
The Role of Viral Infections in the Onset of Autoimmune Diseases


Abstract
1. Introduction
2. Immune Tolerance
3. Inflammation-Induced AID
4. Viral Mechanisms Causing AID
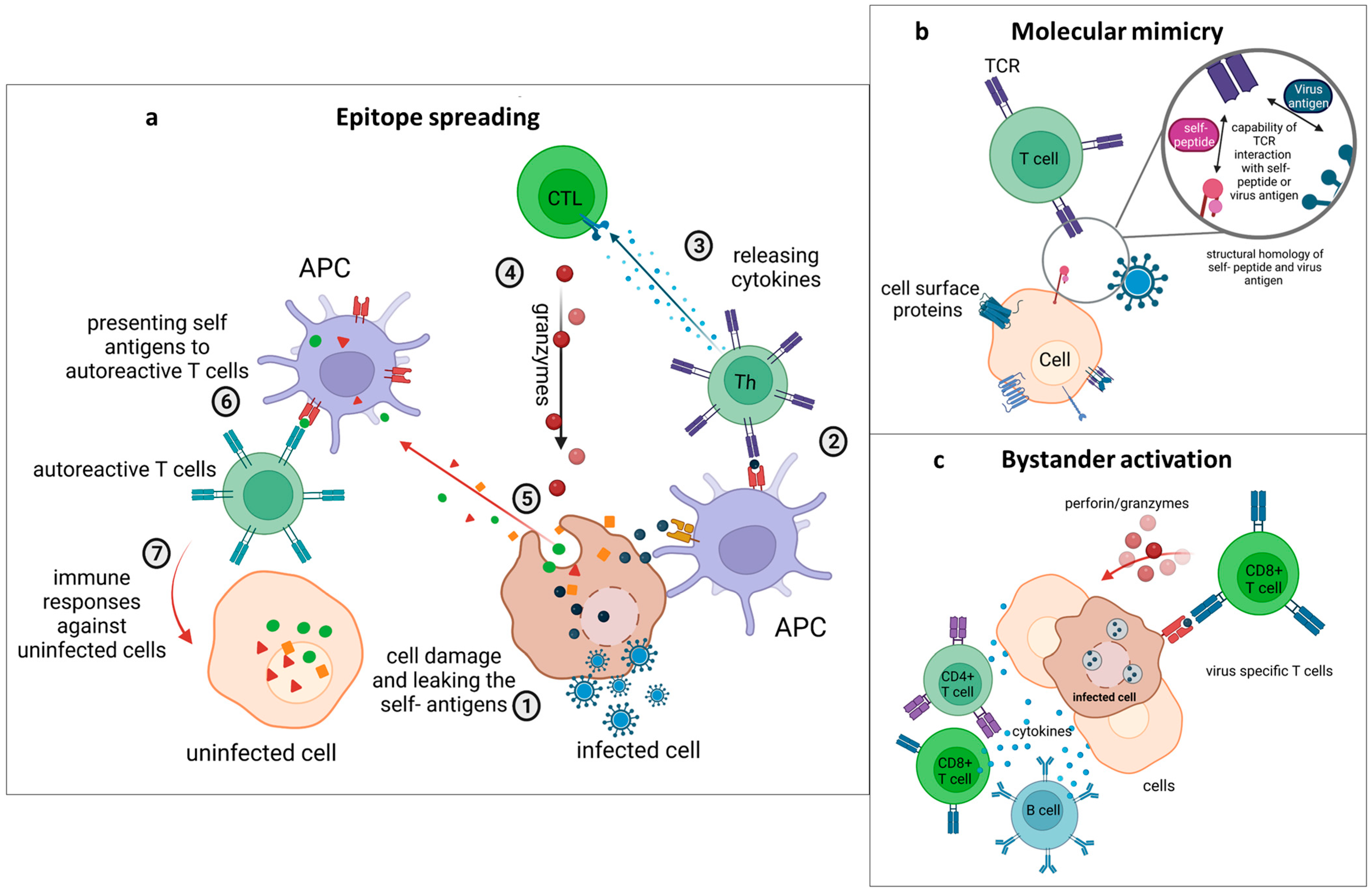
4.1. Epitope Spreading
4.2. Molecular Mimicry
4.3. Bystander Activation
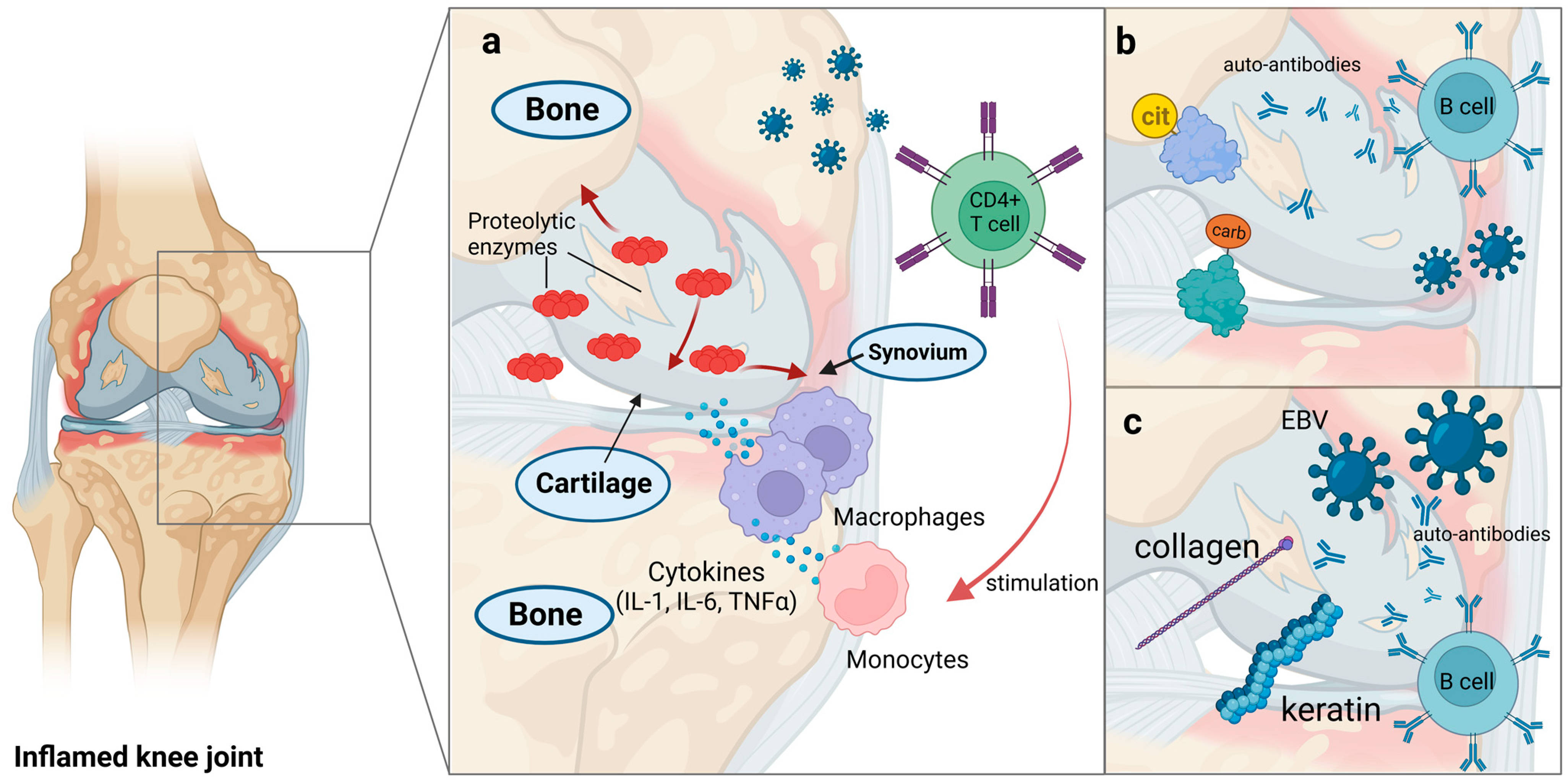
5. Rheumatoid Arthritis

{kind=link}
{kind=link}
{kind=link}
| Autoimmune Disease | Virus | Target Cells or Self-Peptides | Immune Cells or Cytokines | Pathomechanism | Ref. |
|---|---|---|---|---|---|
| Rheumatoid Arthritis | HTLV | Synovial cells | IL1, IL6, TNF-α | Autonomous proliferation of synovial cells and inflammation | [163,164,165] |
| EBV | Collagen and keratin | Autoreactive T cells | Molecular mimicry | [166,167,168] | |
| Multiple Sclerosis | Herpes simplex virus (HSV1, HSV2) | Peripheral sensory nerves, sensory ganglia in CNS | Cytotoxic T lymphocyte, IL-6 | Molecular mimicry of HSV-1 glycoprotein gB epitope and a brain-specific factor | [169,170] |
| Epstein–Barr Virus (EBV) | CNS, myelin basic protein (MBP), anoctamin 2, glial cell adhesion molecule (GlialCAM) | CD4+ and CD8+ T cells | Antibodies against EBV antigens viral capsid antigen (VCA), Epstein–Barr nuclear antigen 1 (EBNA1), and early antigen (EA), Epstein–Barr latent membrane protein 1 (LMP1), molecular mimicry | [127,171,172,173,174,175,176] | |
| Human Herpesvirus 6 (HHV-6) | Oligodendrocyte, MBP | T cells | Molecular mimicry of virus peptide U24 with MBP | [177,178,179] | |
| Varicella-Zoster Virus (VZV) | Ganglia in CNS, peripheral blood mononuclear cells (PBMCs) | CD4+ and CD8+ T cells | - | [180] | |
| Human Endogenous Retroviruses (HERVs) | CNS in white matter lesion, (PBMCs) | TNF-α | Induction of free radicals, ER stress | [169,170,181,182] | |
| Systemic Lupus Erythematosus | EBV | B- and epithelial cells; autoantibodies: SmB and Ro60 | EBV-specific T cells | Molecular mimicry | [183,184,185] |
| Diabetes Mellitus Type 1 | Coxsackie B4 | GAD65 in the beta-cells of pancreas | IFN-γ/type-1-IFN, IL-4 | Molecular mimicry | [186] |
| Rubella | GAD65/67 of the pancreatic islets | CD4+ and CD8+ T cells | Molecular mimicry | [187,188] | |
| Rotavirus | Tyrosine phosphatase IA-2 | Type I IFN | Molecular mimicry and Bystander activation | [189,190] | |
| Cytomegalovirus | GAD65 | GAD65 specific T-cells | Molecular mimicry | [191] |
6. Systemic Lupus Erythematosus
7. Multiple Sclerosis
8. Diabetes Mellitus Type 1
9. SARS-CoV-2 and AIDs
10. Outlook
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIDs | Autoimmune diseases |
| AIRE | Autoimmune regulator |
| APCs | Antigen presenting cells |
| BBB | Blood brain barrier |
| CMV | Cytomegalovirus |
| CNS | Central nervous system |
| cTECs | Cortical thymic epithelial cells |
| CTL | Cytotoxic T lymphocytes |
| DAMPs | Damage-associated molecular patterns |
| DCs | Dendritic cells |
| DMT1 | Diabetes mellitus type 1 |
| EBV | Epstein–Barr virus |
| ER | Endoplasmic reticulum |
| Fezf2 | Fez family zinc finger protein 2 |
| HERV | Human endogenous retrovirus |
| HHV | Human herpesvirus |
| HIV | Human immunodeficiency virus |
| HSV | Herpes simplex virus |
| IFN | Interferon |
| IL | Interleukin |
| MHC | Major histocompatibility complex |
| MS | Multiple sclerosis |
| mTECs | Medullary thymic epithelial cells |
| NF-kβ | Nuclear factor kappa-β |
| NK | Natural killer |
| PAMPs | Pathogen associated molecular patterns |
| RA | Rheumatoid arthritis |
| SLE | Systemic lupus erythematosus |
| T1D | Type 1 diabetes |
| TCR | T-cell receptor |
| TECs | Thymic epithelial cells |
| TF | Transcription factor |
| TNF | Tumor necrosis factor |
| TRAs | Tissue restricted antigens |
| Tregs | Regulatory T cells |
| VZV | Varicella-zoster virus |
References
- Sogkas, G.; Atschekzei, F.; Adriawan, I.R.; Dubrowinskaja, N.; Witte, T.; Schmidt, R.E. Cellular and molecular mechanisms breaking immune tolerance in inborn errors of immunity. Cell. Mol. Immunol. 2021, 18, 1122–1140. [Google Scholar] [CrossRef] [PubMed]
- Mercadante, E.R.; Lorenz, U.M. Breaking Free of Control: How Conventional T Cells Overcome Regulatory T Cell Suppression. Front. Immunol. 2016, 7, 193. [Google Scholar] [CrossRef]
- Cho, J.H.; Gregersen, P.K. Genomics and the multifactorial nature of human autoimmune disease. N. Engl. J. Med. 2011, 365, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Fugger, L.; Jensen, L.T.; Rossjohn, J. Challenges, Progress, and Prospects of Developing Therapies to Treat Autoimmune Diseases. Cell 2020, 181, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, J.; Björses, P.; Perheentupa, J.; Horelli–Kuitunen, N.; Palotie, A.; Peltonen, L.; Lee, Y.S.; Francis, F.; Henning, S.; Thiel, C.; et al. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat. Genet. 1997, 17, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.H.; Rosenberg, F.J.; Straus, S.E.; Dale, J.K.; Middleton, L.A.; Lin, A.Y.; Strober, W.; Lenardo, M.J.; Puck, J.M. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 1995, 81, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Gutierrez-Arcelus, M.; Rich, S.S.; Raychaudhuri, S. Autoimmune diseases—Connecting risk alleles with molecular traits of the immune system. Nat. Rev. Genet. 2016, 17, 160–174. [Google Scholar] [CrossRef]
- Inshaw, J.R.J.; Cutler, A.J.; Burren, O.S.; Stefana, M.I.; Todd, J.A. Approaches and advances in the genetic causes of autoimmune disease and their implications. Nat. Immunol. 2018, 19, 674–684. [Google Scholar] [CrossRef]
- Rich, S.S.; Weitkamp, L.R.; Barbosa, J. Genetic heterogeneity of insulin-dependent (type I) diabetes mellitus: Evidence from a study of extended haplotypes. Am. J. Hum. Genet. 1984, 36, 1015–1023. [Google Scholar]
- Gaffney, P.M.; Kearns, G.M.; Shark, K.B.; Ortmann, W.A.; Selby, S.A.; Malmgren, M.L.; Rohlf, K.E.; Ockenden, T.C.; Messner, R.P.; King, R.A.; et al. A genome-wide search for susceptibility genes in human systemic lupus erythematosus sib-pair families. Proc. Natl. Acad. Sci. USA 1998, 95, 14875–14879. [Google Scholar] [CrossRef]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cezard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; McLeod, R.S.; Griffiths, A.M.; Green, T.; Brettin, T.S.; Stone, V.; Bull, S.B.; et al. Genomewide search in Canadian families with inflammatory bowel disease reveals two novel susceptibility loci. Am. J. Hum. Genet. 2000, 66, 1863–1870. [Google Scholar] [CrossRef]
- Begovich, A.B.; Carlton, V.E.; Honigberg, L.A.; Schrodi, S.J.; Chokkalingam, A.P.; Alexander, H.C.; Ardlie, K.G.; Huang, Q.; Smith, A.M.; Spoerke, J.M.; et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 2004, 75, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Bottini, N.; Musumeci, L.; Alonso, A.; Rahmouni, S.; Nika, K.; Rostamkhani, M.; MacMurray, J.; Meloni, G.F.; Lucarelli, P.; Pellecchia, M.; et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat. Genet. 2004, 36, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Nistico, L.; Buzzetti, R.; Pritchard, L.E.; Van der Auwera, B.; Giovannini, C.; Bosi, E.; Larrad, M.T.; Rios, M.S.; Chow, C.C.; Cockram, C.S.; et al. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian Diabetes Registry. Hum. Mol. Genet. 1996, 5, 1075–1080. [Google Scholar] [CrossRef]
- Anaya, J.M.; Restrepo-Jimenez, P.; Ramirez-Santana, C. The autoimmune ecology: An update. Curr. Opin. Rheumatol. 2018, 30, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Anaya, J.M.; Ramirez-Santana, C.; Alzate, M.A.; Molano-Gonzalez, N.; Rojas-Villarraga, A. The Autoimmune Ecology. Front. Immunol. 2016, 7, 139. [Google Scholar] [CrossRef]
- Hussein, H.M.; Rahal, E.A. The role of viral infections in the development of autoimmune diseases. Crit. Rev. Microbiol. 2019, 45, 394–412. [Google Scholar] [CrossRef]
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; Al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and Autoimmunity: A Review on the Potential Interaction and Molecular Mechanisms. Viruses 2019, 11, 762. [Google Scholar] [CrossRef]
- Fujinami, R.S.; von Herrath, M.G.; Christen, U.; Whitton, J.L. Molecular mimicry, bystander activation, or viral persistence: Infections and autoimmune disease. Clin. Microbiol. Rev. 2006, 19, 80–94. [Google Scholar] [CrossRef]
- Getts, D.R.; Chastain, E.M.; Terry, R.L.; Miller, S.D. Virus infection, antiviral immunity, and autoimmunity. Immunol. Rev. 2013, 255, 197–209. [Google Scholar] [CrossRef]
- Schattner, A.; Rager-Zisman, B. Virus-induced autoimmunity. Rev. Infect. Dis. 1990, 12, 204–222. [Google Scholar] [CrossRef]
- Jiang, W.; Johnson, D.; Adekunle, R.; Heather, H.; Xu, W.; Cong, X.; Wu, X.; Fan, H.; Andersson, L.M.; Robertson, J.; et al. COVID-19 is associated with bystander polyclonal autoreactive B cell activation as reflected by a broad autoantibody production, but none is linked to disease severity. J. Med. Virol. 2023, 95, e28134. [Google Scholar] [CrossRef]
- Novelli, L.; Motta, F.; De Santis, M.; Ansari, A.A.; Gershwin, M.E.; Selmi, C. The JANUS of chronic inflammatory and autoimmune diseases onset during COVID-19—A systematic review of the literature. J. Autoimmun. 2021, 117, 102592. [Google Scholar] [CrossRef]
- Cheng, M.; Anderson, M.S. Thymic tolerance as a key brake on autoimmunity. Nat. Immunol. 2018, 19, 659–664. [Google Scholar] [CrossRef]
- Derbinski, J.; Schulte, A.; Kyewski, B.; Klein, L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat. Immunol. 2001, 2, 1032–1039. [Google Scholar] [CrossRef]
