
The Development and the Validation of a Novel Dissolution Method of Favipiravir Film-Coated Tablets


Abstract
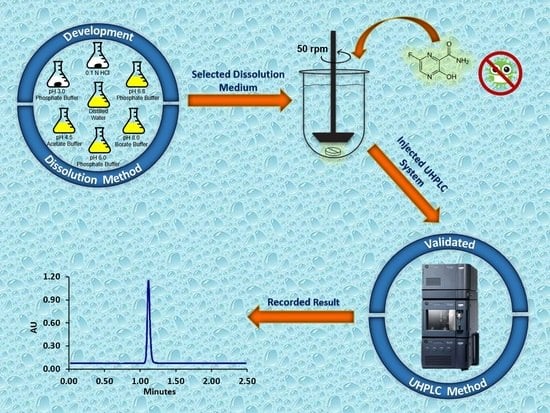
:
1. Introduction
2. Materials and Methods
2.1. Materials, Reagents and Equipments
2.2. Methods
2.2.1. Filter Compatibility
2.2.2. Solubility Determination
- y
- : Area
- x
- : Concentration (mg/mL)
- a
- : Slope
- b
- : Intercept
- ASam
- : Area of Sample
- WSam
- : Weight of Sample (mg)
- S
- : Dilution Coefficient
2.2.3. Selection of Dissolution Volume, Stirring Rate and Apparatus
2.2.4. Dissolution Method Development and Determination of λmax
2.2.5. Dissolution Test Chromatographic Conditions
2.2.6. Dissolution Method Validation
Specificity
Linearity and Range
System Precision
Method Precision—Repeatability
Method Precision—Intermediate Precision
Accuracy
Robustness
Solutions Stability
3. Results and Discussion
3.1. Filter Compatibility
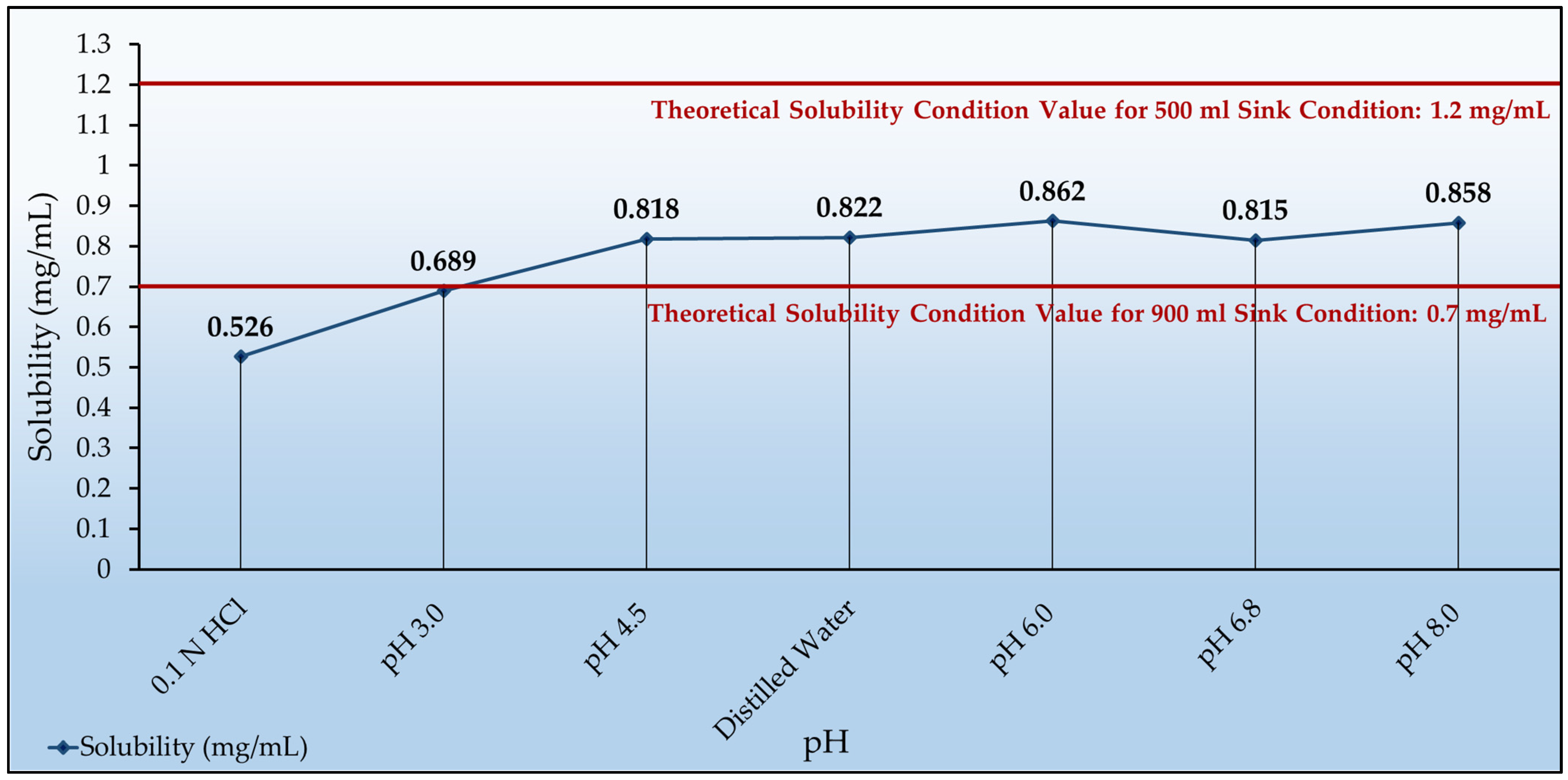
3.2. Solubility Determination
3.3. Selection of Dissolution Volume
3.4. Selection of Stirring Rate and Apparatus
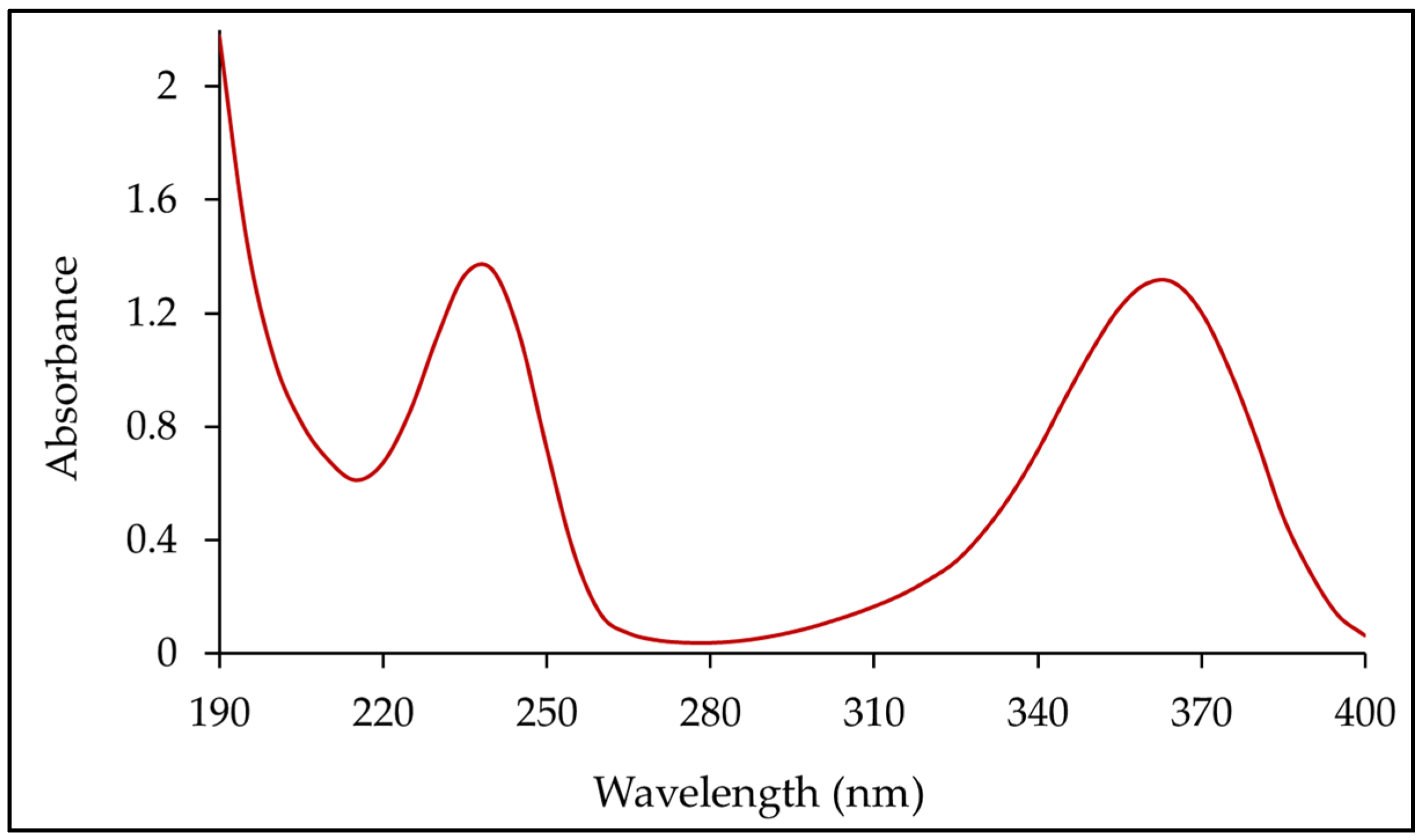
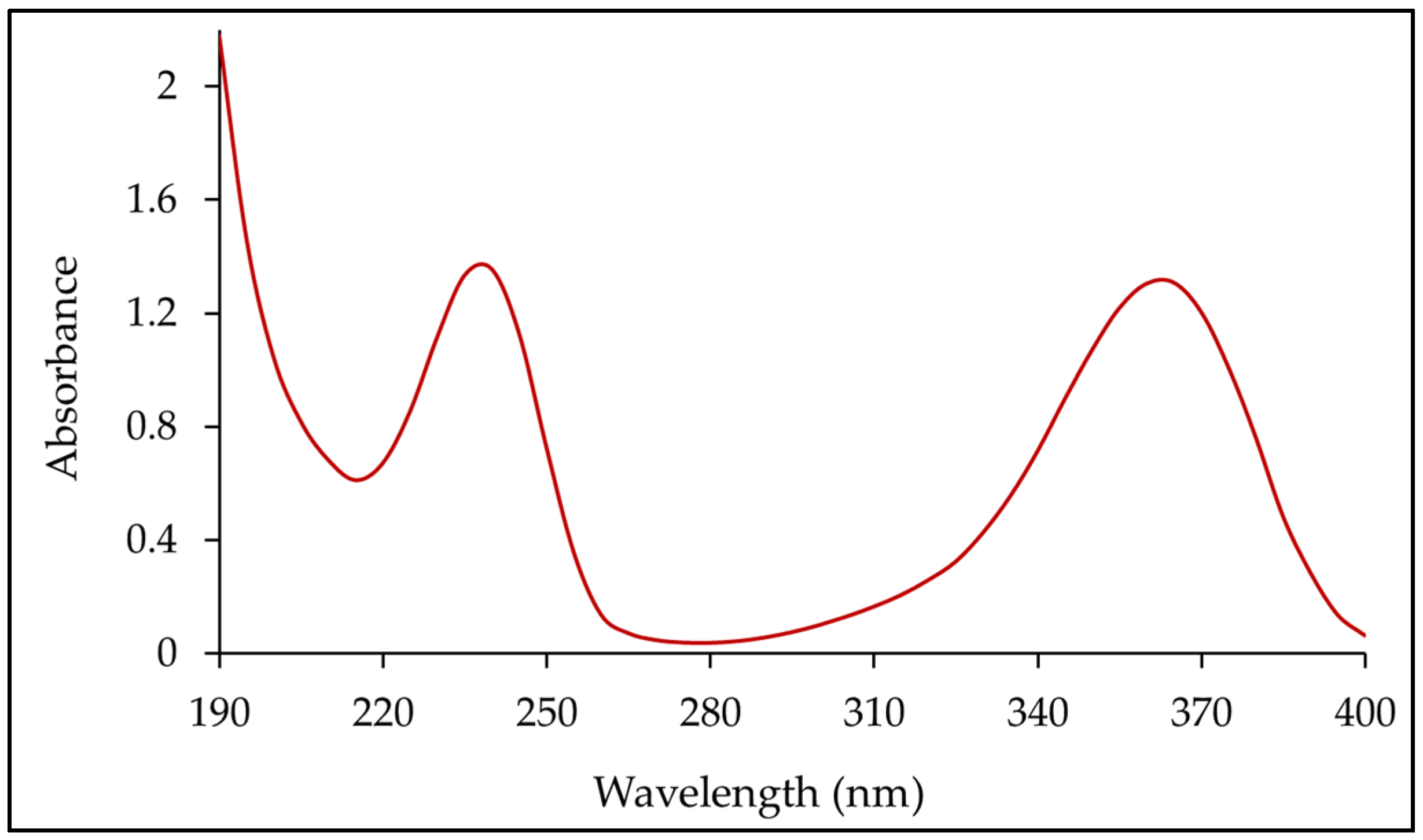
3.5. Dissolution Method Development and Determination of λmax
3.6. Dissolution Method Validation
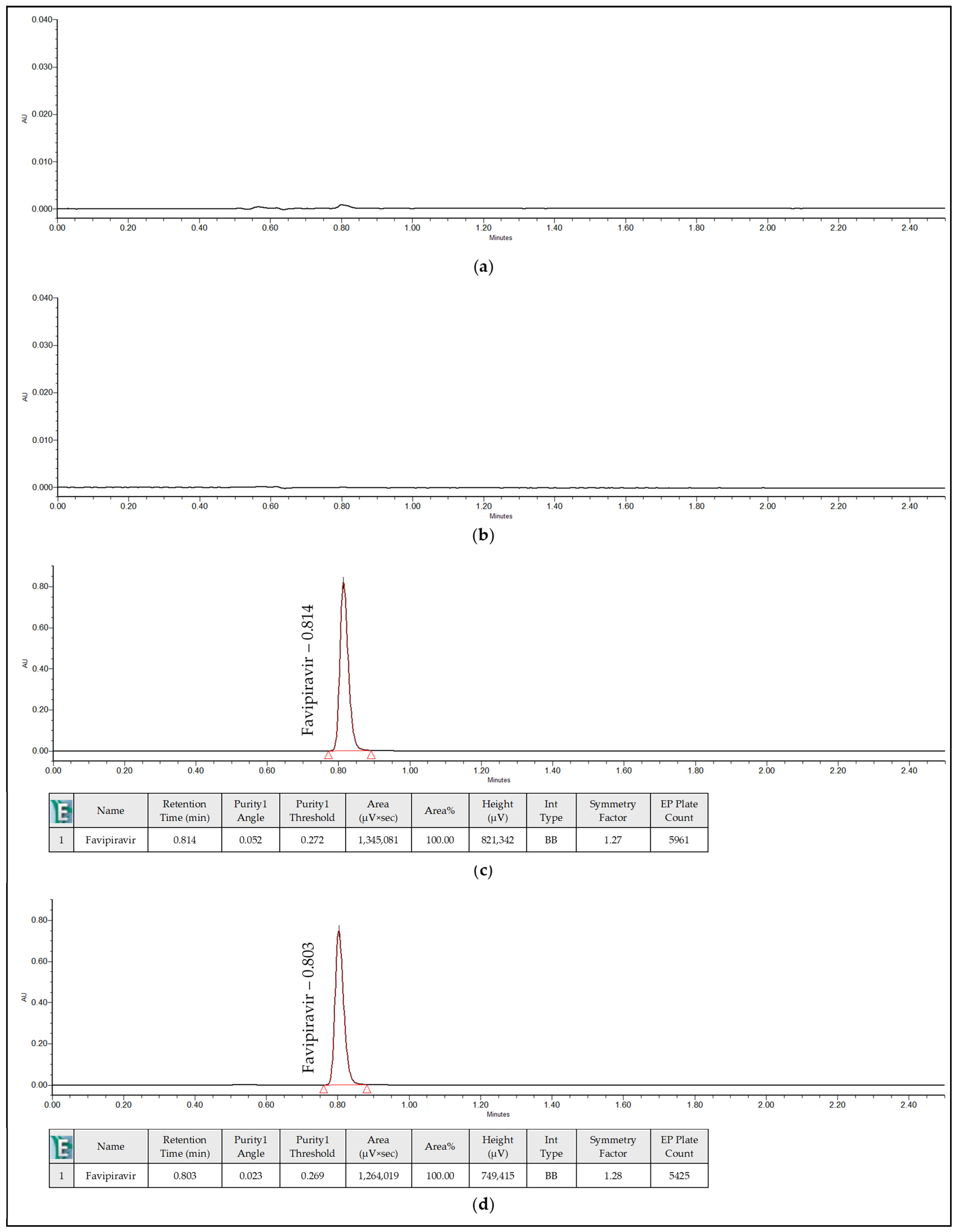
3.6.1. Selectivity
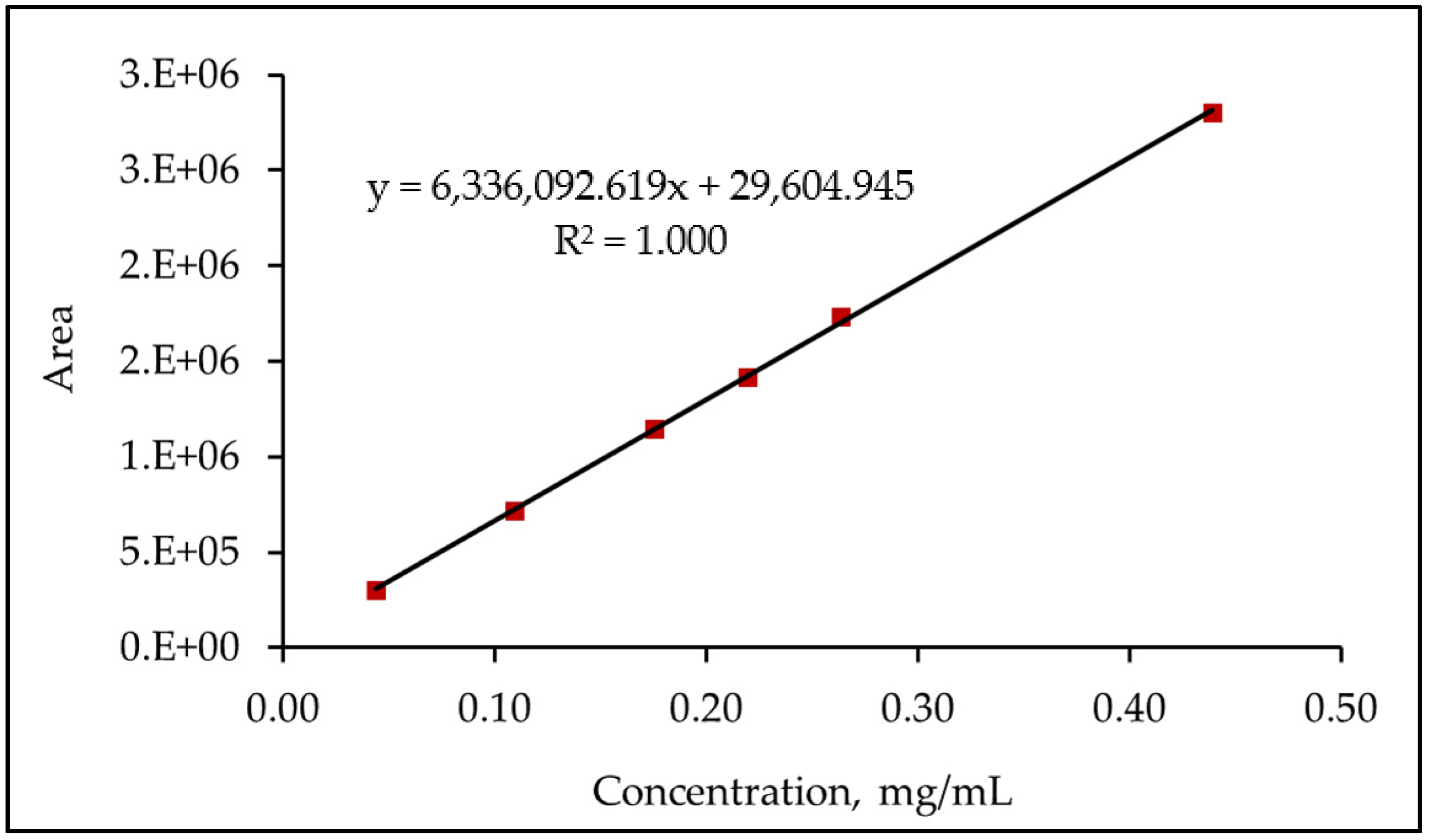
3.6.2. Linearity and Range
3.6.3. System Precision
