
Repurposing of Anti-Malarial Drug Quinacrine for Cancer Treatment: A Review


Abstract
1. Introduction
2. Methods
3. Internalization and Pharmacokinetics of QC
Reported Toxicities of Quinacrine
4. Quinacrine and Cancer
4.1. Quinacrine and DNA Intercalation
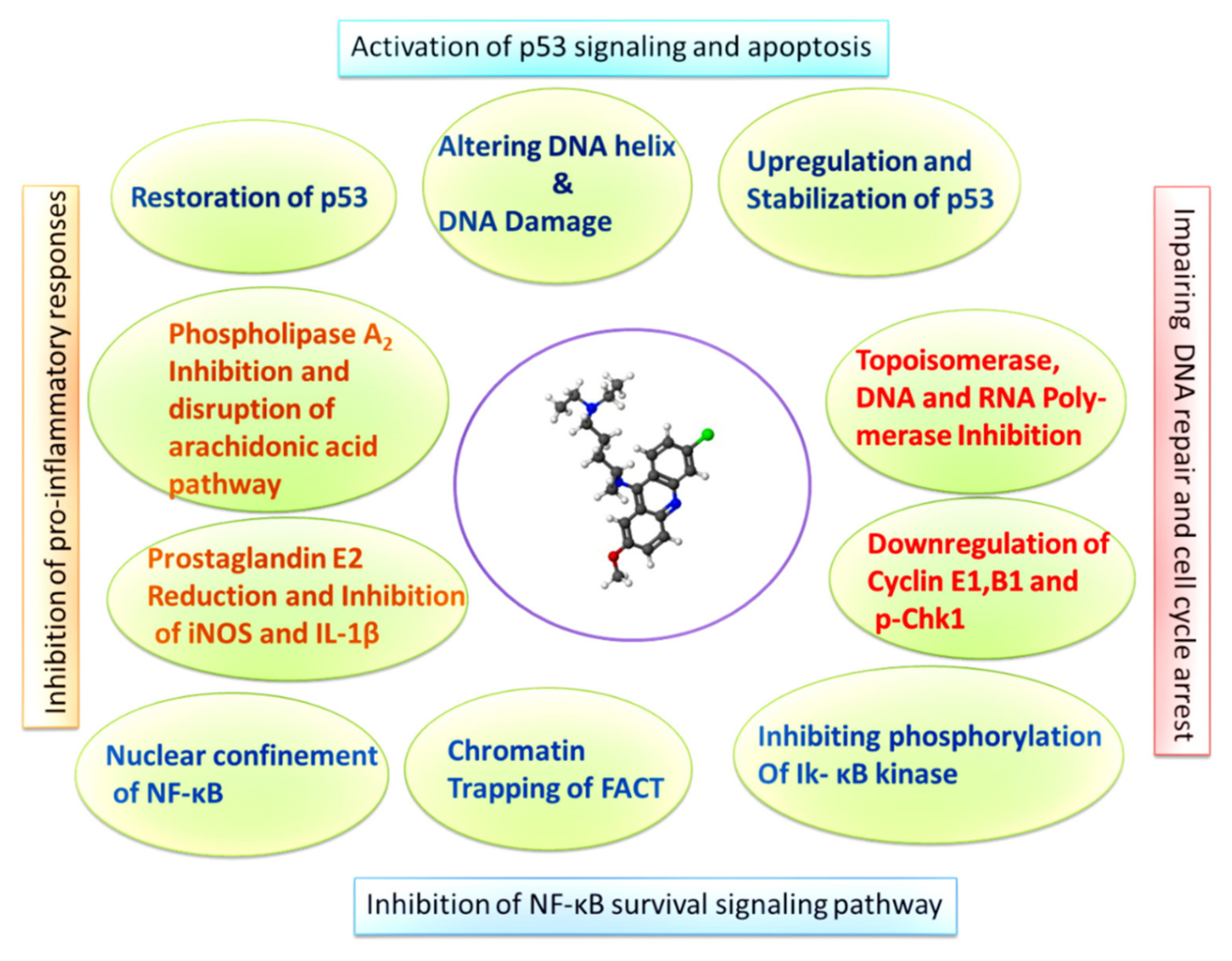
4.2. Quinacrine Mediated Induction of P53 Signaling and Inhibition of NF-κB
4.3. Quinacrine and Inhibition of DNA Replication Enzymes
4.4. Quinacrine Induced Autophagy and Cell Cycle Arrest
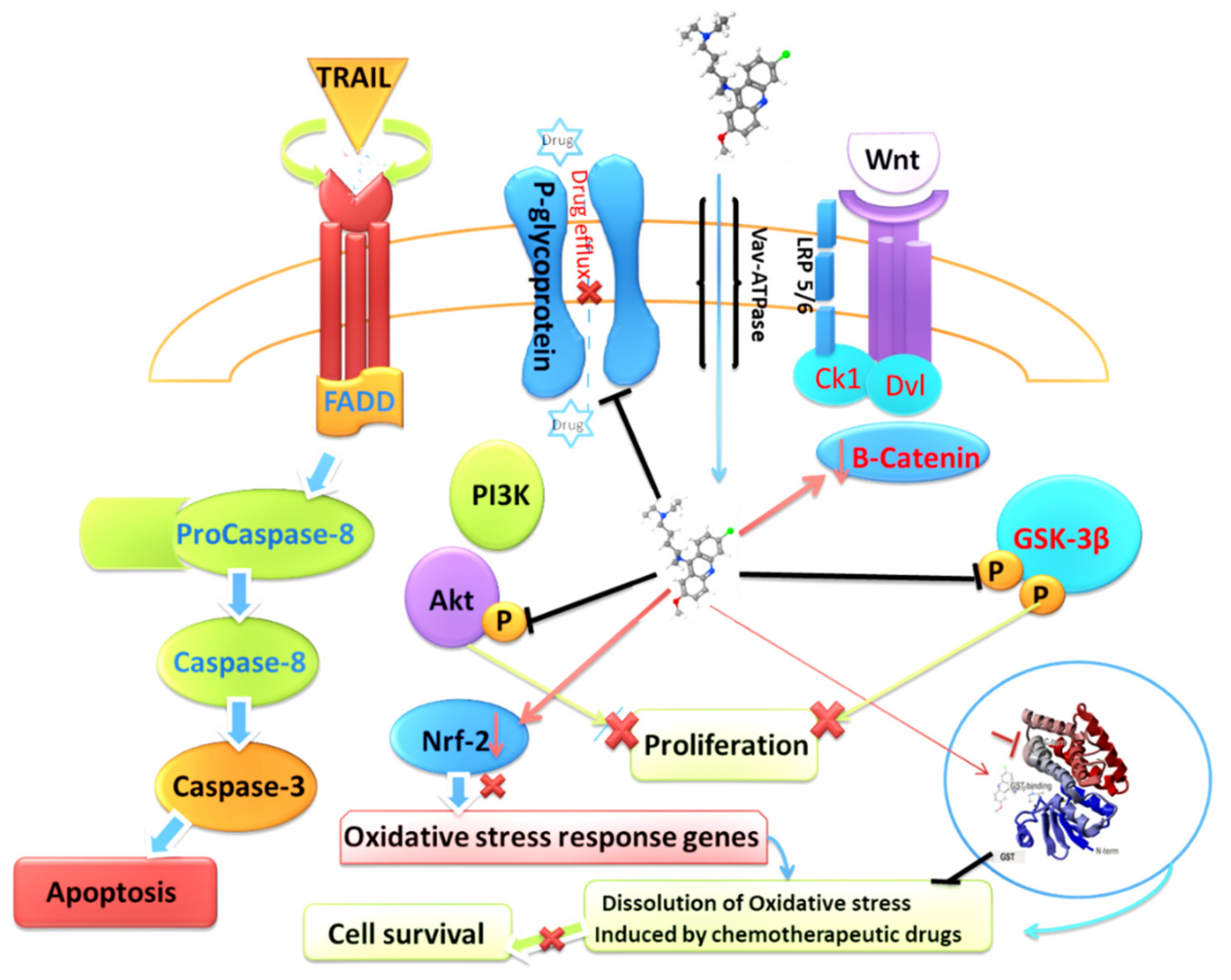
4.5. Quinacrine and TRAIL Sensitivity
4.6. Quinacrine and Chemoresistance
4.7. Synergistic Effects of Quinacrine with Other Chemotherapeutic and Targeted Therapies
5. Quinacrine Nanoparticles in Cancer Treatment
6. Clinical Research Studies of Quinacrine in Cancer Treatment
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Greenwood, D. Conflicts of interest: The genesis of synthetic antimalarial agents in peace and war. J. Antimicrob. Chemother. 1995, 36, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Office of the Surgeon General UCln. The drug treatment of malaria, suppressive and clinical. J. Am. Med. Assoc. 1943, 123, 205–208. [Google Scholar] [CrossRef]
- Joint Report of the Armored Medical Research Laboratory and Commission Tropical Diseases AEB, Preventative Medicine Service, Office of the Surgeon General, US Army, Plasma quinacrine concentration as a function of dosage and environment. Arch Intern. Med. 1946, 78, 64–107. [CrossRef] [PubMed]
- Lee, Y.Q.; Goh, A.S.; Ch’ng, J.H.; Nosten, F.H.; Preiser, P.R.; Pervaiz, S.; Yadav, S.K.; Tan, K.S. A high-content phenotypic screen reveals the disruptive potency of quinacrine and 3′, 4′-dichlorobenzamil on the digestive vacuole of Plasmodium falciparum. Antimicrob. Agents Chemother. 2014, 58, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.C. Treatment of human taeniasis in the Philippines: A review. Southeast Asian J. Trop. Med. Public Health 1991, 22, 271–274. [Google Scholar] [PubMed]
- Lerman, S.J.; Walker, R.A. Treatment of giardiasis: Literature review and recommendations. Clin. Pediatrics 1982, 21, 409–414. [Google Scholar] [CrossRef]
- Feldmann, R.; Salomon, D.; Saurat, J.H. The association of the two antimalarials chloroquine and quinacrine for treatment-resistant chronic and subacute cutaneous lupus erythematosus. Dermatology 1994, 189, 425–427. [Google Scholar] [CrossRef]
- Wallace, D.J. The use of quinacrine (Atabrine) in rheumatic diseases: A reexamination. In Seminars in Arthritis and Rheumatism; WB Saunders: Philadelphia, PA, USA, 1989; Volume 18, pp. 282–296. [Google Scholar]
- Weisblum, B.; De Haseth, P.L. Quinacrine, a chromosome stain specific for deoxyadenylate-deoxythymidylate-rich regions in DNA. Proc. Natl. Acad. Sci. USA 1972, 69, 629–632. [Google Scholar] [CrossRef]
- Latt, S.A. Fluorescent probes of chromosome structure and replication. Can. J. Genet. Cytol. 1977, 19, 603–623. [Google Scholar] [CrossRef]
- Sahar, E.; Latt, S.A. Enhancement of banding patterns in human metaphase chromosomes by energy transfer. Proc. Natl. Acad. Sci. USA 1978, 75, 5650–5654. [Google Scholar] [CrossRef]
- Zipper, J.; Trujillo, V. 25 years of quinacrine sterilization experience in Chile: Review of 2592 cases. Int. J. Gynecol. Obstet. 2003, 83, S23–S29. [Google Scholar] [CrossRef]
- Lippes, J. Quinacrine sterilization (QS): Time for reconsideration. Contraception 2015, 92, 91–95. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taylor, S.A.; Hooton, N.S.; Macarthur, A.M. Quinacrine in the management of malignant pleural effusion. J. Br. Surg. 1977, 64, 52–53. [Google Scholar] [CrossRef] [PubMed]
- Kessel, E. Advances in contraception. Adv. Contracept. 1996, 12, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Shannon, J.A.; Earle, D.P.; Brodie, B.B.; Taggart, J.V.; Berliner, R.W. The pharmacological basis for the rational use of atabrine in the treatment of malaria. J. Pharmacol. Exp. Ther. 1944, 81, 307–330. [Google Scholar]
- Goodman, L.S. Goodman and Gilman’s the Pharmacological Basis of Therapeutics; McGraw-Hill: New York, NY, USA, 1996. [Google Scholar]
- Marceau, F.; Bawolak, M.T.; Bouthillier, J.; Morissette, G. Vacuolar ATPase-mediated cellular concentration and retention of quinacrine: A model for the distribution of lipophilic cationic drugs to autophagic vacuoles. Drug Metab. Dispos. 2009, 37, 2271–2274. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.; Gagné, V.; Fernandes, M.J.; Marceau, F. High affinity capture and concentration of quinacrine in polymorphonuclear neutrophils via vacuolar ATPase-mediated ion trapping: Comparison with other peripheral blood leukocytes and implications for the distribution of cationic drugs. Toxicol. Appl. Pharmacol. 2013, 270, 77–86. [Google Scholar] [CrossRef]