- Farr, A.G.; Dooley, J.L.; Erickson, M. Organization of thymic medullary epithelial heterogeneity: Implications for mechanisms of epithelial differentiation. Immunol. Rev. 2002, 189, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Rattay, K.; Meyer, H.V.; Herrmann, C.; Brors, B.; Kyewski, B. Evolutionary conserved gene co-expression drives generation of self-antigen diversity in medullary thymic epithelial cells. J. Autoimmun. 2016, 67, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Ucar, O.; Rattay, K. Promiscuous Gene Expression in the Thymus: A Matter of Epigenetics, miRNA, and More? Front. Immunol. 2015, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, P.; Reyes, A.; Pinto, S.; Rattay, K.; Nguyen, M.; Kuchler, R.; Huber, W.; Kyewski, B.; Steinmetz, L.M. Single-cell transcriptome analysis reveals coordinated ectopic gene-expression patterns in medullary thymic epithelial cells. Nat. Immunol. 2015, 16, 933–941. [Google Scholar] [CrossRef]
- Anderson, M.S.; Venanzi, E.S.; Klein, L.; Chen, Z.; Berzins, S.P.; Turley, S.J.; von Boehmer, H.; Bronson, R.; Dierich, A.; Benoist, C.; et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 2002, 298, 1395–1401. [Google Scholar] [CrossRef]
- Takaba, H.; Morishita, Y.; Tomofuji, Y.; Danks, L.; Nitta, T.; Komatsu, N.; Kodama, T.; Takayanagi, H. Fezf2 Orchestrates a Thymic Program of Self-Antigen Expression for Immune Tolerance. Cell 2015, 163, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Bautista, J.L.; Cramer, N.T.; Miller, C.N.; Chavez, J.; Berrios, D.I.; Byrnes, L.E.; Germino, J.; Ntranos, V.; Sneddon, J.B.; Burt, T.D.; et al. Single-cell transcriptional profiling of human thymic stroma uncovers novel cellular heterogeneity in the thymic medulla. Nat. Commun. 2021, 12, 1096. [Google Scholar] [CrossRef]
- Sansom, S.N.; Shikama, N.; Zhanybekova, S.; Nusspaumer, G.; Macaulay, I.C.; Deadman, M.E.; Heger, A.; Ponting, C.P.; Holländer, G.A. Population and single cell genomics reveal the Aire-dependency, relief from Polycomb silencing and distribution of self-antigen expression in thymic epithelia. Genome Res. 2014, 24, 1918–1931. [Google Scholar] [CrossRef]
- Bonasio, R.; Scimone, M.L.; Schaerli, P.; Grabie, N.; Lichtman, A.H.; von Andrian, U.H. Clonal deletion of thymocytes by circulating dendritic cells homing to the thymus. Nat. Immunol. 2006, 7, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Vollmann, E.H.; Rattay, K.; Barreiro, O.; Thiriot, A.; Fuhlbrigge, R.A.; Vrbanac, V.; Kim, K.W.; Jung, S.; Tager, A.M.; von Andrian, U.H. Specialized transendothelial dendritic cells mediate thymic T-cell selection against blood-borne macromolecules. Nat. Commun. 2021, 12, 6230. [Google Scholar] [CrossRef]
- Yamano, T.; Nedjic, J.; Hinterberger, M.; Steinert, M.; Koser, S.; Pinto, S.; Gerdes, N.; Lutgens, E.; Ishimaru, N.; Busslinger, M.; et al. Thymic B Cells Are Licensed to Present Self Antigens for Central T Cell Tolerance Induction. Immunity 2015, 42, 1048–1061. [Google Scholar] [CrossRef]
- Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 2017, 17, 281–294. [Google Scholar] [CrossRef]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Savage, P.A.; Klawon, D.E.J.; Miller, C.H. Regulatory T Cell Development. Annu. Rev. Immunol. 2020, 38, 421–453. [Google Scholar] [CrossRef]
- He, X.S.; Gershwin, M.E.; Ansari, A.A. Checkpoint-based immunotherapy for autoimmune diseases—Opportunities and challenges. J. Autoimmun. 2017, 79, 1–3. [Google Scholar] [CrossRef]
- Wykes, M.N.; Lewin, S.R. Immune checkpoint blockade in infectious diseases. Nat. Rev. Immunol. 2018, 18, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef]
- Abramson, J.; Anderson, G. Thymic Epithelial Cells. Annu. Rev. Immunol. 2017, 35, 85–118. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Kuroda, N.; Han, H.; Meguro-Horike, M.; Nishikawa, Y.; Kiyonari, H.; Maemura, K.; Yanagawa, Y.; Obata, K.; Takahashi, S.; et al. Aire controls the differentiation program of thymic epithelial cells in the medulla for the establishment of self-tolerance. J. Exp. Med. 2008, 205, 2827–2838. [Google Scholar] [CrossRef]
- Oven, I.; Brdickova, N.; Kohoutek, J.; Vaupotic, T.; Narat, M.; Peterlin, B.M. AIRE Recruits P-TEFb for Transcriptional Elongation of Target Genes in Medullary Thymic Epithelial Cells. Mol. Cell. Biol. 2007, 27, 8815–8823. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.S.; Venanzi, E.S.; Chen, Z.; Berzins, S.P.; Benoist, C.; Mathis, D. The Cellular Mechanism of Aire Control of T Cell Tolerance. Immunity 2005, 23, 227–239. [Google Scholar] [CrossRef]
- Chentoufi, A.A.; Polychronakos, C. Insulin expression levels in the thymus modulate insulin-specific autoreactive T-cell tolerance: The mechanism by which the IDDM2 locus may predispose to diabetes. Diabetes 2002, 51, 1383–1390. [Google Scholar] [CrossRef]
- Fan, Y.; Rudert, W.A.; Grupillo, M.; He, J.; Sisino, G.; Trucco, M. Thymus-specific deletion of insulin induces autoimmune diabetes. EMBO J. 2009, 28, 2812–2824. [Google Scholar] [CrossRef] [PubMed]
- DeVoss, J.; Hou, Y.; Johannes, K.; Lu, W.; Liou, G.I.; Rinn, J.; Chang, H.; Caspi, R.R.; Fong, L.; Anderson, M.S. Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J. Exp. Med. 2006, 203, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Morse, S.S.; Valinsky, J.E. Mouse thymic virus (MTLV). A mammalian herpesvirus cytolytic for CD4+ (L3T4+) T lymphocytes. J. Exp. Med. 1989, 169, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.J.; Zhao, G.; Penna, V.R.; Park, E.; Lauron, E.J.; Harvey, I.B.; Beatty, W.L.; Plougastel-Douglas, B.; Poursine-Laurent, J.; Fremont, D.H.; et al. A Murine Herpesvirus Closely Related to Ubiquitous Human Herpesviruses Causes T-Cell Depletion. J. Virol. 2017, 91, e02463-16. [Google Scholar] [CrossRef]
- Bigley, T.M.; Yang, L.; Kang, L.I.; Saenz, J.B.; Victorino, F.; Yokoyama, W.M. Disruption of thymic central tolerance by infection with murine roseolovirus induces autoimmune gastritis. J. Exp. Med. 2022, 219, e20211403. [Google Scholar] [CrossRef]
- Arpaia, N.; Green, J.A.; Moltedo, B.; Arvey, A.; Hemmers, S.; Yuan, S.; Treuting, P.M.; Rudensky, A.Y. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 2015, 162, 1078–1089. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Kanamori, M.; Nakatsukasa, H.; Okada, M.; Lu, Q.; Yoshimura, A. Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends Immunol. 2016, 37, 803–811. [Google Scholar] [CrossRef]
- Dominguez-Villar, M.; Hafler, D.A. Regulatory T cells in autoimmune disease. Nat. Immunol. 2018, 19, 665–673. [Google Scholar] [CrossRef]
- Huang, C.; Zhu, H.X.; Yao, Y.; Bian, Z.H.; Zheng, Y.J.; Li, L.; Moutsopoulos, H.M.; Gershwin, M.E.; Lian, Z.X. Immune checkpoint molecules. Possible future therapeutic implications in autoimmune diseases. J. Autoimmun. 2019, 104, 102333. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Huffaker, M.F.; Sanda, S.; Chandran, S.; Chung, S.A.; St Clair, E.W.; Nepom, G.T.; Smilek, D.E. Approaches to Establishing Tolerance in Immune Mediated Diseases. Front. Immunol. 2021, 12, 744804. [Google Scholar] [CrossRef]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- De Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Strowig, T.; Flavell, R.A. Inflammasomes: Far beyond inflammation. Nat. Immunol. 2012, 13, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Kontaki, E.; Boumpas, D.T. Innate immunity in systemic lupus erythematosus: Sensing endogenous nucleic acids. J. Autoimmun. 2010, 35, 206–211. [Google Scholar] [CrossRef]
- Theofilopoulos, A.N.; Gonzalez-Quintial, R.; Lawson, B.R.; Koh, Y.T.; Stern, M.E.; Kono, D.H.; Beutler, B.; Baccala, R. Sensors of the innate immune system: Their link to rheumatic diseases. Nat. Rev. Rheumatol. 2010, 6, 146–156. [Google Scholar] [CrossRef]
- Allam, R.; Pawar, R.D.; Kulkarni, O.P.; Hornung, V.; Hartmann, G.; Segerer, S.; Akira, S.; Endres, S.; Anders, H.J. Viral 5′-triphosphate RNA and non-CpG DNA aggravate autoimmunity and lupus nephritis via distinct TLR-independent immune responses. Eur. J. Immunol. 2008, 38, 3487–3498. [Google Scholar] [CrossRef]
- Wahadat, M.J.; Bodewes, I.L.A.; Maria, N.I.; van Helden-Meeuwsen, C.G.; van Dijk-Hummelman, A.; Steenwijk, E.C.; Kamphuis, S.; Versnel, M.A. Type I IFN signature in childhood-onset systemic lupus erythematosus: A conspiracy of DNA- and RNA-sensing receptors? Arthritis Res. Ther. 2018, 20, 4. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.I.; Lee, K.H.; Joo, Y.H.; Lee, J.M.; Jeon, J.; Jung, H.J.; Shin, M.; Cho, S.; Kim, T.H.; Park, S.; et al. Inflammasomes and autoimmune and rheumatic diseases: A comprehensive review. J. Autoimmun. 2019, 103, 102299. [Google Scholar] [CrossRef] [PubMed]
- Negash, A.A.; Olson, R.M.; Griffin, S.; Gale, M., Jr. Modulation of calcium signaling pathway by hepatitis C virus core protein stimulates NLRP3 inflammasome activation. PLoS Pathog. 2019, 15, e1007593. [Google Scholar] [CrossRef]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef]
- Zhu, S.; Ding, S.; Wang, P.; Wei, Z.; Pan, W.; Palm, N.W.; Yang, Y.; Yu, H.; Li, H.B.; Wang, G.; et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 2017, 546, 667–670. [Google Scholar] [CrossRef]
- Prochnicki, T.; Latz, E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017, 26, 71–93. [Google Scholar] [CrossRef]
- Dang, E.V.; Cyster, J.G. Loss of sterol metabolic homeostasis triggers inflammasomes—How and why. Curr. Opin. Immunol. 2019, 56, 1–9. [Google Scholar] [CrossRef]
- Von Herrath, M.G.; Fujinami, R.S.; Whitton, J.L. Microorganisms and autoimmunity: Making the barren field fertile? Nat. Rev. Microbiol. 2003, 1, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Srinivasappa, J.; Saegusa, J.; Prabhakar, B.S.; Gentry, M.K.; Buchmeier, M.J.; Wiktor, T.J.; Koprowski, H.; Oldstone, M.B.; Notkins, A.L. Molecular mimicry: Frequency of reactivity of monoclonal antiviral antibodies with normal tissues. J. Virol. 1986, 57, 397–401. [Google Scholar] [CrossRef]
- Gebe, J.A.; Falk, B.A.; Rock, K.A.; Kochik, S.A.; Heninger, A.K.; Reijonen, H.; Kwok, W.W.; Nepom, G.T. Low-avidity recognition by CD4+ T cells directed to self-antigens. Eur. J. Immunol. 2003, 33, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Munz, C.; Lunemann, J.D.; Getts, M.T.; Miller, S.D. Antiviral immune responses: Triggers of or triggered by autoimmunity? Nat. Rev. Immunol. 2009, 9, 246–258. [Google Scholar] [CrossRef]