3.6.4. Method Precision—Repeatability
3.6.5. Method Precision—Intermediate Precision
3.6.6. Accuracy
3.6.7. Robustness
3.6.8. Solutions Stability
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.X.; Chen, X.P. Favipiravir: Pharmacokinetics and concerns about clinical trials for 2019-nCoV infection. Clin. Pharmacol. Ther. 2020, 108, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. 2020, 19, 149–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santosh, J.N.; Ramdas, A.P. Current progress, pharmacology and targeted drug development on SARS-CoV-2 pandemic. World J. Pharm. Pharm. Sci. 2020, 9, 1912–1918. [Google Scholar]
- Frediansyah, A.; Tiwaric, R.; Sharund, K.; Dhamae, K.; Harapan, H. Antivirals for COVID-19: A critical review. Clin. Epidemiol. Glob. Health 2021, 9, 90–98. [Google Scholar] [CrossRef]
- Nyarko, R.O.; Boateng, E.; Kahwa, I.; Boateng, P.O. A comparison analysis on remdesivir, favipiravir, hydroxychloroquine, chloroquine and azithromycin in the treatment of corona virus disease 2019 (COVID-19)-A Review. World J. Pharm. Pharm. Sci. 2020, 9, 121–133. [Google Scholar]
- Yaghoubi, A.; Jamehdar, S.A.; Movaqar, A.; Milani, N.; Soleimanpour, S. An effective drug against COVID-19: Reality or dream? Expert Rev. Respir. Med. 2021, 15, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Parkar, J.; Ansari, A.; Vora, A.; Talwar, D.; Tiwaskar, M.; Patil, S.; Barkate, H. Role