1. Introduction
Numerical models for fluid flow usually simulate the conditions for one specific scale in time and space. Physical processes, however, may operate on different scales, and numerical models need to be accurate enough to capture the corresponding phenomena. This can be done by explicit modeling of effects that are connected to each other, or by coupling two or even more models of the scales involved (multiscale modeling). An exemplary phenomenon is the flow of a liquid in a nanochannel: while the flow in the bulk of the channel might still be captured with a continuum model, the same might not be possible for the slip of the fluid along the walls, which arises from molecular wall–fluid interactions. One way to solve this in the context of multiscale modeling is to use a molecular-continuum model, which relies on a molecular dynamics (MD) simulation of the fluid close to the wall, a continuum-based computational fluid dynamics (CFD) model in the bulk and the coupling of both models.
A molecular-continuum scheme consists of two simulation models and the coupling framework, which results in a complex system with many physical and technical parameters. Consequently, the number of possible parameter combinations is very high. However, only few studies have reported on parameter sensitivities. This has two effects. First, it makes it impossible to perfectly reproduce results, which means typically being limited to making coarse-scale, statistical quantitative comparisons only, or even making qualitative comparisons only. Second, it is important to know the influences of all parameters and their interconnections to be able to use a molecular-continuum coupled model for the simulation of other arbitrary setups.
The first molecular-continuum coupling approach was developed by O’Connell and Thompson in 1995 [
1]. Since then, several research groups published papers discussing variations, improvements and applications of similar models. Most have concentrated on novel coupling methodologies, and only a few have commented on the impacts of potential coupling parameters [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12]. A comprehensive review of the general development in the field was given by Mohamed and Mohamad [
13]. One of the main distinctions among coupling methodologies is flux-based vs. state-based coupling. For state-based coupling, state variables, such as mass or velocity, are communicated and applied as boundary conditions to the other domain. In contrast, flux-based coupling exchanges the flux variables. This method has been used to capture fluctuations, which can be important on the mesoscale [
7], but at the same time it is more expensive for incompressible flows [
13] and its stability can be relevant [
14]. State-based coupling is more stable [
14] and easier to implement [
13]. However, calibration of the solvers is often needed to assure agreement in the fluid’s transport properties [
13]. In the following we present a short summary of research regarding the technical parameters of molecular-continuum coupling.
The sizes of the overlapping region and the non-overlapping molecular-dynamics region were studied by Yen et al. [
15]. For their evaluation, a hybrid molecular-continuum model was applied for the simulation of a transient Couette and Poiseuille flow with a slip condition at the wall. A minimum of
pure MD regions was found to be necessary to simulate the correct slip length, where
is the characteristic length scale at the MD scale, considering a single-site Lennard–Jones model. For liquid argon,
has a value of roundabout 0.34 nm. For their setup, the authors found a size of the overlap region of at least
to improve the convergence. They pointed out that a certain minimum size of the inner molecular dynamics region is important for valid and correct simulations with the hybrid model.
An even more intensive study on the optimal design of the overlap layer was conducted by Wang et al. [
16]. Their research was performed using a hybrid molecular-continuum model, coupling LAMMPS and OpenFOAM, for the simulation of a transient Couette flow with slip and no-slip boundary conditions at the wall. Wang et al. split the overlap layer into different functional regions: control (for boundary forces and mass flow modeling); data transfer from continuum to particle region and vice versa; and a buffer region between the data transfer regions. They evaluated the effect of varying the widths of these regions. The best results were obtained when the continuum-to-particle transfer region was twice as wide as the others. Furthermore, they dealt with the topic of how often data from the macroscopic solver need to be sent to the continuum; and how many sampling points should be included to obtain good agreement between both solvers, but at the same time do as few operations as possible. Their main finding was that instead of using all sampling points for the data exchange, just a few need to be included to obtain almost the same accuracy as when using all. Here, the term sampling points refers to the data of one molecular dynamics time step. For more details, the reader should refer to the original paper.
A molecular-continuum coupled simulation can be performed using various open-source software approaches. Besides general frameworks for the coupling of numerical simulation models—for example, preCICE (
https://github.com/precice/precice, accessed on 9 November 2021) specific open-source libraries for molecular-continuum coupling have been developed, such as the CPL-library (
https://github.com/Crompulence/cpl-library, accessed on 9 November 2021), MaMiCo (
https://github.com/HSU-HPC/MaMiCo, accessed on 9 November 2021) and HAC-par [
17]. Since they are customized for these kinds of simulations, they offer many special features (for example, MaMiCo’s multi-instance sampling, which means several quasi-identical, yet randomized molecular dynamics simulations are performed, and hydrodynamic quantities are sampled from this ensemble before it is applied in the continuum simulation) and are generally more suitable for molecular-continuum coupling. For example, the mdFoam+ (
https://github.com/MicroNanoFlows/OpenFOAM-2.4.0-MNF, accessed on 9 November 2021) molecular dynamics model enables the use of OpenFOAM (
https://www.openfoam.com, accessed on 9 November 2021) for a molecular-continuum coupling [
18]. Due to its integration with the OpenFOAM package, it can be used together with a broad range of solvers, pre-processing tools and post-processing tools. However, due to the file-based data exchange in OpenFOAM, which mdFoam+ inherits, and due to issues with input and output in parallelized simulations, mdFoam+ coupled simulations have drawbacks in terms of their parallelized execution on high performance computing clusters.
