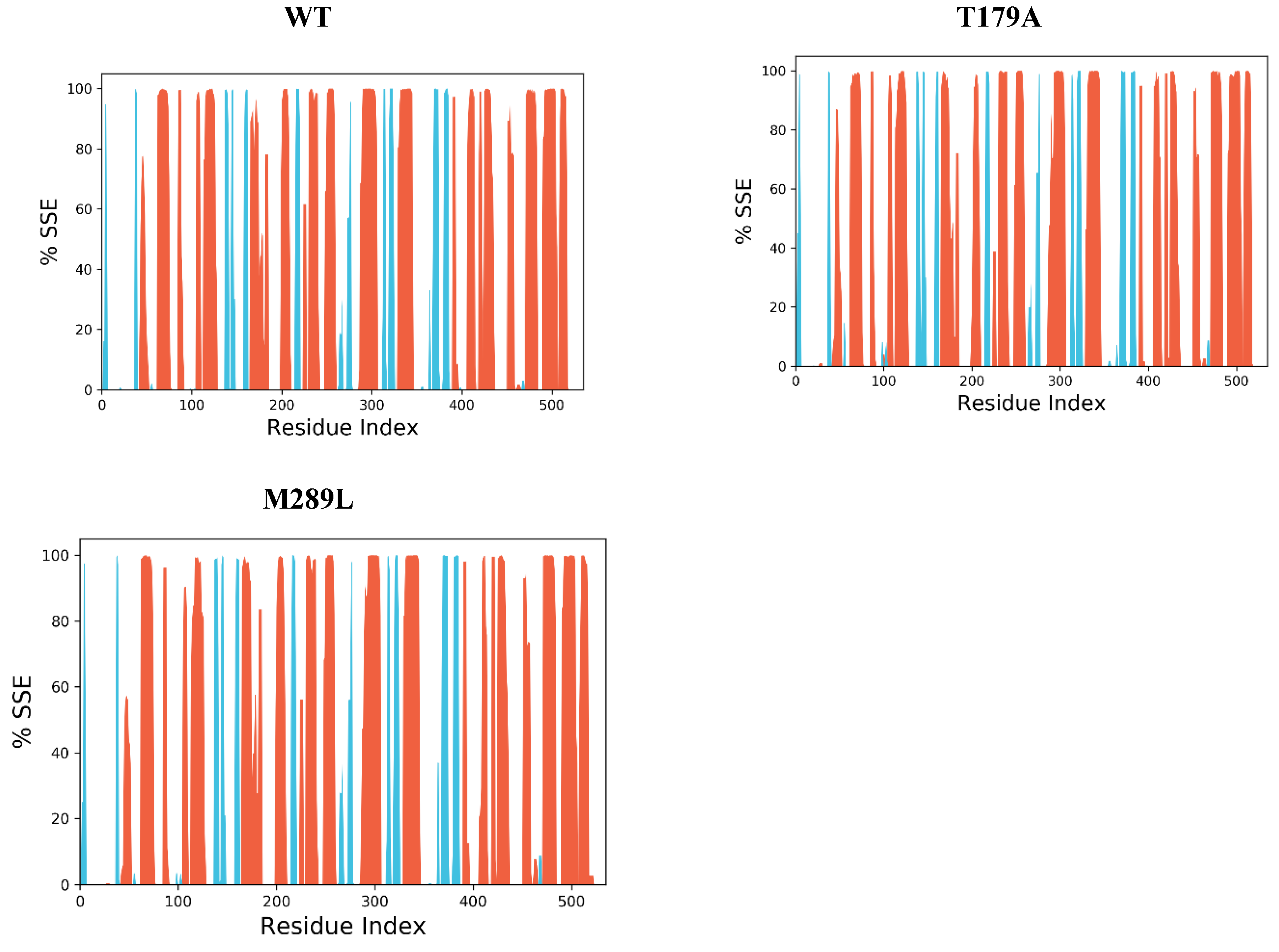
2.9. The Secondary Structure of Protein
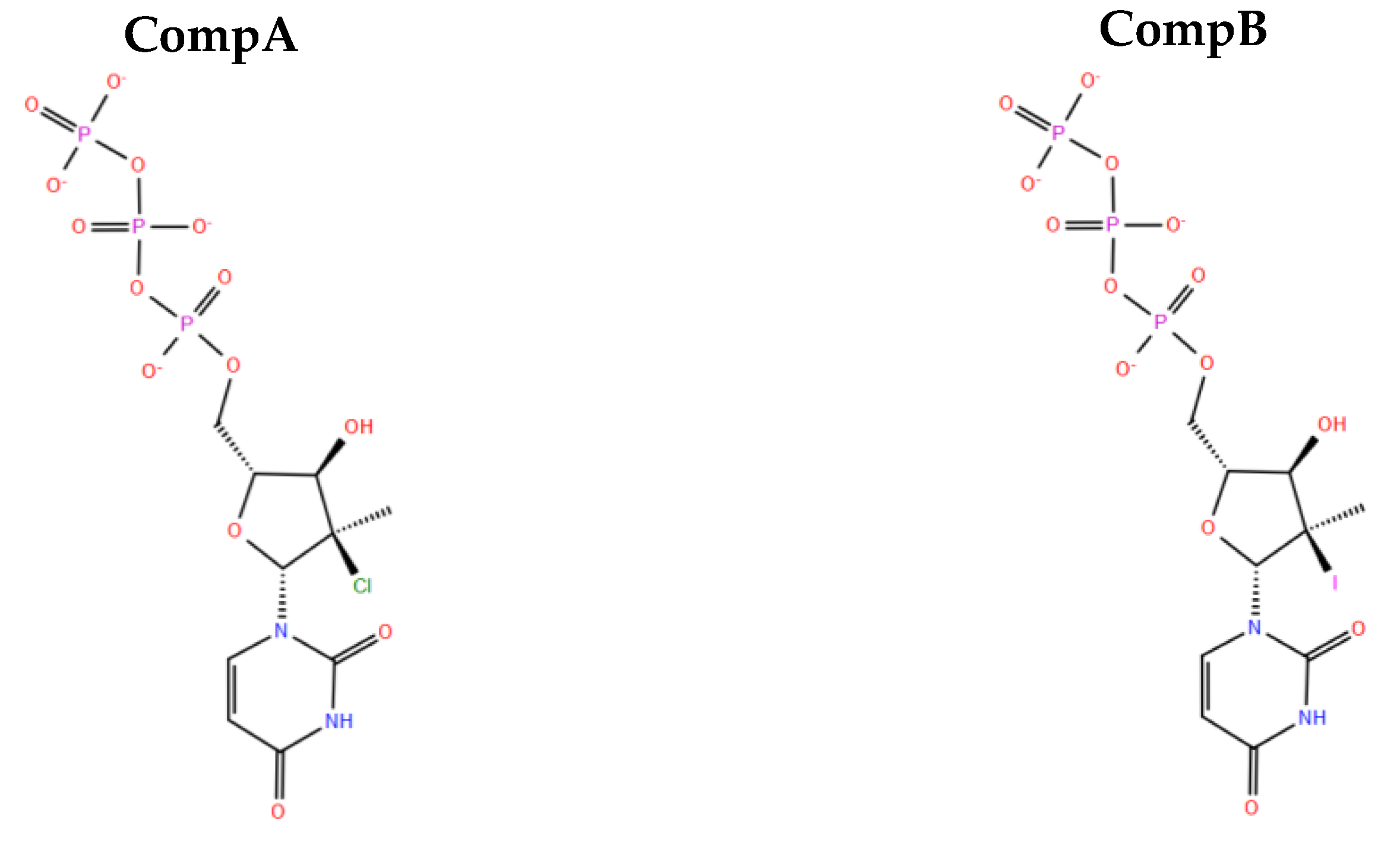
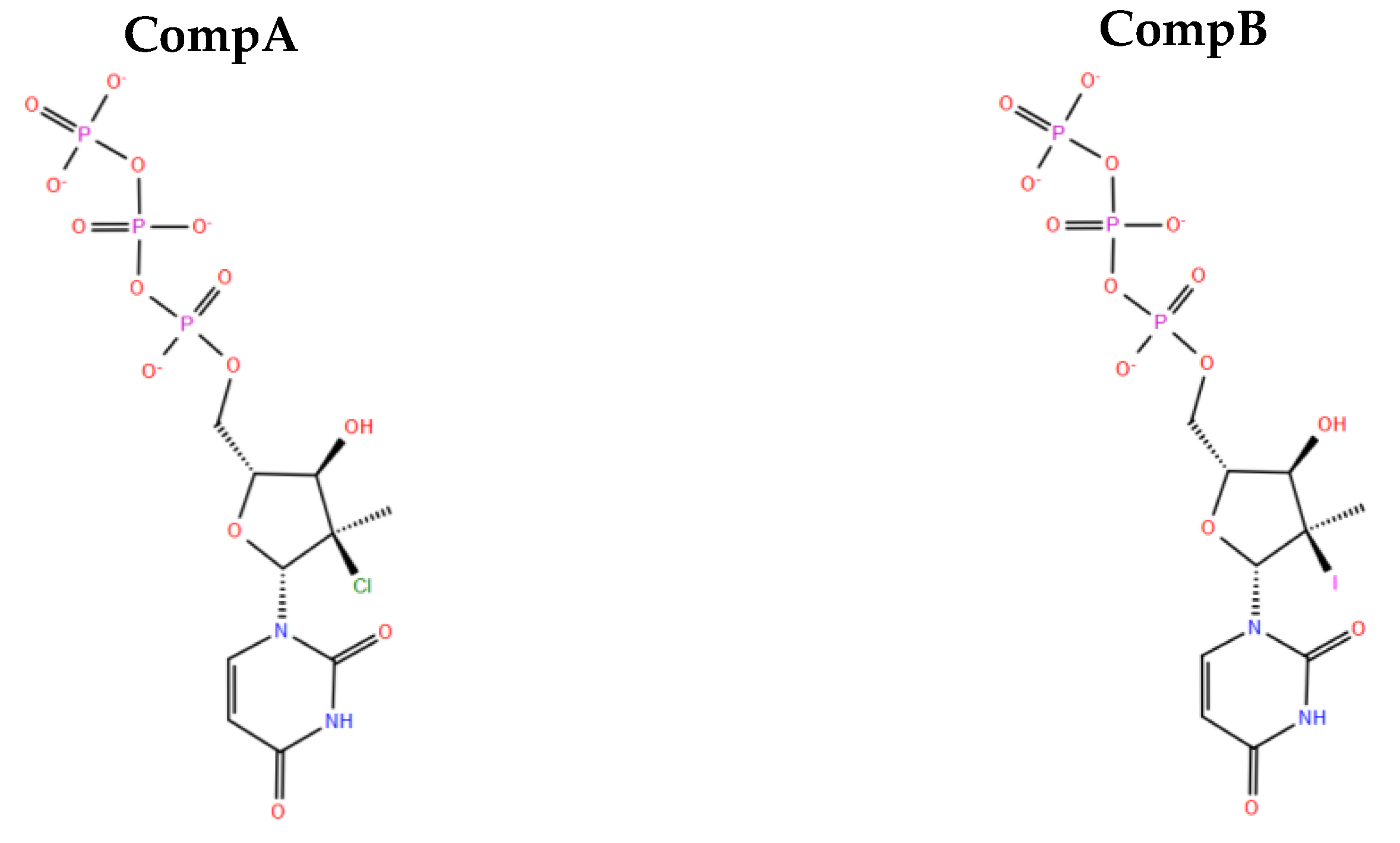
2.10. Designing New Analogues
2.11. Prediction ADME Properties
3. Discussion
4. Materials and Methods
4.1. Molecular Dynamics Simulation
4.2. Analysis of MD Simulation
4.3. Binding Energy Calculations
4.4. ADME Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanafiah, K.M.; Groeger, J.; Flaxman, A.D.; Wiersma, S.T. Global epidemiology of hepatitis C virus infection: New estimates of age-specific antibody to HCV seroprevalence. Hepatology 2013, 57, 1333–1342. [Google Scholar] [CrossRef]
- Rabaan, A.A.; Al-Ahmed, S.H.; Bazzi, A.M.; Alfouzan, W.A.; Alsuliman, S.A.; Aldrazi, F.A.; Haque, S. Overview of hepatitis C infection, molecular biology, and new treatment. J. Infect. Public Health 2020, 13, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, S.; Bukh, J. Current status and future development of infectious cell-culture models for the major genotypes of hepatitis C virus: Essential tools in testing of antivirals and emerging vaccine strategies. Antivir. Res. 2018, 158, 264–287. [Google Scholar] [CrossRef] [PubMed]
- Bukh, J. The history of hepatitis C virus (HCV): Basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J. Hepatol. 2016, 65, S2–S21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2013, 59, 318–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.C.; Simonetti, J.; Fisher, D.G.; Williams, J.; Yamamura, Y.; Rodriguez, N.; Sullivan, D.G.; Gretch, D.R.; McMahon, B.; Williams, K.J. Comparison of different HCV viral load and genotyping assays. J. Clin. Virol. 2003, 28, 27–37. [Google Scholar] [CrossRef]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cozzolino, A.; Cacciapuoti, C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J. Gastroenterol. 2016, 22, 7824–7840. [Google Scholar] [CrossRef] [PubMed]