- Huang, Y.; Okochi, H.; May, B.C.; Legname, G.; Prusiner, S.B.; Benet, L.Z.; Guglielmo, B.J.; Lin, E.T. Quinacrine is mainly metabolized to mono-desethyl quinacrine by CYP3A4/5 and its brain accumulation is limited by P-glycoprotein. Drug Metab. Dispos. 2006, 34, 1136–1144. [Google Scholar] [CrossRef]
- Solitro, A.R.; MacKeigan, J.P. Tissue distribution and tumor concentrations of hydroxychloroquine and quinacrine analogs in mice. bioRxiv 2018, 496018. [Google Scholar]
- Bauer, F. Quinacrine hydrochloride drug eruption (tropical lichenoid dermatitis): Its early and late sequelae and its malignant potential: A review. J. Am. Acad. Dermatol. 1981, 4, 239–248. [Google Scholar] [CrossRef]
- Custer, R.P. Aplastic anemia in soldiers treated with atabrine (quinacrine). Am. J. Med. Sci. 1946, 212, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Fishman, A.P.; Kinsman, J.M. Hypoplastic anemia due to atabrine. Blood 1949, 4, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Paton, M.D.; Riddell, M.J.; Strong, J.A. Aplastic anaemia following mepacrine treatment of lupus erythematosus. Lancet 1955, 265, 281–282. [Google Scholar] [CrossRef]
- Carr, R.E.; Henkind, P.; Rothfield, N.; Siegel, I.M. Ocular toxicity of antimalarial drugs: Long-term follow-up. Am. J. Ophthalmol. 1968, 66, 738–744. [Google Scholar] [CrossRef]
- Ansdell, V.E.; Common, J.D. Corneal changes induced by mepacrinc. J. Trop. Med. Hyg. 1979, 82, 206–207. [Google Scholar]
- Engel, G.L. Quinacrine effects on the central nervous system. JAMA 1966, 197, 515. [Google Scholar] [CrossRef]
- Lindenmayer, J.P.; Vargas, P. Toxic psychosis following use of quinacrine. J. Clin. Psychiatry 1981, 42, 162–164. [Google Scholar]
- Theodore, T.; Kahn, R.L. Toxicity of quinacrine (atabrine) for the central nervous system: III. An experimental study on human subjects. Arch. Neurol. Psychiatry 1946, 56, 284–299. [Google Scholar] [CrossRef]
- Vassey, J.W.; Edmonds, J.; Irvin, J.L.; Green, J.A.; Irvin, E.M. Studies on the administration of quinacrine to tumor-bearing mice. Cancer Res. 1955, 15, 573–578. [Google Scholar]
- Hiller, R.I. A study of quinacrine dihydrochloride in the human breast in vitro and in vivo. Am. J. Surg. 1970, 119, 317–321. [Google Scholar] [CrossRef]
- Borja, E.R.; Pugh, R.P. Single-dose quinacrine (atabrine) and thoracostomy in the control of pleural effusions in patients with neoplastic diseases. Cancer 1973, 31, 899–902. [Google Scholar] [CrossRef]
- Denny, W.A.; Baguley, B.C.; Cain, B.F.; Waring, M.J. Antitumour acridines. In Molecular Aspects of Anti-Cancer Drug Action; Palgrave: London, UK, 1983; pp. 1–34. [Google Scholar]
- Koldsland, S.; Svennevig, J.L.; Lehne, G.; Johnson, E. Chemical pleurodesis in malignant pleural effusions: A randomised prospective study of mepacrine versus bleomycin. Thorax 1993, 48, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Preet, R.; Mohapatra, P.; Mohanty, S.; Sahu, S.K.; Choudhuri, T.; Wyatt, M.D.; Kundu, C.N. Quinacrine has anticancer activity in breast cancer cells through inhibition of topoisomerase activity. Int. J. Cancer 2012, 130, 1660–1670. [Google Scholar] [CrossRef]
- Wu, X.; Wang, Y.; Wang, H.; Wang, Q.; Wang, L.; Miao, J.; Cui, F.; Wang, J. Quinacrine inhibits cell growth and induces apoptosis in human gastric cancer cell line SGC-7901. Curr. Ther. Res. 2012, 73, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Martin, A.; Nirgude, S.; Chaudhary, B.; Mondal, S.; Sarkar, A. Quinacrine inhibits GSTA1 activity and induces apoptosis through G1/S arrest and generation of ROS in human non-small cell lung cancer cell lines. Oncotarget 2020, 11, 1603. [Google Scholar] [CrossRef]
- Harada, M.; Morimoto, K.; Kondo, T.; Hiramatsu, R.; Okina, Y.; Muko, R.; Matsuda, I.; Kataoka, T. Quinacrine inhibits ICAM-1 transcription by blocking DNA binding of the NF-κB subunit p65 and sensitizes human lung adenocarcinoma A549 cells to TNF-α and the Fas ligand. Int. J. Mol. Sci. 2017, 18, 2603. [Google Scholar] [CrossRef]
- Dermawan, J.K.; Gurova, K.; Pink, J.; Dowlati, A.; De, S.; Narla, G.; Sharma, N.; Stark, G.R. Quinacrine Overcomes Resistance to Erlotinib by Inhibiting FACT, NF-κB, and Cell-Cycle Progression in Non–Small Cell Lung Cancer. Mol. Cancer Ther. 2014, 13, 2203–2214. [Google Scholar] [CrossRef]