We carried out a detailed parameter sensitivity study for transient molecular-continuum coupling. We used the macro-micro-coupling tool (MaMiCo), due to its high number of molecular-continuum specific features, good scalability, modular design and correspondingly high level of configurability. The tool allows one to couple arbitrary MD and CFD solvers. Thus, the implemented coupling algorithm, which is abstracted from the underlying solvers, can be reused by other research groups. Uptake of the methods and their application, however, requires a detailed understanding of the parametrization. Therefore, we demonstrate the impacts of various parameters by comparing the results of coupled molecular-continuum simulations with a reference setup. The evaluation incorporates the quantitative differences in velocity and density fields, and a qualitative analysis of the behavior of the fluid. We aimed to develop an understanding of the influences of the parameters and also explain why a specific parameter leads to a certain result in the simulation. Sub-optimal configurations imply quantitatively small errors. These inconsistencies may not be detectable in small coupled simulations, executed over short time periods. For this reason, we simulated a relatively big scenario consisting of 180,000 particles and executed over 150,000 molecular dynamics time steps. Compared to similar studies in this field [
1,
15,
16,
19], our simulation is the biggest one in terms of particle updates, and it is of similar size to others with regard to time steps and spatial dimensions.
Due to the simplicity of its setup, its interesting features (sharp gradients at the beginning, linear flow profile in steady state, etc.) and the existence of an analytical solution, we used start-up Couette flow. For the continuum model, we used a finite-difference solver from the OpenFOAM package. The MD solver SimpleMD from the MaMiCo package was applied, and the coupling of both was accomplished by MaMiCo.
We give an introduction to the coupling method in
Section 2.3. The implementation of the coupling of the OpenFOAM continuum solver and MaMiCo is—as this is also a novel development—described in
Section 2.4. Afterwards, the simulations and their results are presented in
Section 3, followed by a discussion of the lessons we learned during the project regarding coupled molecular-continuum simulations.
2. Materials and Methods
2.1. Molecular Dynamics Simulation
The basic idea of molecular dynamics models [
20] is the simulation of individual molecules or particles on the basis of Newton’s equation of motion and a model for the inter-molecular forces:
with
,
,
,
m denoting position, velocity, force and mass of a molecule
i. We make use of the SimpleMD library [
21,
22], which is restricted to single-site, truncated shifted Lennard–Jones molecules; note that, due to choosing MaMiCo for coupling, other more flexible MD codes such as LAMMPS could be used as well, and corresponding results of our study immediately carry over to other implementations. For the reduction of the computational costs, the interaction of particle pairs with a distance wider than the cutoff radius is neglected.
2.2. Finite Volume Method and OpenFOAM
The continuum part was modeled using a CFD solver from the library OpenFOAM, an open-source, state-of-the-art CFD framework which offers an extensive range of features and has a large user base from different fields. Due to the broad use, the frequent updates and the good functionality, it is an interesting enhancement for MaMiCo to offer an interface for OpenFOAM, which was implemented recently and is also detailed in the following.
We chose icoFoam as the solver from OpenFOAM, as it allows, amongst others, for the simulation of geometrically simple, transiently incompressible, laminar flows, such as our Couette flow. For the application of a different OpenFOAM solver coupled with MaMiCo, technical extensions to the interface would be necessary—for example, the consideration of density variations in the case of a compressible solver.
OpenFOAM is written in object-oriented style using C++. The functions and classes of OpenFOAM are designed to be easily extensible and reusable. According to this concept, the solvers consist of stand-alone executables, which call all the necessary OpenFOAM functionalities. To run a simulation using the icoFoam solver, one only needs to execute the icoFoam script. The scrip starts by reading the case setup from special files (called OpenFOAM dictionaries). For details on the case setup, we refer to the OpenFOAM documentation (
https://cfd.direct/openfoam/user-guide/, accessed on 9 November 2021). Our simulation case was a cubic channel flow. We used the OpenFOAM blockMesh (
https://cfd.direct/openfoam/user-guide/v9-blockmesh/, accessed on 9 November 2021) functionality for the mesh creation.
2.3. Molecular-Continuum Coupling
The coupling of the MD model and the continuum model was established using MaMiCo [
21,
22]. Its purpose is to couple an arbitrary massively parallel MD solver with a continuum solver. The solvers are completely encapsulated. This allows one to exchange one of the solvers without influencing the overall setup. The coupling algorithm is based on the approach by Nie et al. [
9]. An overview of the implementation for the coupling with MaMiCo is given below. For further information on MaMiCo and its functionalities, we refer to [
21,
22,
23].