- Mouat, I.C.; Morse, Z.J.; Shanina, I.; Brown, K.L.; Horwitz, M.S. Latent gammaherpesvirus exacerbates arthritis through modification of age-associated B cells. eLife 2021, 10, e67024. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; O’Neill, P.; Naradikian, M.S.; Scholz, J.L.; Cancro, M.P. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 2011, 118, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- Rubtsov, A.V.; Rubtsova, K.; Fischer, A.; Meehan, R.T.; Gillis, J.Z.; Kappler, J.W.; Marrack, P. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood 2011, 118, 1305–1315. [Google Scholar] [CrossRef]
- Ma, S.; Wang, C.; Mao, X.; Hao, Y. B Cell Dysfunction Associated With Aging and Autoimmune Diseases. Front. Immunol. 2019, 10, 318. [Google Scholar] [CrossRef]
- Mouat, I.C.; Goldberg, E.; Horwitz, M.S. Age-associated B cells in autoimmune diseases. Cell. Mol. Life Sci. 2022, 79, 402. [Google Scholar] [CrossRef] [PubMed]
- Cancro, M.P. Age-Associated B Cells. Annu. Rev. Immunol. 2020, 38, 315–340. [Google Scholar] [CrossRef]
- Austin, J.W.; Buckner, C.M.; Kardava, L.; Wang, W.; Zhang, X.; Melson, V.A.; Swanson, R.G.; Martins, A.J.; Zhou, J.Q.; Hoehn, K.B.; et al. Overexpression of T-bet in HIV infection is associated with accumulation of B cells outside germinal centers and poor affinity maturation. Sci. Transl. Med. 2019, 11, eaax0904. [Google Scholar] [CrossRef]
- Woodruff, M.C.; Ramonell, R.P.; Nguyen, D.C.; Cashman, K.S.; Saini, A.S.; Haddad, N.S.; Ley, A.M.; Kyu, S.; Howell, J.C.; Ozturk, T.; et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat. Immunol. 2020, 21, 1506–1516. [Google Scholar] [CrossRef]
- Jenks, S.A.; Cashman, K.S.; Woodruff, M.C.; Lee, F.E.; Sanz, I. Extrafollicular responses in humans and SLE. Immunol. Rev. 2019, 288, 136–148. [Google Scholar] [CrossRef]
- Ricker, E.; Manni, M.; Flores-Castro, D.; Jenkins, D.; Gupta, S.; Rivera-Correa, J.; Meng, W.; Rosenfeld, A.M.; Pannellini, T.; Bachu, M.; et al. Altered function and differentiation of age-associated B cells contribute to the female bias in lupus mice. Nat. Commun. 2021, 12, 4813. [Google Scholar] [CrossRef]
- Scully, E.P.; Haverfield, J.; Ursin, R.L.; Tannenbaum, C.; Klein, S.L. Considering how biological sex impacts immune responses and COVID-19 outcomes. Nat. Rev. Immunol. 2020, 20, 442–447. [Google Scholar] [CrossRef]
- Liu, Z.; Davidson, A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat. Med. 2012, 18, 871–882. [Google Scholar] [CrossRef]
- Lehmann, P.V.; Forsthuber, T.; Miller, A.; Sercarz, E.E. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature 1992, 358, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Vanderlugt, C.J.; Miller, S.D. Epitope spreading. Curr. Opin. Immunol. 1996, 8, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.M.; Black, M.M. Epitope spreading: Protection from pathogens, but propagation of autoimmunity? Clin. Exp. Dermatol. 2001, 26, 427–433. [Google Scholar] [CrossRef]
- Floreani, A.; Leung, P.S.; Gershwin, M.E. Environmental Basis of Autoimmunity. Clin. Rev. Allergy Immunol. 2016, 50, 287–300. [Google Scholar] [CrossRef]
- Cornaby, C.; Gibbons, L.; Mayhew, V.; Sloan, C.S.; Welling, A.; Poole, B.D. B cell epitope spreading: Mechanisms and contribution to autoimmune diseases. Immunol. Lett. 2015, 163, 56–68. [Google Scholar] [CrossRef] [PubMed]
- McRae, B.L.; Vanderlugt, C.L.; Dal Canto, M.C.; Miller, S.D. Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J. Exp. Med. 1995, 182, 75–85. [Google Scholar] [CrossRef]
- Tuohy, V.K.; Yu, M.; Weinstock-Guttman, B.; Kinkel, R.P. Diversity and plasticity of self recognition during the development of multiple sclerosis. J. Clin. Investig. 1997, 99, 1682–1690. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Johnson, J.M.; Tuohy, V.K. A predictable sequential determinant spreading cascade invariably accompanies progression of experimental autoimmune encephalomyelitis: A basis for peptide-specific therapy after onset of clinical disease. J. Exp. Med. 1996, 183, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Vanderlugt, C.L.; Miller, S.D. Epitope spreading in immune-mediated diseases: Implications for immunotherapy. Nat. Rev. Immunol. 2002, 2, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Kohm, A.P.; McMahon, J.S.; Luo, X.; Miller, S.D. Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9-23 epitope and involves functional epitope spreading. J. Autoimmun. 2012, 39, 347–353. [Google Scholar] [CrossRef]
- Miller, S.D.; Vanderlugt, C.L.; Begolka, W.S.; Pao, W.; Yauch, R.L.; Neville, K.L.; Katz-Levy, Y.; Carrizosa, A.; Kim, B.S. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 1997, 3, 1133–1136. [Google Scholar] [CrossRef]
- Miller, S.D.; Katz-Levy, Y.; Neville, K.L.; Vanderlugt, C.L. Virus-induced autoimmunity: Epitope spreading to myelin autoepitopes in Theiler’s virus infection of the central nervous system. Adv. Virus Res. 2001, 56, 199–217. [Google Scholar] [CrossRef]
- Wucherpfennig, K.W. Mechanisms for the induction of autoimmunity by infectious agents. J. Clin. Investig. 2001, 108, 1097–1104. [Google Scholar] [CrossRef]
- Tsunoda, I.; Fujinami, R.S. Two models for multiple sclerosis: Experimental allergic encephalomyelitis and Theiler’s murine encephalomyelitis virus. J. Neuropathol. Exp. Neurol. 1996, 55, 673–686. [Google Scholar] [CrossRef]
- Katz-Levy, Y.; Neville, K.L.; Girvin, A.M.; Vanderlugt, C.L.; Pope, J.G.; Tan, L.J.; Miller, S.D. Endogenous presentation of self myelin epitopes by CNS-resident APCs in Theiler’s virus-infected mice. J. Clin. Investig. 1999, 104, 599–610. [Google Scholar] [CrossRef]
- Katz-Levy, Y.; Neville, K.L.; Padilla, J.; Rahbe, S.; Begolka, W.S.; Girvin, A.M.; Olson, J.K.; Vanderlugt, C.L.; Miller, S.D. Temporal development of autoreactive Th1 responses and endogenous presentation of self myelin epitopes by central nervous system-resident APCs in Theiler’s virus-infected mice. J. Immunol. 2000, 165, 5304–5314. [Google Scholar] [CrossRef] [PubMed]
- Croxford, J.L.; Olson, J.K.; Miller, S.D. Epitope spreading and molecular mimicry as triggers of autoimmunity in the Theiler’s virus-induced demyelinating disease model of multiple sclerosis. Autoimmun. Rev. 2002, 1, 251–260. [Google Scholar] [CrossRef]
- Degn, S.E.; van der Poel, C.E.; Firl, D.J.; Ayoglu, B.; Al Qureshah, F.A.; Bajic, G.; Mesin, L.; Reynaud, C.A.; Weill, J.C.; Utz, P.J.; et al. Clonal Evolution of Autoreactive Germinal Centers. Cell 2017, 170, 913–926.e19. [Google Scholar] [CrossRef]
- Brossart, P. The Role of Antigen Spreading in the Efficacy of Immunotherapies. Clin. Cancer Res. 2020, 26, 4442–4447. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.M.; Colbert, J.D.; Merino, E.; Kriegsman, B.A.; Rock, K.L. The Biology and Underlying Mechanisms of Cross-Presentation of Exogenous Antigens on MHC-I Molecules. Annu. Rev. Immunol. 2017, 35, 149–176. [Google Scholar] [CrossRef] [PubMed]
- Weck, M.M.; Appel, S.; Werth, D.; Sinzger, C.; Bringmann, A.; Grunebach, F.; Brossart, P. hDectin-1 is involved in uptake and cross-presentation of cellular antigens. Blood 2008, 111, 4264–4272. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Lo, J.A.; Kawakubo, M.; Juneja, V.R.; Su, M.Y.; Erlich, T.H.; LaFleur, M.W.; Kemeny, L.V.; Rashid, M.; Malehmir, M.; Rabi, S.A.; et al. Epitope spreading toward wild-type melanocyte-lineage antigens rescues suboptimal immune checkpoint blockade responses. Sci. Transl. Med. 2021, 13, eabd8636. [Google Scholar] [CrossRef]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Molecular mimicry as a mechanism of autoimmune disease. Clin. Rev. Allergy Immunol. 2012, 42, 102–111. [Google Scholar] [CrossRef]
- Rojas, M.; Restrepo-Jimenez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramirez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123. [Google Scholar] [CrossRef]
- Fujinami, R.S.; Oldstone, M.B.; Wroblewska, Z.; Frankel, M.E.; Koprowski, H. Molecular mimicry in virus infection: Crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc. Natl. Acad. Sci. USA 1983, 80, 2346–2350. [Google Scholar] [CrossRef]
- Fujinami, R.S.; Oldstone, M.B. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: Mechanism for autoimmunity. Science 1985, 230, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Barnett, L.A.; Whitton, J.L.; Wada, Y.; Fujinami, R.S. Enhancement of autoimmune disease using recombinant vaccinia virus encoding myelin proteolipid protein. J. Neuroimmunol. 1993, 44, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.S.; Granucci, F.; Yeh, L.; Schaffer, P.A.; Cantor, H. Molecular mimicry by herpes simplex virus-type 1: Autoimmune disease after viral infection. Science 1998, 279, 1344–1347. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.T.; Wiberg, A.; von Herrath, M.G. Viral infections and molecular mimicry in type 1 diabetes. APMIS 2012, 120, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Gauntt, C.J.; Arizpe, H.M.; Higdon, A.L.; Wood, H.J.; Bowers, D.F.; Rozek, M.M.; Crawley, R. Molecular mimicry, anti-coxsackievirus B3 neutralizing monoclonal antibodies, and myocarditis. J. Immunol. 1995, 154, 2983–2995. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.T.; Heinlen, L.D.; Dennis, G.J.; Roebuck, J.; Harley, J.B.; James, J.A. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat. Med. 2005, 11, 85–89. [Google Scholar] [CrossRef]
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.S.; Bartley, C.M.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef]
- Marino Gammazza, A.; Legare, S.; Lo Bosco, G.; Fucarino, A.; Angileri, F.; Conway de Macario, E.; Macario, A.J.; Cappello, F. Human molecular chaperones share with SARS-CoV-2 antigenic epitopes potentially capable of eliciting autoimmunity against endothelial cells: Possible role of molecular mimicry in COVID-19. Cell Stress Chaperones 2020, 25, 737–741. [Google Scholar] [CrossRef]