- Jani, T.S.; DeVecchio, J.; Mazumdar, T.; Agyeman, A.; Houghton, J.A. Inhibition of NF-κB signaling by quinacrine is cytotoxic to human colon carcinoma cell lines and is synergistic in combination with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) or oxaliplatin. J. Biol. Chem. 2010, 285, 19162–19172. [Google Scholar] [CrossRef]
- Mohapatra, P.; Preet, R.; Das, D.; Satapathy, S.R.; Choudhuri, T.; Wyatt, M.D.; Kundu, C.N. Quinacrine-mediated autophagy and apoptosis in colon cancer cells is through a p53-and p21-dependent mechanism. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2012, 20, 81–91. [Google Scholar] [CrossRef]
- Park, S.; Oh, A.Y.; Cho, J.H.; Yoon, M.H.; Woo, T.G.; Kang, S.M.; Lee, H.Y.; Jung, Y.J.; Park, B.J. Therapeutic effect of quinacrine, an antiprotozoan drug, by selective suppression of p-CHK1/2 in p53-negative malignant cancers. Mol. Cancer Res. 2018, 16, 935–946. [Google Scholar] [CrossRef]
- Abdulghani, J.; Gokare, P.; Gallant, J.N.; Dicker, D.; Whitcomb, T.; Cooper, T.; Liao, J.; Derr, J.; Liu, J.; Goldenberg, D.; et al. Sorafenib and quinacrine target anti-apoptotic protein MCL1: A poor prognostic marker in anaplastic thyroid cancer (ATC). Clin. Cancer Res. 2016, 22, 6192–6203. [Google Scholar] [CrossRef] [PubMed]
- Erkoc, P.; Cingöz, A.; Bagci-Onder, T.; Kizilel, S. Quinacrine Mediated Sensitization of Glioblastoma (GBM) Cells to TRAIL through MMP-Sensitive PEG Hydrogel Carriers. Macromol. Biosci. 2017, 17, 1600267. [Google Scholar] [CrossRef]
- Matteoni, S.; Abbruzzese, C.; Matarrese, P.; De Luca, G.; Mileo, A.M.; Miccadei, S.; Schenone, S.; Musumeci, F.; Haas, T.L.; Sette, G.; et al. The kinase inhibitor SI113 induces autophagy and synergizes with quinacrine in hindering the growth of human glioblastoma multiforme cells. J. Exp. Clin. Cancer Res. 2019, 38, 202. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Roy, S.; Lazar, A.J.; Wang, W.L.; McAuliffe, J.C.; Reynoso, D.; McMahon, J.; Taguchi, T.; Floris, G.; Debiec-Rychter, M.; et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc. Natl. Acad. Sci. USA 2010, 107, 14333–14338. [Google Scholar] [CrossRef]
- Preet, R.; Siddharth, S.; Satapathy, S.R.; Das, S.; Nayak, A.; Das, D.; Wyatt, M.D.; Kundu, C.N. Chk1 inhibitor synergizes quinacrine mediated apoptosis in breast cancer cells by compromising the base excision repair cascade. Biochem. Pharmacol. 2016, 105, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, A.; Österroos, A.; Hassan, S.; Gullbo, J.; Rickardson, L.; Jarvius, M.; Nygren, P.; Fryknäs, M.; Höglund, M.; Larsson, R. Drug screen in patient cells suggests quinacrine to be repositioned for treatment of acute myeloid leukemia. Blood Cancer J. 2015, 5, e307. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Katz, S.I.; Abdulghani, J.; Dicker, D.; El-Deiry, W.S. Quinacrine sensitizes hepatocellular carcinoma cells to TRAIL via up-regulating DR5 and eradicating MCL-1. Cancer Res. 2010, 70, 680. [Google Scholar]
- Oien, D.B.; Ray, U.; Pathoulas, C.L.; Jin, L.; Thirusangu, P.; Jung, D.; Kumka, J.E.; Xiao, Y.; Sarkar Bhattacharya, S.; Montoya, D.; et al. Quinacrine Induces Nucleolar Stress in Treatment-Refractory Ovarian Cancer Cell Lines. Cancers 2021, 13, 4645. [Google Scholar] [CrossRef] [PubMed]
- Khurana, A.; Roy, D.; Kalogera, E.; Mondal, S.; Wen, X.; He, X.; Dowdy, S.; Shridhar, V. Quinacrine promotes autophagic cell death and chemosensitivity in ovarian cancer and attenuates tumor growth. Oncotarget 2015, 6, 36354. [Google Scholar] [CrossRef]
- Jung, D.; Khurana, A.; Roy, D.; Kalogera, E.; Bakkum-Gamez, J.; Chien, J.; Shridhar, V. Quinacrine upregulates p21/p27 independent of p53 through autophagy-mediated downregulation of p62-Skp2 axis in ovarian cancer. Sci. Rep. 2018, 8, 2487. [Google Scholar] [CrossRef]
- Liang, R.; Yao, Y.; Wang, G.; Yue, E.; Yang, G.; Qi, X.; Wang, Y.; Zhao, L.; Zheng, T.; Zhang, Y.; et al. Repositioning quinacrine toward treatment of ovarian cancer by rational combination with TRAIL. Front. Oncol. 2020, 10, 1118. [Google Scholar] [CrossRef] [PubMed]
- Thirusangu, P.; Pathoulas, C.L.; Ray, U.; Xiao, Y.; Staub, J.; Jin, L.; Khurana, A.; Shridhar, V. Quinacrine-Induced Autophagy in Ovarian Cancer Triggers Cathepsin-L Mediated Lysosomal/Mitochondrial Membrane Permeabilization and Cell Death. Cancers 2021, 13, 2004. [Google Scholar] [CrossRef] [PubMed]