- Hedskog, C.; Parhy, B.; Chang, S.; Zeuzem, S.; Moreno, C.; Shafran, S.D.; Borgia, S.M.; Asselah, T.; Alric, L.; Abergel, A.; et al. Identification of 19 Novel Hepatitis C Virus Subtypes—Further Expanding HCV Classification. Open Forum Infect. Dis. 2019, 6, ofz076. [Google Scholar] [CrossRef] [PubMed]
- Chayama, K.; Hayes, C.N. Hepatitis C virus: How genetic variability affects pathobiology of disease. J. Gastroenterol. Hepatol. 2011, 26, 83–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.-C.; Yang, C.-H.; Lo, S.-Y. Hepatitis C Viral Replication Complex. Viruses 2021, 13, 520. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.M.; Murakami, E.; Espiritu, C.; Steuer, H.M.M.; Niu, C.; Keilman, M.; Bao, H.; Zennou, V.; Bourne, N.; Julander, J.G.; et al. PSI-7851, a pronucleotide of beta-D-2′-deoxy-2′-fluoro-2′-C-methyluridine monophosphate, is a potent and pan-genotype inhibitor of hepatitis C virus replication. Antimicrob. Agents Chemother. 2010, 54, 3187–3196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.; Pierce, T.; Kowdley, K.V. Review of direct-acting antiviral agents for the treatment of chronic hepatitis C. Expert Opin. Investig. Drugs 2013, 22, 1107–1121. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.M.; Gupta, S.P.; Samanta, S.; Masand, N. Current perspective of HCV NS5B inhibitors: A review. Curr. Med. Chem. 2011, 18, 5564–5597. [Google Scholar] [CrossRef] [PubMed]