of favipiravir in the treatment of COVID-19. Int. J. Infect. Dis. 2021, 102, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, U.; Raju, R.; Udwadia, Z.F. Favipiravir: A new and emerging antiviral option in COVID-19. Med. J. Armed Forces India 2020, 76, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B 2017, 93, 449–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taskeen, M.; Tirunagari, M.; Qureshi, H.K. Biorelevant and quality control dissolution method development and validation of quetiapine fumarate tablets. Acta Pharm. Sci. 2017, 55, 21–37. [Google Scholar] [CrossRef] [Green Version]
- International Conference on Harmonization (ICH) of technical requirements for registration of pharmaceuticals for human use. In Validation of Analytical Procedures: Text and Methodology Q2(R1); ICH: Geneva, Switzerland, 2005.
- United States Pharmacopeia and National Formulary (USP43-NF38); Reagents and Reference Tables, Solutions, Buffer Solutions; United States Pharmacopeial Convention: Rockville, MD, USA, 2020.
- European Pharmacopoeia, 10th ed.; 5.17.1 Recommendations on Dissolution Testing; 2020; Available online: https://www.edqm.eu/en/european_pharmacopoeia_10th_edition (accessed on 26 October 2020).
- United States Pharmacopeial Convention. United States Pharmacopeia and National Formulary (USP43-NF38); 1092, The Dissolution Procedure; United States Pharmacopeial Convention: Rockville, MD, USA, 2020. [Google Scholar]