- Gurova, K.V.; Hill, J.E.; Guo, C.; Prokvolit, A.; Burdelya, L.G.; Samoylova, E.; Khodyakova, A.V.; Ganapathi, R.; Ganapathi, M.; Tararova, N.D.; et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-κB-dependent mechanism of p53 suppression in tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 17448–17453. [Google Scholar] [CrossRef]
- Friedman, J.; Nottingham, L.; Duggal, P.; Pernas, F.G.; Yan, B.; Yang, X.P.; Chen, Z.; Van Waes, C. Deficient TP53 expression, function, and cisplatin sensitivity are restored by quinacrine in head and neck cancer. Clin. Cancer Res. 2007, 13, 6568–6578. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.; Batis, N.; Franke, A.C.; Clancey, G.; Hartley, M.; Ryan, G.; Brooks, J.; Southam, A.D.; Barnes, N.; Parish, J.; et al. Repurposed quinacrine synergizes with cisplatin, reducing the effective dose required for treatment of head and neck squamous cell carcinoma. Oncotarget 2019, 10, 5229. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fox, G.; Popanda, O.; Edler, L.; Thielmann, H.W. Preferential inhibition of DNA polymerases α, δ, and ε from Novikoff hepatoma cells by inhibitors of cell proliferation. J. Cancer Res. Clin. Oncol. 1996, 122, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Chen, Z.; Wang, L.; Peng, D.; Belkhiri, A.; Lockhart, A.C.; El-Rifai, W. A combination of SAHA and Quinacrine is effective in inducing cancer cell death in upper gastrointestinal cancers. Clin. Cancer Res. 2018, 24, 1905–1916. [Google Scholar] [CrossRef]
- Jing, B.; Jin, J.; Xiang, R.; Liu, M.; Yang, L.; Tong, Y.; Xiao, X.; Lei, H.; Liu, W.; Xu, H.; et al. Vorinostat and quinacrine have synergistic effects in T-cell acute lymphoblastic leukemia through reactive oxygen species increase and mitophagy inhibition. Cell Death Dis. 2018, 9, 589. [Google Scholar] [CrossRef]
- Charmantray, F.; Martelli, A. Interest of acridine derivatives in the anticancer chemotherapy. Curr. Pharm. Des. 2001, 7, 1703–1724. [Google Scholar]
- Gurova, K. New hopes from old drugs: Revisiting DNA-binding small molecules as anticancer agents. Future Oncol. 2009, 5, 1685–1704. [Google Scholar] [CrossRef]
- Hossain, M.Z.; Healey, M.A.; Lee, C.; Poh, W.; Yerram, S.R.; Patel, K.; Azad, N.S.; Herman, J.G.; Kern, S.E. DNA-intercalators causing rapid re-expression of methylated and silenced genes in cancer cells. Oncotarget 2013, 4, 298. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ho, W.C.; Dicker, D.T.; MacKinnon, C.; Winkler, J.D.; Marmorstein, R.; El-Deiry, W.S. Acridine derivatives activate p53 and induce tumor cell death through Bax. Cancer Biol. Ther. 2005, 4, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Ehsanian, R.; Van Waes, C.; Feller, S.M. Beyond DNA binding-a review of the potential mechanisms mediating quinacrine’s therapeutic activities in parasitic infections, inflammation, and cancers. Cell Commun. Signal. 2011, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Klein, C.L.; van Kooten, T.G.; Kirkpatrick, C.J. Mechanisms of cell activation by heavy metal ions. J. Biomed. Mater. Res. Off. J. Soc. Biomater. Jpn. Soc. Biomater. Aust. Soc. Biomater. 1998, 42, 443–452. [Google Scholar] [CrossRef]
- König, W.; Schönfeld, W.; Raulf, M.; Köller, M.; Knöller, J.; Scheffer, J.; Brom, J. The neutrophil and leukotrienes-role in health and disease. Eicosanoids 1990, 3, 1–22. [Google Scholar] [PubMed]
- Stuhlmeier, K.M.; Kao, J.J.; Bach, F.H. Arachidonic acid influences proinflammatory gene induction by stabilizing the inhibitor-κBα/nuclear factor-κB (NF-κB) complex, thus suppressing the nuclear translocation of NF-κB. J. Biol. Chem. 1997, 272, 24679–24683. [Google Scholar] [CrossRef]
- Palmetshofer, A.; Robson, S.C.; Nehls, V. Lysophosphatidic acid activates nuclear factor kappa B and induces proinflammatory gene expression in endothelial cells. Thromb. Haemost. 1999, 82, 1532–1537. [Google Scholar] [PubMed]
- Nesher, E.; Safina, A.; Aljahdali, I.; Portwood, S.; Wang, E.S.; Koman, I.; Wang, J.; Gurova, K.V. Role of chromatin damage and chromatin trapping of FACT in mediating the anticancer cytotoxicity of DNA-binding small-molecule drugs. Cancer Res. 2018, 78, 1431–1443. [Google Scholar] [CrossRef]
- Na, S.I.; Lee, M.Y.; Heo, J.S.; Han, H.J. Hydrogen Peroxide Increases [3H]-2-Deoxyglucose uptake via MAPKs, cPLA2, and NF-κB Signaling Pathways in Mouse Embryonic Stem Cells. Cell. Physiol. Biochem. 2007, 20, 1007–1018. [Google Scholar] [CrossRef]