- Angileri, F.; Legare, S.; Marino Gammazza, A.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F. Is molecular mimicry the culprit in the autoimmune haemolytic anaemia affecting patients with COVID-19? Br. J. Haematol. 2020, 190, e92–e93. [Google Scholar] [CrossRef]
- Lucchese, G.; Floel, A. Molecular mimicry between SARS-CoV-2 and respiratory pacemaker neurons. Autoimmun. Rev. 2020, 19, 102556. [Google Scholar] [CrossRef]
- Sedaghat, Z.; Karimi, N. Guillain Barre syndrome associated with COVID-19 infection: A case report. J. Clin. Neurosci. 2020, 76, 233–235. [Google Scholar] [CrossRef]
- Verdoni, L.; Mazza, A.; Gervasoni, A.; Martelli, L.; Ruggeri, M.; Ciuffreda, M.; Bonanomi, E.; D’Antiga, L. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: An observational cohort study. Lancet 2020, 395, 1771–1778. [Google Scholar] [CrossRef]
- Galeotti, C.; Bayry, J. Autoimmune and inflammatory diseases following COVID-19. Nat. Rev. Rheumatol. 2020, 16, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Saif, D.S.; Ibrahem, R.A.; Eltabl, M.A. Prevalence of peripheral neuropathy and myopathy in patients post-COVID-19 infection. Int. J. Rheum. Dis. 2022, 25, 1246–1253. [Google Scholar] [CrossRef]
- Toscano, G.; Palmerini, F.; Ravaglia, S.; Ruiz, L.; Invernizzi, P.; Cuzzoni, M.G.; Franciotta, D.; Baldanti, F.; Daturi, R.; Postorino, P.; et al. Guillain-Barre Syndrome Associated with SARS-CoV-2. N. Engl. J. Med. 2020, 382, 2574–2576. [Google Scholar] [CrossRef] [PubMed]
- Bonometti, R.; Sacchi, M.C.; Stobbione, P.; Lauritano, E.C.; Tamiazzo, S.; Marchegiani, A.; Novara, E.; Molinaro, E.; Benedetti, I.; Massone, L.; et al. The first case of systemic lupus erythematosus (SLE) triggered by COVID-19 infection. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9695–9697. [Google Scholar] [PubMed]
- Hali, F.; Jabri, H.; Chiheb, S.; Hafiani, Y.; Nsiri, A. A concomitant diagnosis of COVID-19 infection and systemic lupus erythematosus complicated by a macrophage activation syndrome: A new case report. Int. J. Dermatol. 2021, 60, 1030–1031. [Google Scholar] [CrossRef]
- Chang, R.; Yen-Ting Chen, T.; Wang, S.I.; Hung, Y.M.; Chen, H.Y.; Wei, C.J. Risk of autoimmune diseases in patients with COVID-19: A retrospective cohort study. EClinicalMedicine 2023, 56, 101783. [Google Scholar] [CrossRef] [PubMed]
- Coscoy, L. Immune evasion by Kaposi’s sarcoma-associated herpesvirus. Nat. Rev. Immunol. 2007, 7, 391–401. [Google Scholar] [CrossRef]
- Miller-Kittrell, M.; Sparer, T.E. Feeling manipulated: Cytomegalovirus immune manipulation. Virol. J. 2009, 6, 4. [Google Scholar] [CrossRef]
- Pacheco, Y.; Acosta-Ampudia, Y.; Monsalve, D.M.; Chang, C.; Gershwin, M.E.; Anaya, J.M. Bystander activation and autoimmunity. J. Autoimmun. 2019, 103, 102301. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Decembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 2012, 12, 558–570. [Google Scholar] [CrossRef]
- Wieland, S.F.; Takahashi, K.; Boyd, B.; Whitten-Bauer, C.; Ngo, N.; de la Torre, J.C.; Chisari, F.V. Human plasmacytoid dendritic cells sense lymphocytic choriomeningitis virus-infected cells in vitro. J. Virol. 2014, 88, 752–757. [Google Scholar] [CrossRef]
- Duke, R.C. Self recognition by T cells. I. Bystander killing of target cells bearing syngeneic MHC antigens. J. Exp. Med. 1989, 170, 59–71. [Google Scholar] [CrossRef]
- Smyth, M.J.; Sedgwick, J.D. Delayed kinetics of tumor necrosis factor-mediated bystander lysis by peptide-specific CD8+ cytotoxic T lymphocytes. Eur. J. Immunol. 1998, 28, 4162–4169. [Google Scholar] [CrossRef]
- Kim, T.S.; Shin, E.C. The activation of bystander CD8+ T cells and their roles in viral infection. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef]
- Lee, H.G.; Cho, M.Z.; Choi, J.M. Bystander CD4+ T cells: Crossroads between innate and adaptive immunity. Exp. Mol. Med. 2020, 52, 1255–1263. [Google Scholar] [CrossRef]
- Ramanathan, S.; Dubois, S.; Chen, X.L.; Leblanc, C.; Ohashi, P.S.; Ilangumaran, S. Exposure to IL-15 and IL-21 enables autoreactive CD8 T cells to respond to weak antigens and cause disease in a mouse model of autoimmune diabetes. J. Immunol. 2011, 186, 5131–5141. [Google Scholar] [CrossRef]
- Calzascia, T.; Pellegrini, M.; Lin, A.; Garza, K.M.; Elford, A.R.; Shahinian, A.; Ohashi, P.S.; Mak, T.W. CD4 T cells, lymphopenia, and IL-7 in a multistep pathway to autoimmunity. Proc. Natl. Acad. Sci. USA 2008, 105, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Tajima, M.; Wakita, D.; Noguchi, D.; Chamoto, K.; Yue, Z.; Fugo, K.; Ishigame, H.; Iwakura, Y.; Kitamura, H.; Nishimura, T. IL-6-dependent spontaneous proliferation is required for the induction of colitogenic IL-17-producing CD8+ T cells. J. Exp. Med. 2008, 205, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- van Aalst, S.; Ludwig, I.S.; van der Zee, R.; van Eden, W.; Broere, F. Bystander activation of irrelevant CD4+ T cells following antigen-specific vaccination occurs in the presence and absence of adjuvant. PLoS ONE 2017, 12, e0177365. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Lee, J.U.; Kim, D.H.; Lim, S.; Kang, I.; Choi, J.M. Pathogenic function of bystander-activated memory-like CD4+ T cells in autoimmune encephalomyelitis. Nat. Commun. 2019, 10, 709. [Google Scholar] [CrossRef] [PubMed]
- Meresse, B.; Chen, Z.; Ciszewski, C.; Tretiakova, M.; Bhagat, G.; Krausz, T.N.; Raulet, D.H.; Lanier, L.L.; Groh, V.; Spies, T.; et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 2004, 21, 357–366. [Google Scholar] [CrossRef]
- Finckh, A.; Gilbert, B.; Hodkinson, B.; Bae, S.C.; Thomas, R.; Deane, K.D.; Alpizar-Rodriguez, D.; Lauper, K. Global epidemiology of rheumatoid arthritis. Nat. Rev. Rheumatol. 2022, 18, 591–602. [Google Scholar] [CrossRef]
- Deane, K.D.; Demoruelle, M.K.; Kelmenson, L.B.; Kuhn, K.A.; Norris, J.M.; Holers, V.M. Genetic and environmental risk factors for rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 3–18. [Google Scholar] [CrossRef]
- Merlino, L.A.; Curtis, J.; Mikuls, T.R.; Cerhan, J.R.; Criswell, L.A.; Saag, K.G.; Iowa Women’s Health, S. Vitamin D intake is inversely associated with rheumatoid arthritis: Results from the Iowa Women’s Health Study. Arthritis Rheum. 2004, 50, 72–77. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef]
- Choy, E. Understanding the dynamics: Pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology 2012, 51 (Suppl. S5), v3–v11. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, S.; Knedla, A.; Tennie, C.; Kampmann, A.; Wunrau, C.; Dinser, R.; Korb, A.; Schnaker, E.M.; Tarner, I.H.; Robbins, P.D.; et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat. Med. 2009, 15, 1414–1420. [Google Scholar] [CrossRef]
- Kurowska-Stolarska, M.; Alivernini, S. Synovial tissue macrophages in joint homeostasis, rheumatoid arthritis and disease remission. Nat. Rev. Rheumatol. 2022, 18, 384–397. [Google Scholar] [CrossRef]
- Roberts, C.A.; Dickinson, A.K.; Taams, L.S. The Interplay Between Monocytes/Macrophages and CD4+ T Cell Subsets in Rheumatoid Arthritis. Front. Immunol. 2015, 6, 571. [Google Scholar] [CrossRef] [PubMed]
- Umekita, K.; Okayama, A. HTLV-1 Infection and Rheumatic Diseases. Front. Microbiol. 2020, 11, 152. [Google Scholar] [CrossRef] [PubMed]
- Schierhout, G.; McGregor, S.; Gessain, A.; Einsiedel, L.; Martinello, M.; Kaldor, J. Association between HTLV-1 infection and adverse health outcomes: A systematic review and meta-analysis of epidemiological studies. Lancet Infect. Dis. 2020, 20, 133–143. [Google Scholar] [CrossRef]
- Masuko-Hongo, K.; Kato, T.; Nishioka, K. Virus-associated arthritis. Best Pract. Res. Clin. Rheumatol. 2003, 17, 309–318. [Google Scholar] [CrossRef]
- Kuwana, Y.; Takei, M.; Yajima, M.; Imadome, K.; Inomata, H.; Shiozaki, M.; Ikumi, N.; Nozaki, T.; Shiraiwa, H.; Kitamura, N.; et al. Epstein-Barr virus induces erosive arthritis in humanized mice. PLoS ONE 2011, 6, e26630. [Google Scholar] [CrossRef]
- Bo, M.; Niegowska, M.; Erre, G.L.; Piras, M.; Longu, M.G.; Manchia, P.; Manca, M.; Passiu, G.; Sechi, L.A. Rheumatoid arthritis patient antibodies highly recognize IL-2 in the immune response pathway involving IRF5 and EBV antigens. Sci. Rep. 2018, 8, 1789. [Google Scholar] [CrossRef]
- Birkenfeld, P.; Haratz, N.; Klein, G.; Sulitzeanu, D. Cross-reactivity between the EBNA-1 p107 peptide, collagen, and keratin: Implications for the pathogenesis of rheumatoid arthritis. Clin. Immunol. Immunopathol. 1990, 54, 14–25. [Google Scholar] [CrossRef]
- Kakalacheva, K.; Munz, C.; Lunemann, J.D. Viral triggers of multiple sclerosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, J.O.; Jacobson, S. Viruses and multiple sclerosis. CNS Neurol. Disord. Drug Targets 2012, 11, 528–544. [Google Scholar] [CrossRef]
- Bjornevik, K.; Munz, C.; Cohen, J.I.; Ascherio, A. Epstein-Barr virus as a leading cause of multiple sclerosis: Mechanisms and implications. Nat. Rev. Neurol. 2023, 19, 160–171. [Google Scholar] [CrossRef]
- Tengvall, K.; Huang, J.; Hellstrom, C.; Kammer, P.; Bistrom, M.; Ayoglu, B.; Lima Bomfim, I.; Stridh, P.; Butt, J.; Brenner, N.; et al. Molecular mimicry between Anoctamin 2 and Epstein-Barr virus nuclear antigen 1 associates with multiple sclerosis risk. Proc. Natl. Acad. Sci. USA 2019, 116, 16955–16960. [Google Scholar] [CrossRef] [PubMed]
- Jog, N.R.; James, J.A. Epstein Barr Virus and Autoimmune Responses in Systemic Lupus Erythematosus. Front. Immunol. 2020, 11, 623944. [Google Scholar] [CrossRef] [PubMed]
- Van Sechel, A.C.; Bajramovic, J.J.; van Stipdonk, M.J.; Persoon-Deen, C.; Geutskens, S.B.; van Noort, J.M. EBV-induced expression and HLA-DR-restricted presentation by human B cells of alpha B-crystallin, a candidate autoantigen in multiple sclerosis. J. Immunol. 1999, 162, 129–135. [Google Scholar] [CrossRef] [PubMed]