- European Medicines Agency. Committee for Medicinal Products for Human Use. Guideline on the Investigation of Bioequivalence. CPMP/EWP/QWP/1401/98 Rev.1 (01.2010); European Medicines Agency: Amsterdam, The Netherlands, 2010. [Google Scholar]
- European Medicines Agency. Committee for Medicinal Products for Human Use. ICH M9 Guideline on Biopharmaceutics Classification System-Based Biowaivers. EMA/CHMP/ICH/493213/2018 (02.2020); European Medicines Agency: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Ploger, G.F.; Hofsass, M.A.; Dressman, J.B. Solubility determination of active pharmaceutical ingredients which have been recently added to the list of essential medicines in the context of the biopharmaceutics classification system-biowaiver. J. Pharm. Sci. 2018, 107, 1478–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. Committee for Medicinal Products for Human Use. Reflection Paper on the Dissolution Specification for Generic Solid Oral Immediate Release Products with Systemic Action. EMA/CHMP/CVMP/QWP/336031/2017 (08.2017); European Medicines Agency: Amsterdam, The Netherlands, 2017. [Google Scholar]
- US Food and Drug Administration, Center for Drug Evaluation and Research. Guidance for Industry, Dissolution Testing of Immediate Release Solid Oral Dosage Forms; US Food and Drug Administration, Center for Drug Evaluation and Research: Rockville, MD, USA, 1997. [Google Scholar]