The overall simulation domain was split into two overlapping parts. One part was assigned to the continuum solver and the other to the MD solver . The intersection of both regions was the overlap . For the data transfer between the two solvers a Cartesian grid-like data structure called macroscopic cells was applied. For sake of simplicity, we used the same setup for the macroscopic cells as for the continuum grid; however, other configurations would be possible, while requiring additional interpolation routines.
Within the overlap region, both solvers needed to be physically consistent. A state-flux coupling of the velocity u and the density was applied here: fluxes from the continuum solver were applied to the MD system, whereas the MD state was sampled and imposed as a boundary condition to the continuum solver. Two things were necessary to establish this: first, the transfer of the data between the two solvers, and second, the correct application of the data as a boundary condition in the respective solver. This included a mapping from the molecular to continuum data and vice versa:
Molecular to continuum: The velocity was sampled over all particles, which were located in the region of a macroscopic cell (which is equal with the continuum cell). The sampled velocity was applied as a Dirichlet boundary condition on the corresponding boundary. Since an incompressible continuum solver was used, the density was not manipulated.
Continuum to molecular: An additional force [
9] was applied to every particle to match the fluid acceleration from the corresponding continuum cell. The continuity of mass was achieved by either application of periodic boundary conditions or inserting or deleting particles in the outermost macroscopic cells of the MD domain in accordance to the mass flow in the continuum at this boundary.
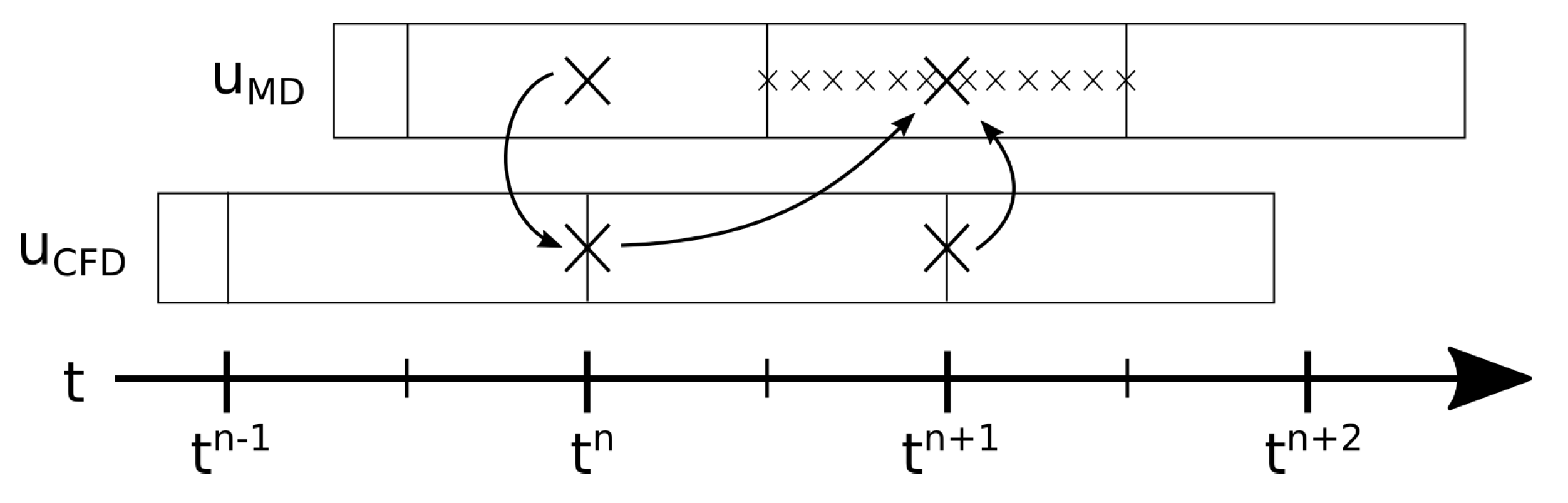
Besides the spatial dimensions of the coupling, the time integration scheme for the coupling shall also be mentioned here. Therefore, we will sketch the process of one coupled time step in the middle of the simulation. The explanation is supported by
Figure 1, which illustrates the cycle. In the initial state, the coupled simulation finished the
n-th timestep (
). More precisely, the continuum flow field was available at time step
n, and MD flow information was available as sampled data for time point
, which were generated from the single MD time points between
,
. The simulation of the next coupled time step
works as follows:
The continuum solver evolves from time step n to . The boundary condition for this calculation is deduced from the flow field of the molecular solver at time step n. Now the flow field for the continuum part at time step is available (excluding the boundary values on the MD boundary).