- Gentles, R.G.; Ding, M.; Bender, J.A.; Bergstrom, C.P.; Grant-Young, K.; Hewawasam, P.; Hudyma, T.; Martin, S.; Nickel, A.; Regueiro-Ren, A.; et al. Discovery and preclinical characterization of the Cyclopropylindolobenzazepine BMS-791325, a potent allosteric inhibitor of the hepatitis C virus NS5B polymerase. J. Med. Chem. 2014, 57, 1855–1879. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S. Recent advances in the molecular biology of hepatitis C virus. J. Mol. Biol. 2001, 313, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Götte, M.; Feld, J.J. Direct-acting antiviral agents for hepatitis C: Structural and mechanistic insights. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-W.; Li, H.; Ren, H.; Hu, P. Global prevalence of pre-existing HCV variants resistant to direct-acting antiviral agents (DAAs): Mining the GenBank HCV genome data. Sci. Rep. 2016, 6, 20310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, A.M.; Espiritu, C.; Bansal, S.; Steuer, H.M.M.; Niu, C.; Zennou, V.; Keilman, M.; Zhu, Y.; Lan, S.; Otto, M.J.; et al. Genotype and Subtype Profiling of PSI-7977 as a Nucleotide Inhibitor of Hepatitis C Virus. Antimicrob. Agents Chemother. 2012, 56, 3359–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawitz, E.; Lalezari, J.P.; Hassanein, T.; Kowdley, K.V.; Poordad, F.F.; Sheikh, A.M.; Afdhal, N.; Bernstein, D.E.; DeJesus, E.; Freilich, B.; et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: A randomised, double-blind, phase 2 trial. Lancet Infect. Dis. 2013, 13, 401–408. [Google Scholar] [CrossRef]
- Jacobson, I.M.; Gordon, S.C.; Kowdley, K.V.; Yoshida, E.M.; Rodriguez-Torres, M.; Sulkowski, M.S.; Shiffman, M.L.; Lawitz, E.; Everson, G.; Bennett, M.; et al. Sofosbuvir for Hepatitis C Genotype 2 or 3 in Patients without Treatment Options. N. Engl. J. Med. 2013, 368, 1867–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorbo, M.C.; Cento, V.; Di Maio, V.C.; Howe, A.Y.; Garcia, F.; Perno, C.F.; Ceccherini-Silberstein, F. Hepatitis C virus drug resistance associated substitutions and their clinical relevance: Update 2018. Drug Resist. Updat. 