- United States Pharmacopeial Convention. United States Pharmacopeia and National Formulary (USP43-NF38); 711, Dissolution; United States Pharmacopeial Convention: Rockville, MD, USA, 2020. [Google Scholar]
- Johnson, L.A. Analysis of variance of parameter estimates: F tests and t tests. Anal. Biochem. 1992, 206, 195–201. [Google Scholar] [CrossRef]
- Vaucher, L.C.; Paim, C.S.; Lange, A.D.; Schapoval, E.E.S. Development and validation of a dissolution test for telithromycin in coated tablets. Quim. Nova 2009, 32, 1329–1333. [Google Scholar] [CrossRef]





{kind=link}
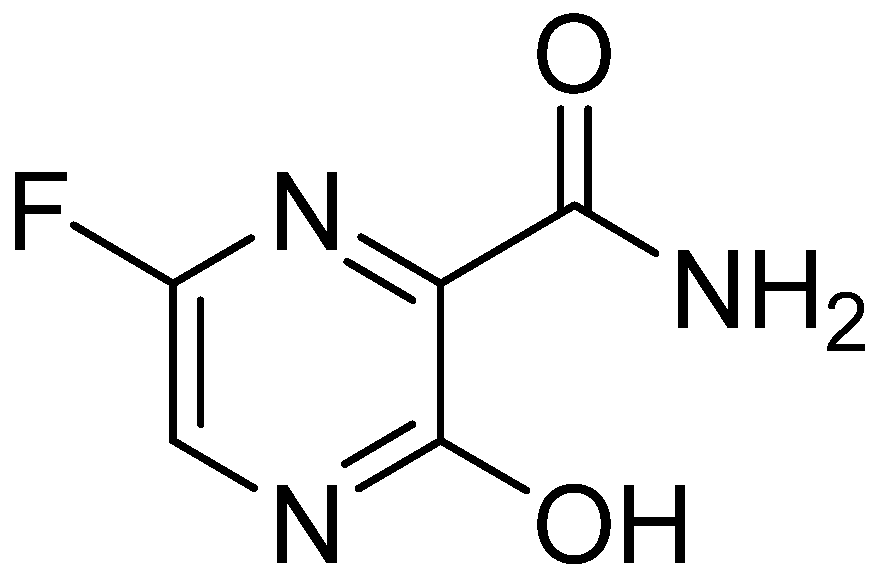{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Filter | Unfiltered | 0.45 µm RC | 0.45 µm PTFE | 0.45 µm PVDF | 0.45 µm Nylon | |
|---|---|---|---|---|---|---|
| Media | ||||||
| 0.1 N HCl | - | 100.6 | 101.2 | 101.1 | 101.6 | |
| Phosphate Buffer with pH 3.0 | - | 100.4 | 100.4 | 100.4 | 100.3 | |
| Acetate Buffer with pH 4.5 | - | 100.0 | 97.3 | 97.3 | 97.6 | |
| Phosphate Buffer with pH 6.0 | - | 97.5 | 97.9 | 98.0 | 98.5 | |
| Phosphate Buffer with pH 6.8 | - | 100.4 | 100.7 | 101.4 | 101.8 | |
| Distilled Water | - | 100.4 | 100.7 | 101.5 | 102.0 | |
| Borate Buffer with pH 8.0 | - | 100.0 | 101.4 | 101.6 | 102.1 | |
| Test Media | Dosage Volume (mL) | Solubility (mg/mL) 1 |
|---|---|---|