The MD solver advances over several MD time steps from continuum time step to . The boundary condition for this process is deduced via extrapolation and interpolation of the two latest flow fields of the continuum solver, which are and . When this is finished, the flow field for the molecular part of the domain is calculated by averaging all time steps from to . The corresponding averages in grid cells, which represent boundaries of the continuum solver, are used as boundary condition on the continuum side, and thus complement the continuum flow field data at .
2.4. Implementation: Coupling OpenFOAM and MaMiCo
The coupling of OpenFOAM and MaMiCo can be implemented in different ways. One could be to use the OpenFOAM function “externalCoupled” (
www.openfoam.com/documentation/guides/latest/doc/guide-fos-field-externalCoupled.html, accessed on 9 November 2021). It couples internal OpenFOAM solvers with external solvers, and it is based on the reading and writing of files. One of MaMiCo’s design principles is, however, the efficient execution of large-scale, concurrently coupled simulations on high performance computing systems. Since concurrently coupled systems require frequent exchange of information, file-based data exchange cannot be part of this coupling. Therefore, the “externalCoupled” tool was not used. Instead, the coupling was designed by the implementation of direct data exchange via a MaMiCo interface to the OpenFOAM solver icoFoam.
The only communication between OpenFOAM and MaMiCo happened through this interface. Thereby, the continuum and the molecular solver were completely encapsulated. The interface is specific for the test case, the Couette startup, we used, but one could adapt it to another test case or to be generic in the future. Due to the small spacial domain, the parallelization of the icoFoam solver would not have been useful. Thus, the interface was implemented for a sequential continuum solver.
The interface for a continuum solver for MaMiCo needs to implement the following independent functionalities:
Initialization of the continuum solver and communication of the general setup to the solver (e.g., time step and domain size).
Trigger the continuum solver to solve the next time step and update the velocity field.
Return the velocity at a certain position in the continuum mesh for the current time step.
Prepare the continuum solver to receive and apply the data from the molecular solver as a boundary condition.
Receive the values for the current time step from the molecular solver and apply them as boundary conditions within the continuum solver fields.
Write output files for the current time step.
These functions enable MaMiCo to control the simulation and the coupling. For the correct initialization of the simulation, functions 1 and 4 is needed. Within one coupled time step, it needs function 2 to advance the continuum solver and functions 3 and 5 to exchange the data between the two solvers. Based on the user’s setup, MaMiCo uses function 6 to get the output from the continuum solver.
For the implementation of these functions in the icoFoam interface for MaMiCo, the icoFoam executable needs to be split. The code of the icoFoam script is copied to the corresponding functionalities of the interface. The initialization goes to function 1, and the update for the flow field variables for the next time step is function 2. Some of the interface’s functionalities need to be written anew. This is the case for the functions connected with the coupling functions 4 and 5, and also function 6, since the output is given in similar manner to the other MaMiCo output. The getter function for the velocity is basically a wrapper of the OpenFOAM getter for the flow field.
3. Results
We simulated a Couette flow startup with the coupled molecular-continuum model described above. The parametric configurations were varied to identify their effects on the overall quality of the simulation result. We start with a default setup, which built a baseline for our experiments and delivered physically consistent output, close to the analytical solution. To simplify considerations, we restricted ourselves to one-dimensionsal parameter variations, i.e., varying only one parameter and keeping all other parameters fixed. The change was analyzed by measuring the error
in time step
, which means the difference between the analytical and the simulated solution for every grid cell:
where
denotes the center point of the
j-th grid cell;
is the simulated velocity from the molecular dynamics in
x-direction at
in time step
;
is the analytical solution for the flow velocity; and
C is the number of grid cells considered. This yielded the specific error for every time step. We further quantified the mean error over time from an average over all MD time steps
N, apart from the 100 first coupling time steps, to exclude initial non-equilibrium perturbations from switching on the coupling:
In the following, we list the technical parameters we analyzed.
Flow velocity magnitude: The analytical solution of the continuum model is only correct for an incompressible fluid. This assumption can be made for fluids with small Mach numbers
[
24]. The Mach number is the relation of the flow speed to the speed of sound. Increasing the overall flow velocity will increase the Mach number and thereby amplify compressibility effects and related modelling error.
Cell size of the continuum solver: The result from the molecular data needs to be sampled before it can be applied in the continuum simulation. To calculate a value, for example, the velocity for a continuum cell, the mean velocity of all particles within the area of this cell is analyzed. Under the condition of a constant density, the number of particles included in the calculation depends on the size of the continuum cell: the bigger the cell, the more particles used for sampling, and the lower the thermal fluctuations; see, e.g., [
25] or the discussion on “Number of MD instances in a multi-instance sampling” below.