- Langer, S.W.; Schmidt, G.; Sørensen, M.; Sehested, M.; Jensen, P.B. Inhibitors of topoisomerase II as pH-dependent modulators of etoposide-mediated cytotoxicity. Clin. Cancer Res. 1999, 5, 2899–2907. [Google Scholar]
- Popanda, O.; Thielmann, H.W. The function of DNA topoisomerases in UV-induced DNA excision repair: Studies with specific inhibitors in permeabilized human fibroblasts. Carcinogenesis 1992, 13, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Fuks, Z.; Smith, K.C. Effect of quinacrine on x-ray sensitivity and the repair of damaged DNA in Escherichia coli K-12. Radiat. Res. 1971, 48, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Voiculetz, N.; Smith, K.C.; Kaplan, H.S. Effect of Quinacrine on survival and DNA repair in X-irradiated Chinese hamster cells. Cancer Res. 1974, 34, 1038–1044. [Google Scholar]
- Werbovetz, K.A.; Lehnert, E.K.; Macdonald, T.L.; Pearson, R.D. Cytotoxicity of acridine compounds for Leishmania promastigotes in vitro. Antimicrob. Agents Chemother. 1992, 36, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Dominick, P.K.; Keppler, B.R.; Legassie, J.D.; Moon, I.K.; Jarstfer, M.B. Nucleic acid-binding ligands identify new mechanisms to inhibit telomerase. Bioorganic Med. Chem. Lett. 2004, 14, 3467–3471. [Google Scholar] [CrossRef]
- Harrison, R.J.; Gowan, S.M.; Kelland, L.R.; Neidle, S. Human telomerase inhibition by substituted acridine derivatives. Bioorganic Med. Chem. Lett. 1999, 9, 2463–2468. [Google Scholar] [CrossRef]
- Sun, H.; Xiang, J.; Li, Q.; Liu, Y.; Li, L.; Shang, Q.; Xu, G.; Tang, Y. Recognize three different human telomeric G-quadruplex conformations by quinacrine. Analyst 2012, 137, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.C.; Ramanathan, S. Quinacrine: Site of inhibition of synchronized cell division in Tetrahymena. Life Sci. 1968, 7, 1053–1062. [Google Scholar] [CrossRef]
- Van Dyke, K.; Lantz, C.; Szustkiewicz, C. Quinacrine: Mechanisms of antimalarial action. Science 1970, 169, 492–493. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Oien, D.B.; Pathoulas, C.L.; Ray, U.; Thirusangu, P.; Kalogera, E.; Shridhar, V. Repurposing quinacrine for treatment-refractory cancer. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2021; Volume 68, pp. 21–30. [Google Scholar]
- Das, S.; Nayak, A.; Siddharth, S.; Nayak, D.; Narayan, S.; Kundu, C.N. TRAIL enhances quinacrine-mediated apoptosis in breast cancer cells through induction of autophagy via modulation of p21 and DR5 interactions. Cell. Oncol. 2017, 40, 593–607. [Google Scholar] [CrossRef]
- Das, S.; Tripathi, N.; Preet, R.; Siddharth, S.; Nayak, A.; Bharatam, P.V.; Kundu, C.N. Quinacrine induces apoptosis in cancer cells by forming a functional bridge between TRAIL-DR5 complex and modulating the mitochondrial intrinsic cascade. Oncotarget 2017, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Marquez, B.; Van Bambeke, F. ABC multidrug transporters: Target for modulation of drug pharmacokinetics and drug-drug interactions. Curr. Drug Targets 2011, 12, 600–620. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Virshup, D.M. Wnt signaling and drug resistance in cancer. Mol. Pharmacol. 2020, 97, 72–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Inaba, M.; Maruyama, E. Reversal of resistance to vincristine in P388 leukemia by various polycyclic clinical drugs, with a special emphasis on quinacrine. Cancer Res. 1988, 48, 2064–2067. [Google Scholar]
- Wu, L.; Chatla, S.; Lin, Q.; Chowdhury, F.A.; Geldenhuys, W.; Du, W. Quinacrine-CASIN combination overcomes chemoresistance in human acute lymphoid leukemia. Nat. Commun. 2021, 12, 6936. [Google Scholar] [CrossRef]
- Carotenuto, P.; Romano, A.; Barbato, A.; Quadrano, P.; Brillante, S.; Volpe, M.; Ferrante, L.; Tammaro, R.; Morleo, M.; De Cegli, R.; et al. Drug Repurposing to Target the Apoptosome in MAPKi-Resistant Melanoma. Cell Rep. 2021. [Google Scholar] [CrossRef]
- Tiberghien, F.; Loor, F. Ranking of P-glycoprotein substrates and inhibitors by a calcein-AM fluorometry screening assay. Anti-Cancer Drugs 1996, 7, 568–578. [Google Scholar] [CrossRef]
- Leader, J.P.; O’Donnell, M.J. Transepithelial transport of fluorescent p-glycoprotein and MRP2 substrates by insect Malpighian tubules: Confocal microscopic analysis of secreted fluid droplets. J. Exp. Biol. 2005, 208, 4363–4376. [Google Scholar] [CrossRef]