| 0.1 N HCl | 380 | 0.526 |
| Phosphate Buffer with pH 3.0 | 290 | 0.689 |
| Phosphate Buffer with pH 6.8 | 245 | 0.815 |
| Acetate Buffer with pH 4.5 | 244 | 0.818 |
| Distilled Water | 243 | 0.822 |
| Borate Buffer with pH 8.0 | 233 | 0.858 |
| Phosphate Buffer with pH 6.0 | 232 | 0.862 |
| Name of Solution | Retention Time (minutes) | Peak Purity | Purity Criteria | |
|---|---|---|---|---|
| Purity Angle | Purity Threshold | |||
| Blank | No peaks | - | - | - |
| Placebo Solution | No peaks | - | - | - |
| Standard Solution | 0.814 | 0.052 | 0.272 | Pass |
| Sample Solution | 0.803 | 0.023 | 0.269 | Pass |
| Level% | Concentration (mg/mL) | Area |
|---|---|---|
| 20 | 0.04394610 | 301,721 |
| 50 | 0.10986525 | 718,160 |
| 80 | 0.17578440 | 1,143,999 |
| 100 | 0.21973050 | 1,417,157 |
| 120 | 0.26367660 | 1,733,645 |
| 200 | 0.43946100 | 2,798,675 |
| Corr. Coefficient | 1.000 | |
| Slope | 6,336,092.9159 | |
| y-intercept | 29,604.9454 | |
| Sample No. | Symmetry Factor | Theorical Plate Count | Retention Time (min) | Area |
|---|---|---|---|---|
| 1 | 1.06 | 6719 | 0.716 | 1,423,910 |
| 2 | 1.07 | 6710 | 0.717 | 1,435,439 |
| 3 | 1.06 | 6693 | 0.716 | 1,420,744 |
| 4 | 1.06 | 6682 | 0.716 | 1,430,521 |
| 5 | 1.06 | 6666 | 0.718 | 1,429,301 |
| 6 | 1.06 | 6701 | 0.717 | 1,429,755 |
| Average | 1.06 | 6695 | 0.717 | 1,428,278 |
| SD | 0.00 | 19.24 | 0.00 | 5203.5 |
| RSD% | 0.38 | 0.29 | 0.11 | 0.36 |