Coupling time step: One coupling time step includes one time step in the continuum and several time steps in the MD simulation, due to the smaller time scale in MD. Increasing the coupling time step results in more MD time steps for sampling, and thus, in smoother data. Making the coupling time step too large, however, also impacts the continuum solver’s stability. In the worst case, this yields oscillations of up to an entire divergence of the coupled simulation. Nie et al. discussed the limits for the time step [
9]. The upper bound is defined by the characteristic time of the flow and the continuum grid. The lower bound is given by the fact that the time step needs to be larger compared to the velocity auto-correlation time, which is
for a Lennard–Jones fluid with density
and temperature
[
9], where
is the characteristic time of the Lennard–Jones potential. Both together yield the following relation, using the spacial discretization
, the time step
and the kinematic viscosity of the fluid
:
which in our dimensionless case results in
.
Number of MD instances in a multi-instance sampling: Multi-instance sampling refers to sampling over an ensemble of quasi-identical, quasi-independent, randomized MD simulations. Due to its Monte Carlo-like nature, the fractional error
of velocity scales with [
25]
where
M is the number of MD instances. This means, doubling the number of instances reduces the error only by a factor of 0.7.
Region for the momentum insertion in the MD model: Both the location and the thickness of the overlap region, in which momentum is transferred from continuum to MD, impact the overall quality of the simulation solution. For example, when using two cell layers instead of one, the MD simulation implicitly also obtains information on the velocity gradient from the continuum simulation, which typically leads to better agreement of the two solvers. Wang et al. found that better results were obtained in their coupled molecular-continuum model, when the area for continuum-to-particle transfer was increased [
16].
Setup for the mass insertion in the MD model: The simulation of a flow over the boundary of the MD domain was accomplished by insertion and deletion of mass. From physical and mathematical point of view, this algorithmic step has a severe impact: mathematically, adding/removing a molecule resembles the incorporation/removal of ordinary differential equations; physically, the non-linear energy landscape must not be perturbed severely [
26], and so forth.
Table 1 summarizes the simulations we performed. The list includes the special parameters analyzed and the error within the MD domain calculated, as described in Equation (
3). The other, general parameters and details for the coupled simulations, which are not listed, can be found in the
Appendix A.
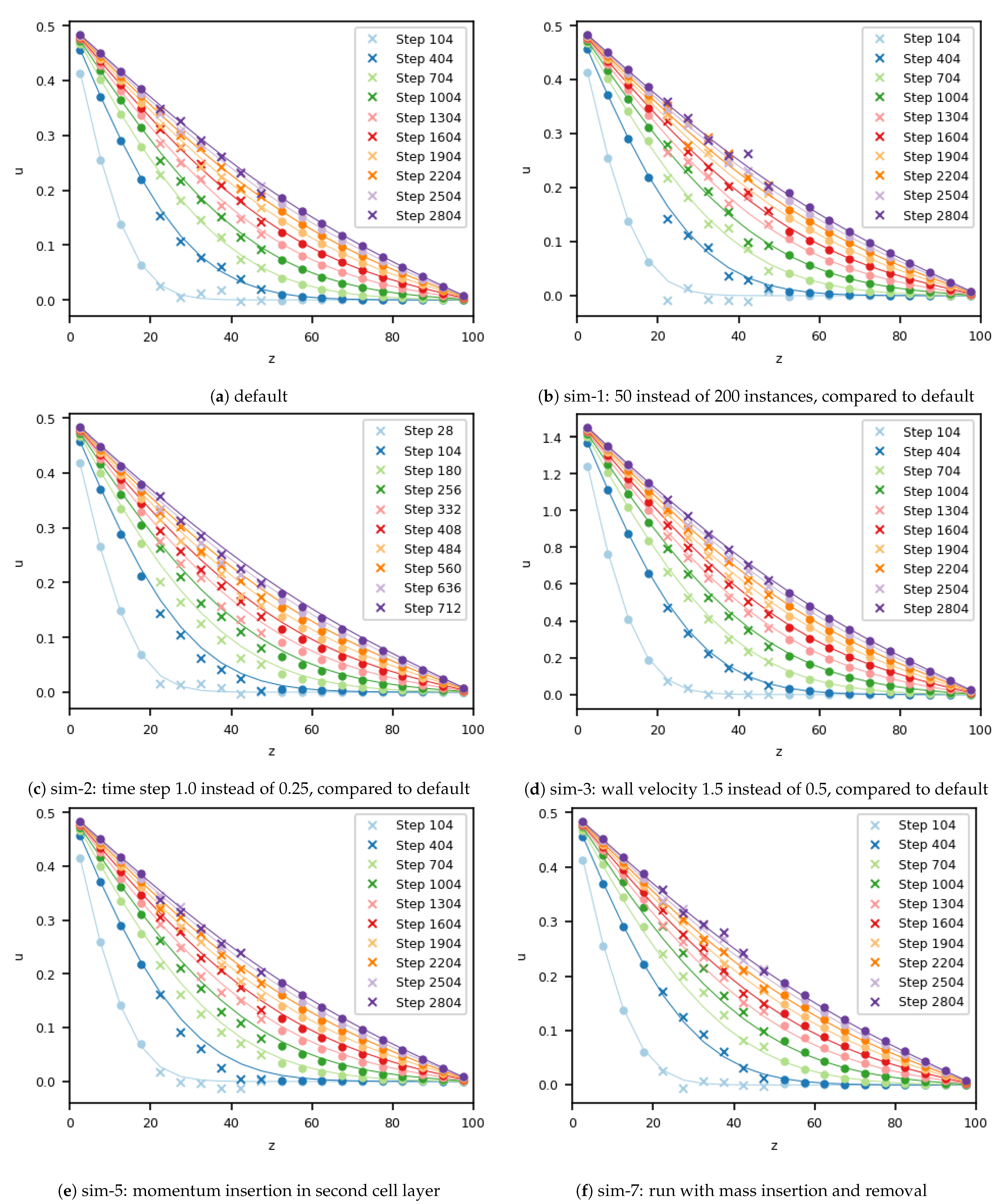
For the default simulation, a “good” setup was chosen, exhibiting a small statistical error of 0.007. Exemplary parts of the velocity field of this simulation are shown in
Figure 2a. As the small error suggests, the velocities for the simulation were in good agreement with the analytical solution during all time steps. Only minor fluctuations from MD can be seen to slightly influence the continuum. Stability of the overall scheme was preserved.