2018, 37, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Arba, M.; Wahyudi, S.T.; Brunt, D.J.; Paradis, N.; Wu, C. Mechanistic insight on the remdesivir binding to RNA-Dependent RNA polymerase (RdRp) of SARS-CoV-2. Comput. Biol. Med. 2021, 129, 104156. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Niu, Y.; Ning, L.; Zhang, Y.; Liu, H.; Yao, X. Computational study on the binding and unbinding mechanism of HCV NS5B with the inhibitor GS-461203 and substrate using conventional and steered molecular dynamics simulations. Chemom. Intell. Lab. Syst. 2016, 156, 72–80. [Google Scholar] [CrossRef]
- Appleby, T.C.; Perry, J.K.; Murakami, E.; Barauskas, O.; Feng, J.; Cho, A.; Fox, D.; Wetmore, D.R.; McGrath, M.E.; Ray, A.S.; et al. Structural basis for RNA replication by the hepatitis C virus polymerase. Science 2015, 347, 771–775. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Delaney, J.S. ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Sofia, M.J.; Furman, P.A. The Discovery of Sofosbuvir: A Liver-Targeted Nucleotide Prodrug for the Treatment and Cure of HCV. In HCV: The Journey from Discovery to a Cure; Sofia, M.J., Ed.; Springer International Publishing: Cham, Switzerland, 2019; Volume I, pp. 141–169. [Google Scholar]
- Sofia, M.J.; Chang, W.; Furman, P.A.; Mosley, R.T.; Ross, B.S. Nucleoside, Nucleotide, and Non-Nucleoside Inhibitors of Hepatitis C Virus NS5B RNA-Dependent RNA-Polymerase. J. Med. Chem. 2011, 55, 2481–2531. [Google Scholar] [CrossRef]