| Sample No. | Favipiravir Release% |
|---|---|
| 1 | 103.1 |
| 2 | 103.4 |
| 3 | 99.9 |
| 4 | 98.8 |
| 5 | 102.7 |
| 6 | 99.7 |
| Average | 101.3 |
| SD | 2.02 |
| RSD% | 1.99 |
| Sample No. | Favipiravir Release% | |
|---|---|---|
| Analyst-1 Day-1 Instrument-1 Column-1 | Analyst-2 Day-2 Instrument-2 Column-2 | |
| 1 | 103.1 | 98.6 |
| 2 | 103.4 | 97.8 |
| 3 | 99.9 | 95.4 |
| 4 | 98.8 | 103.3 |
| 5 | 102.7 | 99.7 |
| 6 | 99.7 | 100.3 |
| Overall Average | 100.2 | |
| Overall SD | 2.49 | |
| Overall RSD% | 2.49 | |
| F-Test of Significance | 0.05 | |
| p-value | 0.521 | |
| Level% | Sample No. | Recovery% | Average | RSD% |
|---|---|---|---|---|
| 10 | 1 | 103.4 | 103.5 | 0.29 |
| 2 | 103.7 | |||
| 3 | 103.3 | |||
| 100 | 1 | 103.9 | 103.6 | 0.67 |
| 2 | 104.2 | |||
| 3 | 102.8 | |||
| 120 | 1 | 102.5 | 102.8 | 0.39 |
| 2 | 102.9 | |||
| 3 | 103.1 | |||
| Overall Average and RSD% | 103.3 | 0.53 | ||
| Analysis Name | Favipiravir Release% | Retention Time (min) | Symmetry Factor | Theorical Plate Count |
|---|---|---|---|---|
| Repeatability | 101.3 | 0.717 | 1.06 | 6695 |
| Wavelength: 223 nm | 100.0 | 0.712 | 1.06 | 6535 |
| Wavelength: 227 nm | 100.0 | 0.712 | 1.06 | 6541 |
| Flow Rate: 0.3 mL/min | 99.3 | 0.948 | 1.05 | 7482 |
| Flow Rate: 0.5 mL/min | 99.8 | 0.571 | 1.06 | 5553 |
| Column Temperature: 33 °C | 99.9 | 0.715 | 1.06 | 6592 |