For simulation 1, the number of MD instances was reduced from 200 to 50. Due to this, the fluctuations within the MD simulation were increased, as can be seen in
Figure 2b. Again, the fluctuations from the molecular dynamics domain had an influence on the velocity in the cells of the continuum that were close to the boundary. Regardless of this effect, the coupled simulation remained stable, and the correct steady state was reached. The error increased by a factor of two compared with the default simulation, which is in line with kinetic theory.
The next parameter under consideration was the coupling time step, which was increased by a factor 4 to 1.0 for simulation 2. The effect of this variation can be seen in
Figure 2c. At first glance, the velocity field seems to be in good agreement with the analytical solution. Compared to the default simulation, where the difference was random because of the fluctuating velocity, the velocity in this case was systematically slower than it should have been, compared to the analytical velocity. Due to the longer time step, the temporal evolution of the sudden startup of the Couette flow could not be resolved sufficiently; consequently, this continuum phenomenon also manifested in the coupled case. Close to the stationary state, in the analytical in simulation results the difference still exists. Velocities from both the continuum and the MD solver did not recover from the initial time step size-induced error.
In simulation 3, we analyzed the influence of a higher overall velocity. Therefore, we increased the velocity of the moving wall in
x-direction from 0.5 to 1.5. The resulting velocity field is plotted in
Figure 2d. Due to the higher velocity, which did not influence the statistical fluctuations of the molecules, the signal-to-noise ratio, as defined by Hadjiconstantinou, was enlarged compared to the baseline case [
25]. This change gives the impression that the agreement of the simulated and analytical solutions improved, even though the quantitative error within the domain of the MD was higher, at 0.015, compared to the simulation with a slower velocity. Due to the higher velocity magnitude, the Mach number
was three times higher compared to the baseline scenario, which is still slightly below the usual bound
that is used to justify the assumption of incompressibility. Nevertheless, the observed error must be attributed to the influence of compressibility.
Next, we considered the size of the overlap region for momentum insertion. Until this case, two cells were used. This was reduced to one cell, and the position of this cell layer was varied. We started by only inserting momentum in the outermost cell layer of the overlap region in terms of the MD domain (simulation 4). The velocity profile is not shown, due to its similarity to the ones from the default case. Additionally, the error in the MD region is of comparable size for both setups.
In simulation 5, only one cell layer was used for momentum transfer, as in simulation 4. This time, however, this cell layer was placed in the middle of the overlap region, which means in the second layer of overlapping cells. The simulated velocity for this situation can be found in
Figure 2e. In contrast to simulation 4, this setup differed from the baseline case. A lag in the profile, compared to the analytical solution, could be already observed for the 404-th coupling time step. This lag stayed until the 1304-th time step, and from there on, slowly decreased, but remained until the end of the simulation.
By moving the momentum insertion from the outermost to the middle cell layer of the overlap domain, the error increased. We therefore shifted the one-cell-momentum-transfer region by one more cell layer into the third overlap layer, to better understand this effect. This setup was chosen for simulation 6. The results indicate that the error decreased. The error in this simulation was 0.008; i.e., this setup was slightly worse than the baseline case and simulation 4. However, again the analytical and the simulated velocity profiles are in good agreement (not shown in
Figure 2). To conclude regarding the last three simulations, decreasing the overlap layer to, e.g., only one cell, increased the probability of error and resulted in higher sensitivity of the overall coupling.