- Bagaglio, S.; Uberti-Foppa, C.; Morsica, G. Resistance Mechanisms in Hepatitis C Virus: Implications for Direct-Acting Antiviral Use. Drugs 2017, 77, 1043–1055. [Google Scholar] [CrossRef]
- Migliaccio, G.; Tomassini, J.E.; Carroll, S.S.; Tomei, L.; Altamura, S.; Bhat, B.; Bartholomew, L.; Bosserman, M.R.; Ceccacci, A.; Colwell, L.F.; et al. Characterization of Resistance to Non-obligate Chain-terminating Ribonucleoside Analogs That Inhibit Hepatitis C Virus Replication in Vitro. J. Biol. Chem. 2003, 278, 49164–49170. [Google Scholar] [CrossRef] [Green Version]
- Svarovskaia, E.S.; Dvory-Sobol, H.; Parkin, N.T.; Hebner, C.M.; Gontcharova, V.; Martin, R.; Ouyang, W.; Han, B.; Xu, S.; Ku, K.; et al. Infrequent Development of Resistance in Genotype 1–6 Hepatitis C Virus–Infected Subjects Treated With Sofosbuvir in Phase 2 and 3 Clinical Trials. Clin. Infect. Dis. 2014, 59, 1666–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Doehle, B.; Rajyaguru, S.; Han, B.; Barauskas, O.; Feng, J.; Perry, J.; Dvory-Sobol, H.; Svarovskaia, E.S.; Miller, M.D.; et al. In vitro selection of resistance to sofosbuvir in HCV replicons of genotype 1 to 6. Antivir. Ther. 2017, 22, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Staroń, J.; Pietruś, W.; Bugno, R.; Kurczab, R.; Satała, G.; Warszycki, D.; Lenda, T.; Wantuch, A.; Hogendorf, A.S.; Hogendorf, A.; et al. Tuning the activity of known drugs via the introduction of halogen atoms, a case study of SERT ligands—Fluoxetine and fluvoxamine. Eur. J. Med. Chem. 2021, 220, 113533. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yang, Z.; Liu, Y.; Lu, Y.; Chen, K.; Zhu, W. Halogen Bond: Its Role beyond Drug–Target Binding Affinity for Drug Discovery and Development. J. Chem. Inf. Modeling 2014, 54, 69–78. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Kumar, V.; Liu, H.; Wu, C. Drug repurposing against SARS-CoV-2 receptor binding domain using ensemble-based virtual screening and molecular dynamics simulations. Comput. Biol. Med. 2021, 135, 104634. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC’06: 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Trajectory | Mean RMSD of Protein Cα Atoms (Å) | Mean RMSD of Ligand (Å) |
|---|---|---|---|
| WT | Trajectory 1 | 2.039 ± 0.218 | 1.079 ± 0.216 |
| Trajectory 2 | 2.242 ± 0.216 | 1.724 ± 0.478 | |
| T179A | Trajectory 1 | 2.710 ± 0.366 | 3.150 ± 0.325 |
| Trajectory 2 | 2.552 ± 0.338 | 1.948 ± 0.384 | |
| M289L | Trajectory 1 | 2.883 ± 0.479 | 2.115 ± 0.425 |
| Trajectory 2 | 3.417 ± 0.544 | 1.928 ± 0.619 |
| Systems | WT | T179A | M289L |
|---|---|---|---|
| ΔGbind | −45.5 ± 5.8 | −35.0 ± 5.3 | −54.0 ± 7.7 |
| ΔΔGbind | 0.0 | 10.5 | 8.5 |
| ΔEvdw | −6.0 ± 6.8 | −2.3 ± 6.1 | 4.5 ± 7.7 |
| ΔΔEvdw | 0.0 | 3.7 | 10.5 |
| ΔEele | −35.3 ± 6.4 | −28.7 ± 5.8 | −55.1 ± 10.2 |
| ΔΔEele | 0.0 | 6.6 | 19.8 |
| ΔElipo | −4.3 ± 0.3 | −4.0 ± 0.9 | −3.4 ± 0.4 |
| ΔΔElipo | 0.0 | 0.3 | 0.9 |
| Systems | T179A | T179A-CompA | T179A-CompB |
|---|---|---|---|
| ΔGbind | −35.0 ± 5.3 | −74.0 ± 6.6 | −73.2 ± 6.9 |
| ΔΔGbind | 0.0 | 39 | 38.2 |
| ΔEvdw | −2.3 ± 6.1 | −13.3 ± 6.9 | −14.4 ± 6.3 |
| ΔΔEvdw | 0.0 | 11.0 | 12.1 |
| ΔEele | −28.7 ± 5.8 | −53.6 ± 4.4 | −53.0 ± 6.0 |
| ΔΔEele | 0.0 | 24.9 | 24.3 |
| ΔElipo | −4.0 ± 0.9 | −7.1 ± 0.4 | −5.8 ± 0.4 |
| ΔΔElipo | 0.0 | 3.1 | 1.8 |
| Compound | Log S (ESOL) | GI Absorption | BBB Permeant | CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 | Lipinksi Rule | PAINS |
|---|---|---|---|---|---|---|---|---|---|---|
| Sof a | −3.27 Soluble | Low | No | No | No | No | No | Yes | No; 2 violations: MW > 500, NorO > 10 | 0 alert |