- van Nierop, G.P.; Mautner, J.; Mitterreiter, J.G.; Hintzen, R.Q.; Verjans, G.M. Intrathecal CD8 T-cells of multiple sclerosis patients recognize lytic Epstein-Barr virus proteins. Mult. Scler. 2016, 22, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Gabibov, A.G.; Belogurov, A.A., Jr.; Lomakin, Y.A.; Zakharova, M.Y.; Avakyan, M.E.; Dubrovskaya, V.V.; Smirnov, I.V.; Ivanov, A.S.; Molnar, A.A.; Gurtsevitch, V.E.; et al. Combinatorial antibody library from multiple sclerosis patients reveals antibodies that cross-react with myelin basic protein and EBV antigen. FASEB J. 2011, 25, 4211–4221. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M.; Cuomo, L.; Zompetta, C.; Ruggieri, S.; Frati, L.; Faggioni, A.; Ragona, G. Human herpesvirus 6 and multiple sclerosis: A study of T cell cross-reactivity to viral and myelin basic protein antigens. J. Med. Virol. 2002, 68, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Tejada-Simon, M.V.; Zang, Y.C.; Hong, J.; Rivera, V.M.; Zhang, J.Z. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann. Neurol. 2003, 53, 189–197. [Google Scholar] [CrossRef]
- Cheng, W.; Ma, Y.; Gong, F.; Hu, C.; Qian, L.; Huang, Q.; Yu, Q.; Zhang, J.; Chen, S.; Liu, Z.; et al. Cross-reactivity of autoreactive T cells with MBP and viral antigens in patients with MS. Front. Biosci. (Landmark Ed.) 2012, 17, 1648–1658. [Google Scholar] [CrossRef]
- Kang, J.H.; Sheu, J.J.; Kao, S.; Lin, H.C. Increased risk of multiple sclerosis following herpes zoster: A nationwide, population-based study. J. Infect. Dis. 2011, 204, 188–192. [Google Scholar] [CrossRef]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Multiple sclerosis: Autoimmunity and viruses. Curr. Opin. Rheumatol. 2013, 25, 496–501. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M.; Murdjeva, M.; Puri, B.K. Do Human Endogenous Retroviruses Contribute to Multiple Sclerosis, and if So, How? Mol. Neurobiol. 2019, 56, 2590–2605. [Google Scholar] [CrossRef]
- James, J.A.; Gross, T.; Scofield, R.H.; Harley, J.B. Immunoglobulin epitope spreading and autoimmune disease after peptide immunization: Sm B/B’-derived PPPGMRPP and PPPGIRGP induce spliceosome autoimmunity. J. Exp. Med. 1995, 181, 453–461. [Google Scholar] [CrossRef]
- Yadav, P.; Tran, H.; Ebegbe, R.; Gottlieb, P.; Wei, H.; Lewis, R.H.; Mumbey-Wafula, A.; Kaplan, A.; Kholdarova, E.; Spatz, L. Antibodies elicited in response to EBNA-1 may cross-react with dsDNA. PLoS ONE 2011, 6, e14488. [Google Scholar] [CrossRef]
- Garzelli, C.; Taub, F.E.; Scharff, J.E.; Prabhakar, B.S.; Ginsberg-Fellner, F.; Notkins, A.L. Epstein-Barr virus-transformed lymphocytes produce monoclonal autoantibodies that react with antigens in multiple organs. J. Virol. 1984, 52, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.L.; Erlander, M.G.; Clare-Salzler, M.; Atkinson, M.A.; Maclaren, N.K.; Tobin, A.J. Autoimmunity to two forms of glutamate decarboxylase in insulin-dependent diabetes mellitus. J. Clin. Investig. 1992, 89, 283–292. [Google Scholar] [CrossRef]
- Ou, D.; Mitchell, L.A.; Metzger, D.L.; Gillam, S.; Tingle, A.J. Cross-reactive rubella virus and glutamic acid decarboxylase (65 and 67) protein determinants recognised by T cells of patients with type I diabetes mellitus. Diabetologia 2000, 43, 750–762. [Google Scholar] [CrossRef]
- Karounos, D.G.; Wolinsky, J.S.; Thomas, J.W. Monoclonal antibody to rubella virus capsid protein recognizes a beta-cell antigen. J. Immunol. 1993, 150, 3080–3085. [Google Scholar] [CrossRef]
- Honeyman, M.C.; Stone, N.L.; Harrison, L.C. T-cell epitopes in type 1 diabetes autoantigen tyrosine phosphatase IA-2: Potential for mimicry with rotavirus and other environmental agents. Mol. Med. 1998, 4, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Honeyman, M.C.; Coulson, B.S.; Stone, N.L.; Gellert, S.A.; Goldwater, P.N.; Steele, C.E.; Couper, J.J.; Tait, B.D.; Colman, P.G.; Harrison, L.C. Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes 2000, 49, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, H.S.; Schloot, N.C.; van Veelen, P.A.; Willemen, S.J.; Franken, K.L.; van Rood, J.J.; de Vries, R.R.; Chaudhuri, A.; Behan, P.O.; Drijfhout, J.W.; et al. Cytomegalovirus in autoimmunity: T cell crossreactivity to viral antigen and autoantigen glutamic acid decarboxylase. Proc. Natl. Acad. Sci. USA 2001, 98, 3988–3991. [Google Scholar] [CrossRef] [PubMed]
- Trouw, L.A.; Haisma, E.M.; Levarht, E.W.; van der Woude, D.; Ioan-Facsinay, A.; Daha, M.R.; Huizinga, T.W.; Toes, R.E. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum. 2009, 60, 1923–1931. [Google Scholar] [CrossRef]
- Wu, F.; Gao, J.; Kang, J.; Wang, X.; Niu, Q.; Liu, J.; Zhang, L. B Cells in Rheumatoid Arthritis: Pathogenic Mechanisms and Treatment Prospects. Front. Immunol. 2021, 12, 750753. [Google Scholar] [CrossRef]
- Plenge, R.M.; Padyukov, L.; Remmers, E.F.; Purcell, S.; Lee, A.T.; Karlson, E.W.; Wolfe, F.; Kastner, D.L.; Alfredsson, L.; Altshuler, D.; et al. Replication of putative candidate-gene associations with rheumatoid arthritis in >4000 samples from North America and Sweden: Association of susceptibility with PTPN22, CTLA4, and PADI4. Am. J. Hum. Genet. 2005, 77, 1044–1060. [Google Scholar] [CrossRef]
- Hayer, S.; Redlich, K.; Korb, A.; Hermann, S.; Smolen, J.; Schett, G. Tenosynovitis and osteoclast formation as the initial preclinical changes in a murine model of inflammatory arthritis. Arthritis Rheum. 2007, 56, 79–88. [Google Scholar] [CrossRef]
- Kazantseva, M.G.; Hung, N.A.; Highton, J.; Hessian, P.A. MMP expression in rheumatoid inflammation: The rs11568818 polymorphism is associated with MMP-7 expression at an extra-articular site. Genes Immun. 2013, 14, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Takayanagi, H. Mechanisms of joint destruction in rheumatoid arthritis—Immune cell-fibroblast-bone interactions. Nat. Rev. Rheumatol. 2022, 18, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.; Silman, A.; Barrett, E.; Symmons, D. Low frequency of recent parvovirus infection in a population-based cohort of patients with early inflammatory polyarthritis. Ann. Rheum. Dis. 1998, 57, 375–377. [Google Scholar] [CrossRef]
- Saal, J.G.; Krimmel, M.; Steidle, M.; Gerneth, F.; Wagner, S.; Fritz, P.; Koch, S.; Zacher, J.; Sell, S.; Einsele, H.; et al. Synovial Epstein-Barr virus infection increases the risk of rheumatoid arthritis in individuals with the shared HLA-DR4 epitope. Arthritis Rheum. 1999, 42, 1485–1496. [Google Scholar] [CrossRef]
- Khasnis, A.A.; Schoen, R.T.; Calabrese, L.H. Emerging viral infections in rheumatic diseases. Semin. Arthritis Rheum. 2011, 41, 236–246. [Google Scholar] [CrossRef]
- Takahashi, Y.; Murai, C.; Shibata, S.; Munakata, Y.; Ishii, T.; Ishii, K.; Saitoh, T.; Sawai, T.; Sugamura, K.; Sasaki, T. Human parvovirus B19 as a causative agent for rheumatoid Arthritis. Proc. Natl. Acad. Sci. USA 1998, 95, 8227–8232. [Google Scholar] [CrossRef] [PubMed]
- Arleevskaya, M.I.; Kravtsova, O.A.; Lemerle, J.; Renaudineau, Y.; Tsibulkin, A.P. How Rheumatoid Arthritis Can Result from Provocation of the Immune System by Microorganisms and Viruses. Front. Microbiol. 2016, 7, 1296. [Google Scholar] [CrossRef] [PubMed]
- Alspaugh, M.A.; Henle, G.; Lennette, E.T.; Henle, W. Elevated levels of antibodies to Epstein-Barr virus antigens in sera and synovial fluids of patients with rheumatoid Arthritis. J. Clin. Investig. 1981, 67, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Westergaard, M.W.; Draborg, A.H.; Troelsen, L.; Jacobsen, S.; Houen, G. Isotypes of Epstein-Barr virus antibodies in rheumatoid arthritis: Association with rheumatoid factors and citrulline-dependent antibodies. BioMed Res. Int. 2015, 2015, 472174. [Google Scholar] [CrossRef]
- Fechtner, S.; Berens, H.; Bemis, E.; Johnson, R.L.; Guthridge, C.J.; Carlson, N.E.; Demoruelle, M.K.; Harley, J.B.; Edison, J.D.; Norris, J.A.; et al. Antibody Responses to Epstein-Barr Virus in the Preclinical Period of Rheumatoid Arthritis Suggest the Presence of Increased Viral Reactivation Cycles. Arthritis Rheumatol. 2022, 74, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Sherina, N.; Hreggvidsdottir, H.S.; Bengtsson, C.; Hansson, M.; Israelsson, L.; Alfredsson, L.; Lundberg, K. Low levels of antibodies against common viruses associate with anti-citrullinated protein antibody-positive rheumatoid arthritis; implications for disease aetiology. Arthritis Res. Ther. 2017, 19, 219. [Google Scholar] [CrossRef]
- Ball, R.J.; Avenell, A.; Aucott, L.; Hanlon, P.; Vickers, M.A. Systematic review and meta-analysis of the sero-epidemiological association between Epstein-Barr virus and rheumatoid Arthritis. Arthritis Res. Ther. 2015, 17, 274. [Google Scholar] [CrossRef]
- Takeda, T.; Mizugaki, Y.; Matsubara, L.; Imai, S.; Koike, T.; Takada, K. Lytic Epstein-Barr virus infection in the synovial tissue of patients with rheumatoid Arthritis. Arthritis Rheum. 2000, 43, 1218–1225. [Google Scholar] [CrossRef]
- Li, S.; Yu, Y.; Yue, Y.; Zhang, Z.; Su, K. Microbial Infection and Rheumatoid Arthritis. J. Clin. Cell. Immunol. 2013, 4, 174. [Google Scholar] [CrossRef]
- Vassilopoulos, D.; Calabrese, L.H. Virally associated arthritis 2008: Clinical, epidemiologic, and pathophysiologic considerations. Arthritis Res. Ther. 2008, 10, 215. [Google Scholar] [CrossRef]
- Lazar, S.; Kahlenberg, J.M. Systemic Lupus Erythematosus: New Diagnostic and Therapeutic Approaches. Annu. Rev. Med. 2023, 74, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Long, H.; Lu, Q. Recent advances in understanding pathogenesis and therapeutic strategies of Systemic Lupus Erythematosus. Int. Immunopharmacol. 2020, 89 Pt A, 107028. [Google Scholar] [CrossRef]