- Preet, R.; Mohapatra, P.; Das, D.; Satapathy, S.R.; Choudhuri, T.; Wyatt, M.D.; Kundu, C.N. Lycopene synergistically enhances quinacrine action to inhibit Wnt-TCF signaling in breast cancer cells through APC. Carcinogenesis 2013, 34, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Gasparian, A.V.; Zhuang, Z.; Bosykh, D.A.; Komar, A.A.; Gudkov, A.V.; Gurova, K.V. 9-Aminoacridine-based anticancer drugs target the PI3K/AKT/mTOR, NF-κB and p53 pathways. Oncogene 2009, 28, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Kim, C.W.; Lee, D.H.; Lee, J.S.; Oh, E.T.; Park, H.J. Quinacrine-mediated inhibition of Nrf2 reverses hypoxia-induced 5-fluorouracil resistance in colorectal cancer. Int. J. Mol. Sci. 2019, 20, 4366. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, S.; Sabzichi, M.; Rashidi, M.; Mohammadian, J.; Mahmoudi, S.; Maroufi, N.F.; Ramezani, F.; Ghorbani, M.; Mohammadi, M.; Pirouzpanah, M.; et al. Sensitization of A-549 lung cancer cells to Cisplatin by Quinacrine-loaded lipidic nanoparticles via suppressing Nrf2 mediated defense mechanism. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- De Souza, P.L.; Castillo, M.; Myers, C.E. Enhancement of paclitaxel activity against hormone-refractory prostate cancer cells in vitro and in vivo by quinacrine. Br. J. Cancer 1997, 75, 1593–1600. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, H.J.; Yu, Y.; Zhang, Y.; Li, R.J.; Ju, R.J.; Wang, X.X.; Sun, M.G.; Shi, J.F.; Lu, W.L. Mitochondrial targeting liposomes incorporating daunorubicin and quinacrine for treatment of relapsed breast cancer arising from cancer stem cells. Biomaterials 2012, 33, 565–582. [Google Scholar] [CrossRef]
- Gallant, J.N.; Allen, J.E.; Smith, C.D.; Dicker, D.T.; Wang, W.; Dolloff, N.G.; Navaraj, A.; El-Deiry, W.S. Quinacrine synergizes with 5-fluorouracil and other therapies in colorectal cancer. Cancer Biol. Ther. 2011, 12, 239–251. [Google Scholar] [CrossRef]
- Eriksson, A.; Chantzi, E.; Fryknäs, M.; Gullbo, J.; Nygren, P.; Gustafsson, M.; Höglund, M.; Larsson, R. Towards repositioning of quinacrine for treatment of acute myeloid leukemia–Promising synergies and in vivo effects. Leuk. Res. 2017, 63, 41–46. [Google Scholar] [CrossRef]
- Kalogera, E.; Roy, D.; Khurana, A.; Mondal, S.; Weaver, A.L.; He, X.; Dowdy, S.C.; Shridhar, V. Quinacrine in endometrial cancer: Repurposing an old antimalarial drug. Gynecol. Oncol. 2017, 146, 187–195. [Google Scholar] [CrossRef]
- Zhang, P.; Li, N.; Kiang, K.M.; Zhu, Z.; Leung, G.W.; Cheng, S.Y.; Leung, G.K. Quinacrine enhances temozolomide cytotoxicity in temozolomide-sensitive and-resistant glioblastoma cells. Glioma 2018, 1, 175. [Google Scholar]
- Bhateja, P.; Dowlati, A.; Sharma, N. Phase I study of the combination of quinacrine and erlotinib in patients with locally advanced or metastatic non small cell lung cancer. Investig. New Drugs 2018, 36, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Winer, A.; Denlinger, C.S.; Vijayvergia, N.; Cohen, S.J.; Astaturov, I.; Dotan, E.; Gallant, J.N.; Wang, E.W.; Kunkel, M.; Lim, B.; et al. First-in-Human Phase 1b Trial of Quinacrine Plus Capecitabine in Patients With Refractory Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2021, 20, e43–e52. [Google Scholar] [CrossRef]
- Hembram, K.C.; Chatterjee, S.; Sethy, C.; Nayak, D.; Pradhan, R.; Molla, S.; Bindhani, B.K.; Kundu, C.N. Comparative and mechanistic study on the anticancer activity of quinacrine-based silver and gold hybrid nanoparticles in head and neck cancer. Mol. Pharm. 2019, 16, 3011–3023. [Google Scholar] [CrossRef] [PubMed]
- Hembram, K.C.; Dash, S.R.; Das, B.; Sethy, C.; Chatterjee, S.; Bindhani, B.K.; Kundu, C.N. Quinacrine based gold hybrid nanoparticles caused apoptosis through modulating replication fork in oral cancer stem cells. Mol. Pharm. 2020, 17, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Etman, S.M.; Mehanna, R.A.; Bary, A.A.; Elnaggar, Y.S.; Abdallah, O.Y. Undaria pinnatifida fucoidan nanoparticles loaded with quinacrine attenuate growth and metastasis of pancreatic cancer. Int. J. Biol. Macromol. 2021, 170, 284–297. [Google Scholar] [CrossRef]
- Nayak, A.; Satapathy, S.R.; Das, D.; Siddharth, S.; Tripathi, N.; Bharatam, P.V.; Kundu, C. Nanoquinacrine induced apoptosis in cervical cancer stem cells through the inhibition of hedgehog-GLI1 cascade: Role of GLI-1. Sci. Rep. 2016, 6, 20600. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, N.S.; Vaidya, B.; Gupta, V. Nano-synergistic combination of Erlotinib and Quinacrine for non-small cell lung cancer (NSCLC) therapeutics–Evaluation in biologically relevant in-vitro models. Mater. Sci. Eng. C 2021, 121, 111891. [Google Scholar] [CrossRef]