| Sof-Cl a | −3.53 Soluble | Low | No | No | No | No | No | Yes | No; 2 violations: MW > 500, NorO > 10 | 0 alert |
| Sof-I a | −4.44 Moderately soluble | Low | No | No | No | No | No | Yes | No; 2 violations: MW > 500, NorO > 10 | 0 alert |
| GS-461203 | 0.57 Highly soluble | Low | No | No | No | No | No | No | Yes; 1 violation: NorO > 10 | 0 alert |
| Comp A | 0.31 Highly soluble | Low | No | No | No | No | No | No | No; 2 violations: MW > 500, NorO > 10 | 0 alert |
| Comp B | −0.61 Very soluble | Low | No | No | No | No | No | No | No; 2 violations: MW > 500, NorO > 10 | 0 alert |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arba, M.; Wahyudi, S.T.; Zubair, M.S.; Brunt, D.; Singh, M.; Wu, C. Binding of GS-461203 and Its Halogen Derivatives to HCV Genotype 2a RNA Polymerase Drug Resistance Mutants. Sci. Pharm. 2022, 90, 26. https://doi.org/10.3390/scipharm90020026
Arba M, Wahyudi ST, Zubair MS, Brunt D, Singh M, Wu C. Binding of GS-461203 and Its Halogen Derivatives to HCV Genotype 2a RNA Polymerase Drug Resistance Mutants. Scientia Pharmaceutica. 2022; 90(2):26. https://doi.org/10.3390/scipharm90020026
Chicago/Turabian StyleArba, Muhammad, Setyanto Tri Wahyudi, Muhammad Sulaiman Zubair, Dylan Brunt, Mursalin Singh, and Chun Wu. 2022. "Binding of GS-461203 and Its Halogen Derivatives to HCV Genotype 2a RNA Polymerase Drug Resistance Mutants" Scientia Pharmaceutica 90, no. 2: 26. https://doi.org/10.3390/scipharm90020026
APA StyleArba, M., Wahyudi, S. T., Zubair, M. S., Brunt, D., Singh, M., & Wu, C. (2022). Binding of GS-461203 and Its Halogen Derivatives to HCV Genotype 2a RNA Polymerase Drug Resistance Mutants. Scientia Pharmaceutica, 90(2), 26. https://doi.org/10.3390/scipharm90020026