- Crow, M.K.; Olferiev, M.; Kirou, K.A. Targeting of type I interferon in systemic autoimmune diseases. Transl. Res. 2015, 165, 296–305. [Google Scholar] [CrossRef]
- Stetson, D.B. Endogenous retroelements and autoimmune disease. Curr. Opin. Immunol. 2012, 24, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Aringer, M.; Johnson, S.R. Classifying and diagnosing systemic lupus erythematosus in the 21st century. Rheumatology 2020, 59 (Suppl. S5), v4–v11. [Google Scholar] [CrossRef]
- Silverman, G.J.; Srikrishnan, R.; Germar, K.; Goodyear, C.S.; Andrews, K.A.; Ginzler, E.M.; Tsao, B.P. Genetic imprinting of autoantibody repertoires in systemic lupus erythematosus patients. Clin. Exp. Immunol. 2008, 153, 102–116. [Google Scholar] [CrossRef]
- Riemekasten, G.; Hahn, B.H. Key autoantigens in SLE. Rheumatology 2005, 44, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Holyst, M.M.; Hill, D.L.; Hoch, S.O.; Hoffman, R.W. Analysis of human T cell and B cell responses against U small nuclear ribonucleoprotein 70-kd, B, and D polypeptides among patients with systemic lupus erythematosus and mixed connective tissue disease. Arthritis Rheum. 1997, 40, 1493–1503. [Google Scholar] [CrossRef]
- Kaul, A.; Gordon, C.; Crow, M.K.; Touma, Z.; Urowitz, M.B.; van Vollenhoven, R.; Ruiz-Irastorza, G.; Hughes, G. Systemic lupus erythematosus. Nat. Rev. Dis. Prim. 2016, 2, 16039. [Google Scholar] [CrossRef]
- Nelson, P.; Rylance, P.; Roden, D.; Trela, M.; Tugnet, N. Viruses as potential pathogenic agents in systemic lupus erythematosus. Lupus 2014, 23, 596–605. [Google Scholar] [CrossRef]
- Illescas-Montes, R.; Corona-Castro, C.C.; Melguizo-Rodriguez, L.; Ruiz, C.; Costela-Ruiz, V.J. Infectious processes and systemic lupus erythematosus. Immunology 2019, 158, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Crispin, J.C.; Kyttaris, V.C.; Terhorst, C.; Tsokos, G.C. T cells as therapeutic targets in SLE. Nat. Rev. Rheumatol. 2010, 6, 317–325. [Google Scholar] [CrossRef]
- Katsiari, C.G.; Liossis, S.N.; Sfikakis, P.P. The pathophysiologic role of monocytes and macrophages in systemic lupus erythematosus: A reappraisal. Semin. Arthritis Rheum. 2010, 39, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Bijl, M.; Reefman, E.; Horst, G.; Limburg, P.C.; Kallenberg, C.G. Reduced uptake of apoptotic cells by macrophages in systemic lupus erythematosus: Correlates with decreased serum levels of complement. Ann. Rheum. Dis. 2006, 65, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Herrada, A.A.; Escobedo, N.; Iruretagoyena, M.; Valenzuela, R.A.; Burgos, P.I.; Cuitino, L.; Llanos, C. Innate Immune Cells’ Contribution to Systemic Lupus Erythematosus. Front. Immunol. 2019, 10, 772. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.J.; Canete, P.F.; Wang, H.; Medhavy, A.; Bones, J.; Roco, J.A.; He, Y.; Qin, Y.; Cappello, J.; Ellyard, J.I.; et al. TLR7 gain-of-function genetic variation causes human lupus. Nature 2022, 605, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Quaglia, M.; Merlotti, G.; De Andrea, M.; Borgogna, C.; Cantaluppi, V. Viral Infections and Systemic Lupus Erythematosus: New Players in an Old Story. Viruses 2021, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Rigante, D.; Esposito, S. Infections and Systemic Lupus Erythematosus: Binding or Sparring Partners? Int. J. Mol. Sci. 2015, 16, 17331–17343. [Google Scholar] [CrossRef]
- Niller, H.H.; Wolf, H.; Ay, E.; Minarovits, J. Epigenetic dysregulation of epstein-barr virus latency and development of autoimmune disease. Adv. Exp. Med. Biol. 2011, 711, 82–102. [Google Scholar] [PubMed]
- Pender, M.P. Infection of autoreactive B lymphocytes with EBV, causing chronic autoimmune diseases. Trends Immunol. 2003, 24, 584–588. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Robertson, J.M.; James, J.A. Preclinical systemic lupus erythematosus. Rheum. Dis. Clin. N. Am. 2014, 40, 621–635. [Google Scholar] [CrossRef]
- Eriksson, C.; Kokkonen, H.; Johansson, M.; Hallmans, G.; Wadell, G.; Rantapaa-Dahlqvist, S. Autoantibodies predate the onset of systemic lupus erythematosus in northern Sweden. Arthritis Res. Ther. 2011, 13, R30. [Google Scholar] [CrossRef]
- Guo, G.; Ye, S.; Xie, S.; Ye, L.; Lin, C.; Yang, M.; Shi, X.; Wang, F.; Li, B.; Li, M.; et al. The cytomegalovirus protein US31 induces inflammation through mono-macrophages in systemic lupus erythematosus by promoting NF-kappaB2 activation. Cell Death Dis. 2018, 9, 104. [Google Scholar] [CrossRef]
- Rajadhyaksha, A.; Mehra, S. Dengue fever evolving into systemic lupus erythematosus and lupus nephritis: A case report. Lupus 2012, 21, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Ramagopalan, S.V.; Dobson, R.; Meier, U.C.; Giovannoni, G. Multiple sclerosis: Risk factors, prodromes, and potential causal pathways. Lancet Neurol. 2010, 9, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.C.; Jenkins, R.; Leary, S.M.; Stevenson, V.L.; Losseff, N.A.; Crum, W.R.; Harvey, R.J.; Rossor, M.N.; Miller, D.H.; Thompson, A.J. Progressive cerebral atrophy in MS: A serial study using registered, volumetric MRI. Neurology 2000, 54, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Alfredsson, L.; Olsson, T. Lifestyle and Environmental Factors in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9, a028944. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.M.; Noyce, A.J.; Bestwick, J.; Belete, D.; Giovannoni, G.; Dobson, R. Gene-Environment Interactions in Multiple Sclerosis: A UK Biobank Study. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1007. [Google Scholar] [CrossRef]
- Moutsianas, L.; Jostins, L.; Beecham, A.H.; Dilthey, A.T.; Xifara, D.K.; Ban, M.; Shah, T.S.; Patsopoulos, N.A.; Alfredsson, L.; Anderson, C.A.; et al. Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat. Genet. 2015, 47, 1107–1113. [Google Scholar] [CrossRef]
- Attfield, K.E.; Jensen, L.T.; Kaufmann, M.; Friese, M.A.; Fugger, L. The immunology of multiple sclerosis. Nat. Rev. Immunol. 2022, 22, 734–750. [Google Scholar] [CrossRef] [PubMed]
- Petermann, F.; Korn, T. Cytokines and effector T cell subsets causing autoimmune CNS disease. FEBS Lett. 2011, 585, 3747–3757. [Google Scholar] [CrossRef]
- Rensing-Ehl, A.; Malipiero, U.; Irmler, M.; Tschopp, J.; Constam, D.; Fontana, A. Neurons induced to express major histocompatibility complex class I antigen are killed via the perforin and not the Fas (APO-1/CD95) pathway. Eur. J. Immunol. 1996, 26, 2271–2274. [Google Scholar] [CrossRef]
- Denic, A.; Wootla, B.; Rodriguez, M. CD8+ T cells in multiple sclerosis. Expert Opin. Ther. Targets 2013, 17, 1053–1066. [Google Scholar] [CrossRef]
- Meuth, S.G.; Herrmann, A.M.; Simon, O.J.; Siffrin, V.; Melzer, N.; Bittner, S.; Meuth, P.; Langer, H.F.; Hallermann, S.; Boldakowa, N.; et al. Cytotoxic CD8+ T cell-neuron interactions: Perforin-dependent electrical silencing precedes but is not causally linked to neuronal cell death. J. Neurosci. 2009, 29, 15397–15409. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Engelhardt, B. Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroinflammation. Vasc. Biol. 2020, 2, H1–H18. [Google Scholar] [CrossRef]
- Galea, I. The blood-brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Tahmasebinia, F.; Pourgholaminejad, A. The role of Th17 cells in auto-inflammatory neurological disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79 Pt B, 408–416. [Google Scholar] [CrossRef]
- Miljkovic, D.; Spasojevic, I. Multiple sclerosis: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2013, 19, 2286–2334. [Google Scholar] [CrossRef] [PubMed]
- Derfuss, T.; Meinl, E. Identifying autoantigens in demyelinating diseases: Valuable clues to diagnosis and treatment? Curr. Opin. Neurol. 2012, 25, 231–238. [Google Scholar] [CrossRef]
- De Vos, A.F.; van Meurs, M.; Brok, H.P.; Boven, L.A.; Hintzen, R.Q.; van der Valk, P.; Ravid, R.; Rensing, S.; Boon, L.; t Hart, B.A.; et al. Transfer of central nervous system autoantigens and presentation in secondary lymphoid organs. J. Immunol. 2002, 169, 5415–5423. [Google Scholar] [CrossRef]
- Haghayegh Jahromi, N.; Marchetti, L.; Moalli, F.; Duc, D.; Basso, C.; Tardent, H.; Kaba, E.; Deutsch, U.; Pot, C.; Sallusto, F.; et al. Intercellular Adhesion Molecule-1 (ICAM-1) and ICAM-2 Differentially Contribute to Peripheral Activation and CNS Entry of Autoaggressive Th1 and Th17 Cells in Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2019, 10, 3056. [Google Scholar] [CrossRef] [PubMed]
- Schlager, C.; Korner, H.; Krueger, M.; Vidoli, S.; Haberl, M.; Mielke, D.; Brylla, E.; Issekutz, T.; Cabanas, C.; Nelson, P.J.; et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature 2016, 530, 349–353. [Google Scholar] [CrossRef]
- Becher, B.; Bechmann, I.; Greter, M. Antigen presentation in autoimmunity and CNS inflammation: How T lymphocytes recognize the brain. J. Mol. Med. 2006, 84, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Calabresi, P.A.; Arnold, D.; Markowitz, C.; Shafer, S.; Kasper, L.H.; Waubant, E.; Gazda, S.; Fox, R.J.; Panzara, M.; et al. Rituximab in relapsing-remitting multiple sclerosis: A 72-week, open-label, phase I trial. Ann. Neurol. 2008, 63, 395–400. [Google Scholar] [CrossRef]
- Kappos, L.; Li, D.; Calabresi, P.A.; O’Connor, P.; Bar-Or, A.; Barkhof, F.; Yin, M.; Leppert, D.; Glanzman, R.; Tinbergen, J.; et al. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar] [CrossRef]
- Li, R.; Patterson, K.R.; Bar-Or, A. Reassessing B cell contributions in multiple sclerosis. Nat. Immunol. 2018, 19, 696–707. [Google Scholar] [CrossRef]
- Kinnunen, T.; Chamberlain, N.; Morbach, H.; Cantaert, T.; Lynch, M.; Preston-Hurlburt, P.; Herold, K.C.; Hafler, D.A.; O’Connor, K.C.; Meffre, E. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J. Clin. Investig. 2013, 123, 2737–2741. [Google Scholar] [CrossRef]