Next, we analyzed the impact of mass insertion and deletion at all outer boundaries of the MD domain. The details on the insertion and deletion of mass can be found in the
Appendix A together with all the details of the simulation setup. To emphasize the impact of particle insertion and removal, we further modified the MD boundary condition and changed the periodic boundaries into reflecting ones. This setup is referred to as simulation 7, and velocity profiles can be found in
Figure 2f. In the simulation with these parameters, the error in the velocity field was around 0.010, close to the accuracy of the default setup (without mass transfer). As stated before, a small loss in accuracy was expected. In simulation 7, the insertion or deletion of particles modifies the mass in the system, and therefore, the validity of the density requires analysis. The scenario was initialized with a density of 0.8130. In the default simulation, the density after 3000 coupling cycles was 0.8115 in the inner part of the MD domain, which corresponds to a deviation of ca. 0.1%, due to minor perturbations at the outer boundaries evolving from the boundary forcing. The error in the density field of the simulation 7 increased by a factor of 5 to 0.49%, but still remained small. The particle distribution within the molecular dynamics domain was homogeneous. Due to the periodic boundary condition for the MD domain in the default simulation, density remained constant and no discrepancies were observed along the channel.
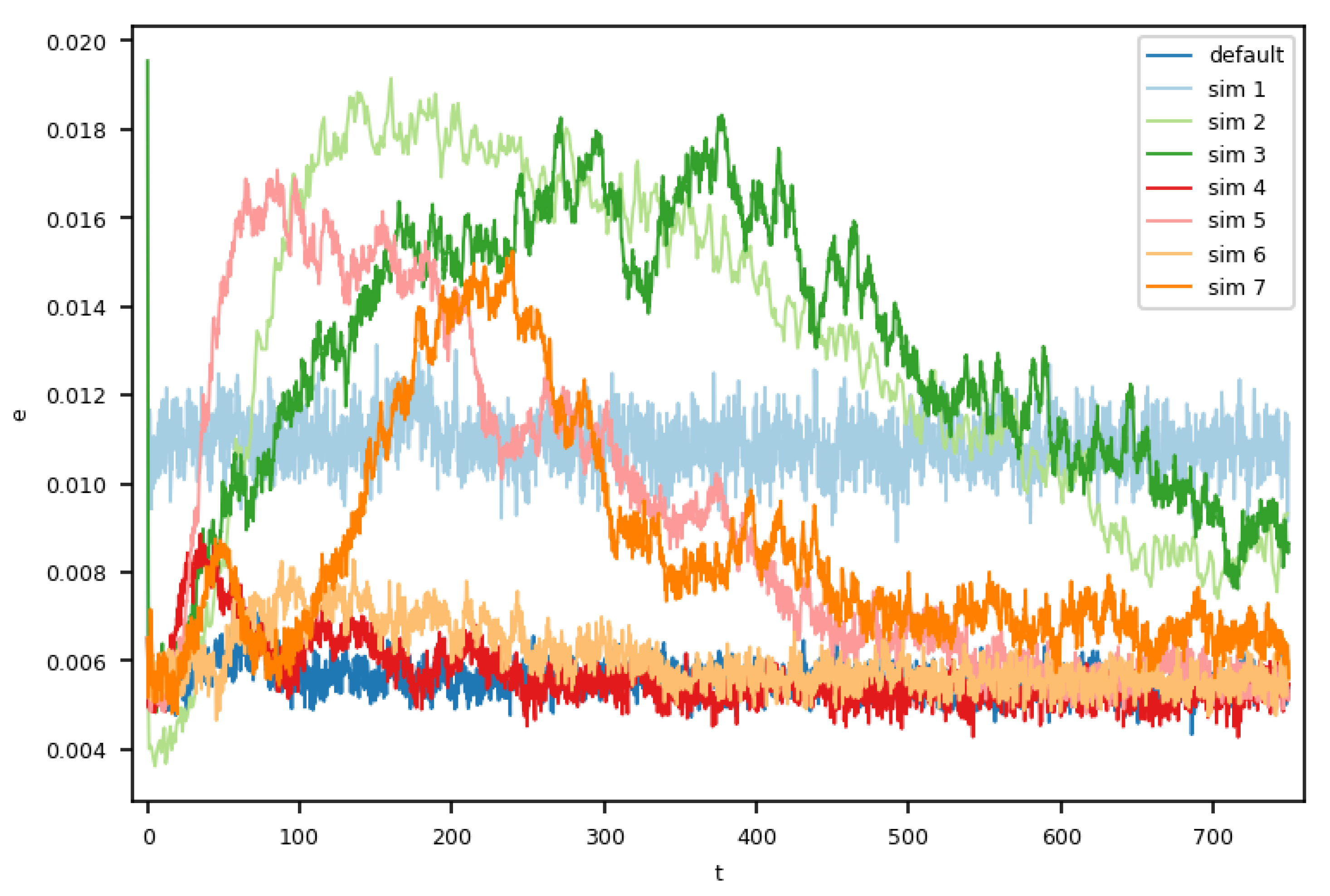
Finally, we analyzed the temporal evolution of the error and related fluctuations within the molecular dynamics domain by comparing of all eight setups, as shown in
Figure 3, where we plotted
(see Formula (
2)). The default case showed an almost constant error over time. Simulations 4 and 6, which only used the outermost or innermost cell for the momentum insertion, had slightly increased error during their respective initial phases. From
onward, the error rates of simulations 4 and 6 were comparable to that in the default case.