- Vaidya, B.; Kulkarni, N.S.; Shukla, S.K.; Parvathaneni, V.; Chauhan, G.; Damon, J.K.; Sarode, A.; Garcia, J.V.; Kunda, N.; Mitragotri, S.; et al. Development of inhalable quinacrine loaded bovine serum albumin modified cationic nanoparticles: Repurposing quinacrine for lung cancer therapeutics. Int. J. Pharm. 2020, 577, 118995. [Google Scholar] [CrossRef]
- Hickman, J.A.; Jones, M.C. Treatment of neoplastic pleural effusions with local instillations of quinacrine (mepacrine) hydrochloride. Thorax 1970, 25, 226–229. [Google Scholar] [CrossRef][Green Version]
- Bayly, T.C.; Kisner, D.L.; Sybert, A.; Macdonald, J.S.; Tsou, E.; Schein, P.S. Tetracycline and quinacrine in the control of malignant pleural effusions. A randomized trial. Cancer 1978, 41, 1188–1192. [Google Scholar] [CrossRef]
- Agrenius, V.; Chmielewska, J.; Widstrom, O.; Blomback, M. Increased coagulation activity of the pleura after tube drainage and quinacrine instillation in malignant pleural effusion. Eur. Respir. J. 1991, 4, 1135–1139. [Google Scholar] [PubMed]
- Agrenius, V.; Chmielewska, J.; Widstrom, O.L.; Blomback, M. Pleural fibrinolytic activity is decreased in inflammation as demonstrated in quinacrine pleurodesis treatment of malignant pleural effusion. Am. Rev. Respir. Dis. 1989, 140, 1381–1385. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Sl No. | Type of Cancer | Drugs Studied | Effects | Reference |
|---|---|---|---|---|
| 1 | NSCLC | Quinacrine | Inhibits GSTA1 activity, ROS generation, and mitochondrial damage | [38] |
| 2 | NSCLC | Quinacrine | Suppresses NF-κB activity and ICAM-1 transcription | [39] |
| 3 | NSCLC | Quinacrine and Erlotinib | Inhibits FACT and NF-κB activity and cell cycle arrest and restores sensitivity to Erlotinib | [40] |
| 4 | CC | Quinacrine and TRAIL | Downregulation of c-FLIP and Mcl-1, sensitizes cells to TRAIL induced apoptosis | [41] |
| 5 | CC | Quinacrine | Upregulates p53 and p21 expression, simultaneous downregulation of p62 SQSTM leading to autophagic cell death | [42] |
| 6 | CC | Quinacrine | Downregulates p-Chk1/2, increases binding activity between p-Chk1/2 and β-TrCP leading to its degradation, G2/M arrest, apoptosis | [43] |
| 7 | ATC | Quinacrine and Sorafenib | Eradicates Mcl-1 expression, arrests proliferation and apoptosis induction | [44] |
| 8 | GBM | Quinacrine and TRAIL | Enhancement of TRAIL sensitivity and apoptosis induction | [45] |
| 9 | GBM | Quinacrine and telomozideQuinacrine and SI113 | Activates p53 and enhances apoptotic signaling, restores telomozide sensitivity. Downregulates p62 SQSTM and promotes autophagic flux | [46,47] |
| 10 | BC | Quinacrine | Activation of p53 signaling, suppresses NF-κB activity and apoptosis induction | [36] |
| 11 | BC | Quinacrine | Downregulation and suppression of p-CHK1/2 | [43] |
| 12 | BC | Quinacrine and SB218078(Chk1 inhibitor) | Downregulates p-Chk1/2, Cyclin B1, E1 and Cdc25A, arrests cells at G2/M stage, suppress BER, enhances SB218078 effect and apoptosis | [48] |
| 13 | AML | Quinacrine | Inhibits protein synthesis and ribosomal biogenesis, cell death | [49] |
| 14 | HCC | Quinacrine and TRAIL | Upregulates DR5 expression and eradicates anti-apoptotic protein Mcl-1, sensitizes cells to TRAIL mediated cell death | [50] |
| 15 | OC | Quinacrine and Rucaparib | Inhibits ribosomal biogenesis through attenuation of nucleostemin (NS/GNL3) and RPA194 expression, promotes DNA damage sensitizes cells to PARP inhibitor Rucaparib | [51] |
| 16 | OC | Quinacrine and carboplatin | Downregulates p62 SQSTM, enhances autophagic flux and restores sensitivity to carboplatin | [52] |
| 17 | OC | Quinacrine | Downregulates p62-Skp2 axis and simultaneously upregulates p21/p27 leading to autophagic cell death | [53] |
| 18 | OC | Quinacrine and TRAIL | Upregulates DR5 expression and prolongation of its half-life, sensitizes cells to TRAIL and enhances its apoptotic effect | [54] |
| 19 | OC | Quinacrine | Activates Cathespin L and downregulates p62, promotes lysosomal membrane premeability | [55] |
| 20 | RCC | Quinacrine | Activation of p53 signaling and apoptosis induction | [56] |
| 21 | HNSCC | Quinacrine and cisplatin | Restores deficient p53 expression, restores sensitivity to cisplatin and enhances its cytotoxic effect | [57] |
| 22 | HNSCC | Quinacrine and cisplatin | [58] | |