- Kinnunen, T.; Chamberlain, N.; Morbach, H.; Choi, J.; Kim, S.; Craft, J.; Mayer, L.; Cancrini, C.; Passerini, L.; Bacchetta, R.; et al. Accumulation of peripheral autoreactive B cells in the absence of functional human regulatory T cells. Blood 2013, 121, 1595–1603. [Google Scholar] [CrossRef]
- Kinzel, S.; Lehmann-Horn, K.; Torke, S.; Hausler, D.; Winkler, A.; Stadelmann, C.; Payne, N.; Feldmann, L.; Saiz, A.; Reindl, M.; et al. Myelin-reactive antibodies initiate T cell-mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol. 2016, 132, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Sola, P.; Merelli, E.; Marasca, R.; Poggi, M.; Luppi, M.; Montorsi, M.; Torelli, G. Human herpesvirus 6 and multiple sclerosis: Survey of anti-HHV-6 antibodies by immunofluorescence analysis and of viral sequences by polymerase chain reaction. J. Neurol. Neurosurg. Psychiatry 1993, 56, 917–919. [Google Scholar] [CrossRef]
- Wilborn, F.; Schmidt, C.A.; Brinkmann, V.; Jendroska, K.; Oettle, H.; Siegert, W. A potential role for human herpesvirus type 6 in nervous system disease. J. Neuroimmunol. 1994, 49, 213–214. [Google Scholar] [CrossRef]
- Merelli, E.; Bedin, R.; Sola, P.; Barozzi, P.; Mancardi, G.L.; Ficarra, G.; Franchini, G. Human herpes virus 6 and human herpes virus 8 DNA sequences in brains of multiple sclerosis patients, normal adults and children. J. Neurol. 1997, 244, 450–454. [Google Scholar] [CrossRef]
- Leibovitch, E.C.; Caruso, B.; Ha, S.K.; Schindler, M.K.; Lee, N.J.; Luciano, N.J.; Billioux, B.J.; Guy, J.R.; Yen, C.; Sati, P.; et al. Herpesvirus trigger accelerates neuroinflammation in a nonhuman primate model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 11292–11297. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.S.; Maurer, M.A.; Meixlsperger, S.; Lippmann, A.; Cheong, C.; Zuo, J.; Haigh, T.A.; Taylor, G.S.; Munz, C. Robust T-cell stimulation by Epstein-Barr virus-transformed B cells after antigen targeting to DEC-205. Blood 2013, 121, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Raab-Traub, N.; Casali, P.; Cerutti, A. EBV-encoded latent membrane protein 1 cooperates with BAFF/BLyS and APRIL to induce T cell-independent Ig heavy chain class switching. J. Immunol. 2003, 171, 5215–5224. [Google Scholar] [CrossRef] [PubMed]
- Kalchschmidt, J.S.; Bashford-Rogers, R.; Paschos, K.; Gillman, A.C.; Styles, C.T.; Kellam, P.; Allday, M.J. Epstein-Barr virus nuclear protein EBNA3C directly induces expression of AID and somatic mutations in B cells. J. Exp. Med. 2016, 213, 921–928. [Google Scholar] [CrossRef]
- Bray, P.F.; Bloomer, L.C.; Salmon, V.C.; Bagley, M.H.; Larsen, P.D. Epstein-Barr virus infection and antibody synthesis in patients with multiple sclerosis. Arch. Neurol. 1983, 40, 406–408. [Google Scholar] [CrossRef]
- Larsen, P.D.; Bloomer, L.C.; Bray, P.F. Epstein-Barr nuclear antigen and viral capsid antigen antibody titers in multiple sclerosis. Neurology 1985, 35, 435–438. [Google Scholar] [CrossRef]
- Sumaya, C.V.; Myers, L.W.; Ellison, G.W.; Ench, Y. Increased prevalence and titer of Epstein-Barr virus antibodies in patients with multiple sclerosis. Ann. Neurol. 1985, 17, 371–377. [Google Scholar] [CrossRef]
- DeLorenze, G.N.; Munger, K.L.; Lennette, E.T.; Orentreich, N.; Vogelman, J.H.; Ascherio, A. Epstein-Barr virus and multiple sclerosis: Evidence of association from a prospective study with long-term follow-up. Arch. Neurol. 2006, 63, 839–844. [Google Scholar] [CrossRef]
- Munger, K.L.; Levin, L.I.; O’Reilly, E.J.; Falk, K.I.; Ascherio, A. Anti-Epstein-Barr virus antibodies as serological markers of multiple sclerosis: A prospective study among United States military personnel. Mult. Scler. 2011, 17, 1185–1193. [Google Scholar] [CrossRef]
- Sundstrom, P.; Juto, P.; Wadell, G.; Hallmans, G.; Svenningsson, A.; Nystrom, L.; Dillner, J.; Forsgren, L. An altered immune response to Epstein-Barr virus in multiple sclerosis: A prospective study. Neurology 2004, 62, 2277–2282. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, J.W.; Hatfield, L.M.; Vu, T. Epstein-Barr virus neutralizing and early antigen antibodies in multiple sclerosis. Eur. J. Neurol. 2010, 17, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Lunemann, J.D.; Tintore, M.; Messmer, B.; Strowig, T.; Rovira, A.; Perkal, H.; Caballero, E.; Munz, C.; Montalban, X.; Comabella, M. Elevated Epstein-Barr virus-encoded nuclear antigen-1 immune responses predict conversion to multiple sclerosis. Ann. Neurol. 2010, 67, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Pender, M.P.; Csurhes, P.A.; Burrows, J.M.; Burrows, S.R. Defective T-cell control of Epstein-Barr virus infection in multiple sclerosis. Clin. Transl. Immunol. 2017, 6, e126. [Google Scholar] [CrossRef]
- Serafini, B.; Rosicarelli, B.; Franciotta, D.; Magliozzi, R.; Reynolds, R.; Cinque, P.; Andreoni, L.; Trivedi, P.; Salvetti, M.; Faggioni, A.; et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J. Exp. Med. 2007, 204, 2899–2912. [Google Scholar] [CrossRef]
- Jog, N.R.; McClain, M.T.; Heinlen, L.D.; Gross, T.; Towner, R.; Guthridge, J.M.; Axtell, R.C.; Pardo, G.; Harley, J.B.; James, J.A. Epstein Barr virus nuclear antigen 1 (EBNA-1) peptides recognized by adult multiple sclerosis patient sera induce neurologic symptoms in a murine model. J. Autoimmun. 2020, 106, 102332. [Google Scholar] [CrossRef]
- Ji, Q.; Castelli, L.; Goverman, J.M. MHC class I-restricted myelin epitopes are cross-presented by Tip-DCs that promote determinant spreading to CD8+ T cells. Nat. Immunol. 2013, 14, 254–261. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Op de Beeck, A.; Eizirik, D.L. Viral infections in type 1 diabetes mellitus--why the beta cells? Nat. Rev. Endocrinol. 2016, 12, 263–273. [Google Scholar] [CrossRef]
- Smyth, D.J.; Cooper, J.D.; Bailey, R.; Field, S.; Burren, O.; Smink, L.J.; Guja, C.; Ionescu-Tirgoviste, C.; Widmer, B.; Dunger, D.B.; et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat. Genet. 2006, 38, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef]
- Jun, H.S.; Yoon, J.W. The role of viruses in type I diabetes: Two distinct cellular and molecular pathogenic mechanisms of virus-induced diabetes in animals. Diabetologia 2001, 44, 271–285. [Google Scholar] [CrossRef]
- Watad, A.; Azrielant, S.; Bragazzi, N.L.; Sharif, K.; David, P.; Katz, I.; Aljadeff, G.; Quaresma, M.; Tanay, G.; Adawi, M.; et al. Seasonality and autoimmune diseases: The contribution of the four seasons to the mosaic of autoimmunity. J. Autoimmun. 2017, 82, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Moltchanova, E.V.; Schreier, N.; Lammi, N.; Karvonen, M. Seasonal variation of diagnosis of Type 1 diabetes mellitus in children worldwide. Diabet. Med. 2009, 26, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, S.R.; Foskett, D.B.; Maxwell, A.J.; Ward, E.J.; Faulkner, C.L.; Luo, J.Y.X.; Rawlinson, W.D.; Craig, M.E.; Kim, K.W. Viruses and Type 1 Diabetes: From Enteroviruses to the Virome. Microorganisms 2021, 9, 1519. [Google Scholar] [CrossRef] [PubMed]
- Filippi, C.M.; von Herrath, M.G. Viral trigger for type 1 diabetes: Pros and cons. Diabetes 2008, 57, 2863–2871. [Google Scholar] [CrossRef]
- Andreoletti, L.; Hober, D.; Hober-Vandenberghe, C.; Belaich, S.; Vantyghem, M.C.; Lefebvre, J.; Wattre, P. Detection of coxsackie B virus RNA sequences in whole blood samples from adult patients at the onset of type I diabetes mellitus. J. Med. Virol. 1997, 52, 121–127. [Google Scholar] [CrossRef]
- Hyoty, H.; Hiltunen, M.; Knip, M.; Laakkonen, M.; Vahasalo, P.; Karjalainen, J.; Koskela, P.; Roivainen, M.; Leinikki, P.; Hovi, T.; et al. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes 1995, 44, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Jubelt, B.; Lipton, H.L. Enterovirus/picornavirus infections. Handb. Clin. Neurol. 2014, 123, 379–416. [Google Scholar] [CrossRef]
- Lo, C.W.; Wu, K.G.; Lin, M.C.; Chen, C.J.; Ho, D.M.; Tang, R.B.; Chan, Y.J. Application of a molecular method for the classification of human enteroviruses and its correlation with clinical manifestations. J. Microbiol. Immunol. Infect. 2010, 43, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Ifie, E.; Russell, M.A.; Dhayal, S.; Leete, P.; Sebastiani, G.; Nigi, L.; Dotta, F.; Marjomaki, V.; Eizirik, D.L.; Morgan, N.G.; et al. Unexpected subcellular distribution of a specific isoform of the Coxsackie and adenovirus receptor, CAR-SIV, in human pancreatic beta cells. Diabetologia 2018, 61, 2344–2355. [Google Scholar] [CrossRef] [PubMed]
- Shafren, D.R.; Bates, R.C.; Agrez, M.V.; Herd, R.L.; Burns, G.F.; Barry, R.D. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J. Virol. 1995, 69, 3873–3877. [Google Scholar] [CrossRef] [PubMed]
- Dotta, F.; Censini, S.; van Halteren, A.G.; Marselli, L.; Masini, M.; Dionisi, S.; Mosca, F.; Boggi, U.; Muda, A.O.; Del Prato, S.; et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc. Natl. Acad. Sci. USA 2007, 104, 5115–5120. [Google Scholar] [CrossRef]
- Nekoua, M.P.; Alidjinou, E.K.; Hober, D. Persistent coxsackievirus B infection and pathogenesis of type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2022, 18, 503–516. [Google Scholar] [CrossRef]
- Chehadeh, W.; Kerr-Conte, J.; Pattou, F.; Alm, G.; Lefebvre, J.; Wattre, P.; Hober, D. Persistent infection of human pancreatic islets by coxsackievirus B is associated with alpha interferon synthesis in beta cells. J. Virol. 2000, 74, 10153–10164. [Google Scholar] [CrossRef]