| Column Temperature: 37 °C | 99.5 | 0.711 | 1.06 | 6556 |
| Stirring Rate: 45 rpm | 88.5 | 0.714 | 1.06 | 6485 |
| Stirring Rate: 55 rpm | 99.4 | 0.713 | 1.07 | 6458 |
| Injection Time (Hours) | Standard Solution | Sample Solution | ||
|---|---|---|---|---|
| Area | Similarity% | Area | Similarity% | |
| Initial | 1,474,726 | - | 1,462,137 | - |
| 24 | 1,466,496 | 99.4 | 1,449,968 | 99.2 |
| 48 | 1,450,231 | 98.3 | 1,433,548 | 98.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Göktuğ, Ö.; Altaş, E.; Kayar, G.; Gökalp, M. The Development and the Validation of a Novel Dissolution Method of Favipiravir Film-Coated Tablets. Sci. Pharm. 2022, 90, 3. https://doi.org/10.3390/scipharm90010003
Göktuğ Ö, Altaş E, Kayar G, Gökalp M. The Development and the Validation of a Novel Dissolution Method of Favipiravir Film-Coated Tablets. Scientia Pharmaceutica. 2022; 90(1):3. https://doi.org/10.3390/scipharm90010003
Chicago/Turabian StyleGöktuğ, Özge, Ecem Altaş, Gönül Kayar, and Mine Gökalp. 2022. "The Development and the Validation of a Novel Dissolution Method of Favipiravir Film-Coated Tablets" Scientia Pharmaceutica 90, no. 1: 3. https://doi.org/10.3390/scipharm90010003
APA StyleGöktuğ, Ö., Altaş, E., Kayar, G., & Gökalp, M. (2022). The Development and the Validation of a Novel Dissolution Method of Favipiravir Film-Coated Tablets. Scientia Pharmaceutica, 90(1), 3. https://doi.org/10.3390/scipharm90010003