| 23 | Novikoff’s Hepatoma | Quinacrine | Inhibits DNA polymerase activity of malignant cells | [59] |
| 24 | GIST | SAHA and Quinacrine | Enhances | [60] |
| 25 | GIST | |||
| 26 | ALL | Vorinostat and Quinacrine | Multifold enhanced ROS generation and mitochondrial damage | [61] |
| Reported Molecular Target | QC Effect | Downstream Effect on Functionality/Pathway of Target | Reference |
|---|---|---|---|
| Gluthathione-S Transferase A1 | Inhibitor | Inhibits glutathione conjugation with electrophilic compounds and subsequent dissolution of oxidative stress caused by free radicals and alkylating drugs | [38] |
| P53 | Activation, stabilization, and upregulation | Activates the p53 signaling and stabilizes p53 leading to apoptosis | [56,64,65] |
| NF-κB | Downregulation | Inhibits NF-κB transcription activity and activation of downstream signaling, preventing initiation of cell survival mechanisms | [64,69,70] |
| Phospholipase A2 | Inhibitor | Interferes with release of arachidonic acid and prevents precursor synthesis for eicosanoids | [57,66,67] |
| FACT | Downregulation | Trapping FACT onto chromatin and preventing activation of downstream survival signaling molecules such as NF-κB | [39,71,72] |
| Topoisomerases I and II | Inhibitor | Impairs the helical binding activity of topoisomerases and interferes with DNA replication and repair | [40,73,74,75,76] |
| Telomerase | Inhibitor | Inhibits telomerase replication by preventing the formation of G-quadruplex structures in telomeres | [77,78,79] |
| DNA Polymerases α,δ, and ε | Inhibitor | Selectively inhibits the DNA Polymerase activity in malignant cells | [80,81,82] |
| RNA Polymerase II | Inhibitor | Interferes with ribosomal biogenesis and prevents transcription | [49,59] |
| P62/SQSTM | Downregulation | Downregulates the autophagosome cargo protein and tiggers accumulation of LC3B-II in autophagic vesicles | [42,51] |
| P21/p27 | Upregulation | Induces upregulation of p21/p27 and prevents its degradation by Skp-1 | [52] |
| CHK | Downregulation | Depleting the levels of phosphorylated Chk-1 and RPA and interefering with BER | [43,48,53] |
| Sl No. | Drug | Nature of Combination with QC | Mode of Study | Type of Cancer | Reference |
|---|---|---|---|---|---|
| 1 | Sorafenib | Synergistic | In Vitro and In Vivo models | Anaplastic thyroid cancer | [58] |
| 2 | Vorinostat (SAHA) | Synergistic | In Vitro and In Vivo models | Gastrointestinal cancer, T-cell acute lymphoblastic leukemia | [94,95] |
| 3 | 5- FluoroUracil | Synergistic | In Vitro and In Vivo models | Colorectal cancer | [60] |
| 4 | Paclitaxel | Synergistic | In Vitro and In Vivo models | Prostate cancer | [97,99] |
| 5 | Daunorobucin | Synergistic | Breast cancer | [98] | |
| 6 | Cisplatin | Synergistic | In Vitro | Head and neck squamous cell carcinoma, Endometrial cancer | [93,96,99] |
| 7 | Erlotinib | Synergistic | In Vitro and In Vivo models, and Phase I Clinical study | Non-small cell lung cancer | [72,105] |
| 8 | Capecitabine | Synergistic | Phase I Clinical stuy | Colorectal cancer | [106] |
| 9 | Cytarabine | Synergistic | In Vitro and In Vivo models | Acute Myeloid Leukemia (ALL) | [61,91] |
| 10 | Vincristine | Synergistic | In Vitro and In vivo models | Leukemia | [90] |
| 11 | Carboplatin | Synergistic | In Vitro and In vivo models | Ovarian cancer | [52] |
| 12 | Temozolomide | Synergistic | In Vitro | Glioblastoma Multiforme (GBM) | [104] |
| 13 | SI113 (SGK inhibitor) | Synergistic | In Vitro | Glioblastoma Multiforme (GBM) | [46] |
| 14 | Rucaparib | Synergistic | In Vitro | Ovarian cancer | [51] |
| 15 | Imatinib | Synergistic | In Vitro and In vivo models | Gastrointestinal Tumor (GIST) | [47] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, M.; Sarkar, A. Repurposing of Anti-Malarial Drug Quinacrine for Cancer Treatment: A Review. Sci. Pharm. 2022, 90, 12. https://doi.org/10.3390/scipharm90010012
Kumar M, Sarkar A. Repurposing of Anti-Malarial Drug Quinacrine for Cancer Treatment: A Review. Scientia Pharmaceutica. 2022; 90(1):12. https://doi.org/10.3390/scipharm90010012
Chicago/Turabian StyleKumar, Makhan, and Angshuman Sarkar. 2022. "Repurposing of Anti-Malarial Drug Quinacrine for Cancer Treatment: A Review" Scientia Pharmaceutica 90, no. 1: 12. https://doi.org/10.3390/scipharm90010012
APA StyleKumar, M., & Sarkar, A. (2022). Repurposing of Anti-Malarial Drug Quinacrine for Cancer Treatment: A Review. Scientia Pharmaceutica, 90(1), 12. https://doi.org/10.3390/scipharm90010012