- Nekoua, M.P.; Bertin, A.; Sane, F.; Alidjinou, E.K.; Lobert, D.; Trauet, J.; Hober, C.; Engelmann, I.; Moutairou, K.; Yessoufou, A.; et al. Pancreatic beta cells persistently infected with coxsackievirus B4 are targets of NK cell-mediated cytolytic activity. Cell. Mol. Life Sci. 2020, 77, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Vehik, K.; Lynch, K.F.; Wong, M.C.; Tian, X.; Ross, M.C.; Gibbs, R.A.; Ajami, N.J.; Petrosino, J.F.; Rewers, M.; Toppari, J.; et al. Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat. Med. 2019, 25, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Oikarinen, M.; Tauriainen, S.; Honkanen, T.; Oikarinen, S.; Vuori, K.; Kaukinen, K.; Rantala, I.; Maki, M.; Hyoty, H. Detection of enteroviruses in the intestine of type 1 diabetic patients. Clin. Exp. Immunol. 2008, 151, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Sane, F.; Caloone, D.; Gmyr, V.; Engelmann, I.; Belaich, S.; Kerr-Conte, J.; Pattou, F.; Desailloud, R.; Hober, D. Coxsackievirus B4 can infect human pancreas ductal cells and persist in ductal-like cell cultures which results in inhibition of Pdx1 expression and disturbed formation of islet-like cell aggregates. Cell. Mol. Life Sci. 2013, 70, 4169–4180. [Google Scholar] [CrossRef] [PubMed]
- Elshebani, A.; Olsson, A.; Westman, J.; Tuvemo, T.; Korsgren, O.; Frisk, G. Effects on isolated human pancreatic islet cells after infection with strains of enterovirus isolated at clinical presentation of type 1 diabetes. Virus Res. 2007, 124, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Hodik, M.; Skog, O.; Lukinius, A.; Isaza-Correa, J.M.; Kuipers, J.; Giepmans, B.N.; Frisk, G. Enterovirus infection of human islets of Langerhans affects beta-cell function resulting in disintegrated islets, decreased glucose stimulated insulin secretion and loss of Golgi structure. BMJ Open Diabetes Res. Care 2016, 4, e000179. [Google Scholar] [CrossRef] [PubMed]
- Oikarinen, S.; Krogvold, L.; Edwin, B.; Buanes, T.; Korsgren, O.; Laiho, J.E.; Oikarinen, M.; Ludvigsson, J.; Skog, O.; Anagandula, M.; et al. Characterisation of enterovirus RNA detected in the pancreas and other specimens of live patients with newly diagnosed type 1 diabetes in the DiViD study. Diabetologia 2021, 64, 2491–2501. [Google Scholar] [CrossRef]
- Brilot, F.; Chehadeh, W.; Charlet-Renard, C.; Martens, H.; Geenen, V.; Hober, D. Persistent infection of human thymic epithelial cells by coxsackievirus B4. J. Virol. 2002, 76, 5260–5265. [Google Scholar] [CrossRef]
- Jaidane, H.; Caloone, D.; Lobert, P.E.; Sane, F.; Dardenne, O.; Naquet, P.; Gharbi, J.; Aouni, M.; Geenen, V.; Hober, D. Persistent infection of thymic epithelial cells with coxsackievirus B4 results in decreased expression of type 2 insulin-like growth factor. J. Virol. 2012, 86, 11151–11162. [Google Scholar] [CrossRef]
- Jaidane, H.; Gharbi, J.; Lobert, P.E.; Lucas, B.; Hiar, R.; M’Hadheb, M.B.; Brilot, F.; Geenen, V.; Aouni, M.; iHober, D. Prolonged viral RNA detection in blood and lymphoid tissues from coxsackievirus B4 E2 orally-inoculated Swiss mice. Microbiol. Immunol. 2006, 50, 971–974. [Google Scholar] [CrossRef]
- Nyalwidhe, J.O.; Gallagher, G.R.; Glenn, L.M.; Morris, M.A.; Vangala, P.; Jurczyk, A.; Bortell, R.; Harlan, D.M.; Wang, J.P.; Nadler, J.L. Coxsackievirus-Induced Proteomic Alterations in Primary Human Islets Provide Insights for the Etiology of Diabetes. J. Endocr. Soc. 2017, 1, 1272–1286. [Google Scholar] [CrossRef]
- Marroqui, L.; Dos Santos, R.S.; Op de Beeck, A.; Coomans de Brachene, A.; Marselli, L.; Marchetti, P.; Eizirik, D.L. Interferon-alpha mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 2017, 60, 656–667. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Sammeth, M.; Bouckenooghe, T.; Bottu, G.; Sisino, G.; Igoillo-Esteve, M.; Ortis, F.; Santin, I.; Colli, M.L.; Barthson, J.; et al. The human pancreatic islet transcriptome: Expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012, 8, e1002552. [Google Scholar] [CrossRef]
- Van Belle, T.L.; Coppieters, K.T.; Von Herrath, M.G. Type 1 Diabetes: Etiology, Immunology, and Therapeutic Strategies. Physiol. Rev. 2011, 91, 79–118. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Bowman, M.A.; Campbell, L.; Darrow, B.L.; Kaufman, D.L.; Maclaren, N.K. Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J. Clin. Investig. 1994, 94, 2125–2129. [Google Scholar] [CrossRef]
- Tian, J.; Lehmann, P.V.; Kaufman, D.L. T cell cross-reactivity between coxsackievirus and glutamate decarboxylase is associated with a murine diabetes susceptibility allele. J. Exp. Med. 1994, 180, 1979–1984. [Google Scholar] [CrossRef]
- Horwitz, M.S.; Bradley, L.M.; Harbertson, J.; Krahl, T.; Lee, J.; Sarvetnick, N. Diabetes induced by Coxsackie virus: Initiation by bystander damage and not molecular mimicry. Nat. Med. 1998, 4, 781–785. [Google Scholar] [CrossRef]
- Richter, W.; Mertens, T.; Schoel, B.; Muir, P.; Ritzkowsky, A.; Scherbaum, W.A.; Boehm, B.O. Sequence homology of the diabetes-associated autoantigen glutamate decarboxylase with coxsackie B4-2C protein and heat shock protein 60 mediates no molecular mimicry of autoantibodies. J. Exp. Med. 1994, 180, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Schloot, N.C.; Willemen, S.J.; Duinkerken, G.; Drijfhout, J.W.; de Vries, R.R.; Roep, B.O. Molecular mimicry in type 1 diabetes mellitus revisited: T-cell clones to GAD65 peptides with sequence homology to Coxsackie or proinsulin peptides do not crossreact with homologous counterpart. Hum. Immunol. 2001, 62, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Blomqvist, M.; Juhela, S.; Erkkila, S.; Korhonen, S.; Simell, T.; Kupila, A.; Vaarala, O.; Simell, O.; Knip, M.; Ilonen, J. Rotavirus infections and development of diabetes-associated autoantibodies during the first 2 years of life. Clin. Exp. Immunol. 2002, 128, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Makela, M.; Oling, V.; Marttila, J.; Waris, M.; Knip, M.; Simell, O.; Ilonen, J. Rotavirus-specific T cell responses and cytokine mRNA expression in children with diabetes-associated autoantibodies and type 1 diabetes. Clin. Exp. Immunol. 2006, 145, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Unanue, E.R. Antigen presentation in the autoimmune diabetes of the NOD mouse. Annu. Rev. Immunol. 2014, 32, 579–608. [Google Scholar] [CrossRef]
- Sutherland, A.P.; Van Belle, T.; Wurster, A.L.; Suto, A.; Michaud, M.; Zhang, D.; Grusby, M.J.; von Herrath, M. Interleukin-21 is required for the development of type 1 diabetes in NOD mice. Diabetes 2009, 58, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Spolski, R.; Kashyap, M.; Robinson, C.; Yu, Z.; Leonard, W.J. IL-21 signaling is critical for the development of type I diabetes in the NOD mouse. Proc. Natl. Acad. Sci. USA 2008, 105, 14028–14033. [Google Scholar] [CrossRef] [PubMed]
- Varchetta, S.; Mele, D.; Oliviero, B.; Mantovani, S.; Ludovisi, S.; Cerino, A.; Bruno, R.; Castelli, A.; Mosconi, M.; Vecchia, M.; et al. Unique immunological profile in patients with COVID-19. Cell. Mol. Immunol. 2021, 18, 604–612. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Steenblock, C.; Richter, S.; Berger, I.; Barovic, M.; Schmid, J.; Schubert, U.; Jarzebska, N.; von Massenhausen, A.; Linkermann, A.; Schurmann, A.; et al. Viral infiltration of pancreatic islets in patients with COVID-19. Nat. Commun. 2021, 12, 3534. [Google Scholar] [CrossRef]
- Xie, Y.; Al-Aly, Z. Risks and burdens of incident diabetes in long COVID: A cohort study. Lancet Diabetes Endocrinol. 2022, 10, 311–321. [Google Scholar] [CrossRef]
- Muller, J.A.; Gross, R.; Conzelmann, C.; Kruger, J.; Merle, U.; Steinhart, J.; Weil, T.; Koepke, L.; Bozzo, C.P.; Read, C.; et al. SARS-CoV-2 infects and replicates in cells of the human endocrine and exocrine pancreas. Nat. Metab. 2021, 3, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Catriona, C.; Paolo, P. SARS-CoV-2 induced post-translational protein modifications: A trigger for developing autoimmune diabetes? Diabetes Metab. Res. Rev. 2022, 38, e3508. [Google Scholar] [CrossRef]
- Bestion, E.; Halfon, P.; Mezouar, S.; Mege, J.L. Cell and Animal Models for SARS-CoV-2 Research. Viruses 2022, 14, 1507. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.; Acharya, A.; Mohan, M.; Ng, C.L.; Reid, S.P.; Byrareddy, S.N. Animal models for SARS-CoV-2 research: A comprehensive literature review. Transbound. Emerg. Dis. 2021, 68, 1868–1885. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K. In Vitro and Animal Models for SARS-CoV-2 research. Trends Pharmacol. Sci. 2020, 41, 513–517. [Google Scholar] [CrossRef]
- McCray, P.B., Jr.; Pewe, L.; Wohlford-Lenane, C.; Hickey, M.; Manzel, L.; Shi, L.; Netland, J.; Jia, H.P.; Halabi, C.; Sigmund, C.D.; et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J. Virol. 2007, 81, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chan, J.F.; Yuen, K.Y. Animal models in SARS-CoV-2 research. Nat. Methods 2022, 19, 392–394. [Google Scholar] [CrossRef]
- Munoz-Fontela, C.; Dowling, W.E.; Funnell, S.G.P.; Gsell, P.S.; Riveros-Balta, A.X.; Albrecht, R.A.; Andersen, H.; Baric, R.S.; Carroll, M.W.; Cavaleri, M.; et al. Animal models for COVID-19. Nature 2020, 586, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Park, J.G.; Pino, P.A.; Akhter, A.; Alvarez, X.; Torrelles, J.B.; Martinez-Sobrido, L. Animal Models of COVID-19: Transgenic Mouse Model. Methods Mol. Biol. 2022, 2452, 259–289. [Google Scholar] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sundaresan, B.; Shirafkan, F.; Ripperger, K.; Rattay, K. The Role of Viral Infections in the Onset of Autoimmune Diseases. Viruses 2023, 15, 782. https://doi.org/10.3390/v15030782
Sundaresan B, Shirafkan F, Ripperger K, Rattay K. The Role of Viral Infections in the Onset of Autoimmune Diseases. Viruses. 2023; 15(3):782. https://doi.org/10.3390/v15030782
Chicago/Turabian StyleSundaresan, Bhargavi, Fatemeh Shirafkan, Kevin Ripperger, and Kristin Rattay. 2023. "The Role of Viral Infections in the Onset of Autoimmune Diseases" Viruses 15, no. 3: 782. https://doi.org/10.3390/v15030782
APA StyleSundaresan, B., Shirafkan, F., Ripperger, K., & Rattay, K. (2023). The Role of Viral Infections in the Onset of Autoimmune Diseases. Viruses, 15(3), 782. https://doi.org/10.3390/v15030782