As expected, the setup using less instances for the multi-instance sampling was similar to the baseline case in the temporal evolution by showing a constant, yet higher error rate.
In the other four setups, we observed an initial increase in the error, which after a while decreased again, and in some cases, stabilized, though still exceeding the error of the default case. The higher error rates at the beginning agree with our analyses of Couette profiles. We also conducted a deeper analysis of the thermal fluctuations in our Couette flows with varying MD ensemble sizes, which were generally in good agreement with statistical mechanics; refer to [
27] for details.
Furthermore, we wanted to comment on the influence of the continuum cell size by increasing it by a factor of 1.5. For this purpose, we increased the entire setup size (cf.
Table 2), and positioned the (lower, left, front corner of the) MD domain at an offset (30,30,15). All other parameters stayed constant and can be found in the
Appendix A.
To match the overall number of samples in simulations 8 and 9 [
25], we ran the simulations at resolutions 5.0 and 7.5, respectively, with modified MD ensembles. Both the simulated velocity profiles had good agreement with the analytical solution (not shown here). The setup sim-9 exhibited a slightly smaller (approximately 3%) but comparable error.
4. Discussion and Lessons Learned
In this contribution, we analyzed hybrid transient molecular-continuum simulations with various setups of technical parameters. The comparison was based mostly on the quantitative and qualitative differences in the velocity field.
Based on our observations reported in the previous sections and all the work we conducted beyond, we formulated some key lessons learned for establishing a stable and accurate two-way coupled molecular-continuum flow simulation. Our focus here was put on Couette flow, which is a one-dimensional flow scenario, but it still allowed us to draw conclusions even for fully three-dimensional systems. In the future, we aim to apply this molecular-continuum simulation technology to systems that focus on relevant scenarios from process engineering and thermodynamics, such as evaporation, or multiphase scenarios.
4.1. Lesson 1: Averaging Matters!
Despite the usual literature in the field, which often states that the coupling direction molecular→continuum is “straight-forward” via simple averaging, we want to stress few key observations. Statistical error analysis tells us that (a) independent samples and (b) a sufficiently big set of samples are required, which easily yields very computationally intensive setups—computationally intensive in the sense of spatial, temporal or ensemble averaging (multi-instance sampling). While 1D-flows such as Couette flow can be tackled through sampling over the other two dimensions in a 3D setup, our default simulation setup was chosen under the implicit requirement of a fully 3D flow scenario. I.e., with our default simulation and simulation 1, we could get a rough answer on how many samples we actually required, which were not fully independent, as we sampled over all time steps. Nevertheless, considering the given flow scenario (flow velocities, Mach number, etc.) with density
, macroscopic cells of size
and 50 MD time steps per coupling time step, our multi-instance approach yielded approximately 100 million/25 million molecule samples in the default simulation/simulation 1, per cell. This justifies (a) the use of high-performance and supercomputing for general molecular-continuum setups and (b) the need for more enhanced algorithms to reduce Brownian noise, such as algorithms incorporating noise filters; see also, amongst others, [
23,
28].
All these points are mentioned here with regard to a coupling with an incompressible or a weakly compressible solver. Based on other research [
4], these challenges are expected to be less pronounced for fully compressible systems.
While providing significant benefits in terms of reducing the computational effort, noise filters, however, introduce even more parameters to tune. This renders actual a priori error estimation more complex. More work in this direction is necessary and also under current investigation by our group.
4.2. Lesson 2: Only Long-Term Simulation Runs Tell the Truth!
As observed in, e.g., our study on the location and thickness of the overlap layer, actual stability and accuracy cannot necessarily be detected from short-term simulations. Artifacts can manifest themselves only slightly in the beginning but become more pronounced in the long-term. Therefore, long-term runs are advisable to numerically demonstrate the stability and accuracy of a molecular-continuum method.
4.3. Lesson 3: Choose the Overlap Layer Big Enough!
While reported in the literature before (see, e.g., [
16]), we confirmed with our studies that the size and shape of the overlap region has a severe impact on the molecular-continuum simulation quality.
4.4. Lesson 4: Continuum Scales Matter!
As mentioned in Lesson 1, the number of samples required for stable and accurate coupling can easily grow to millions when using molecules. If ultra-fine resolution is also required on the continuum side, e.g., to resolve geometries, this will become an issue, even if the simulation scenario allows for a longer coupling time step—which can easily not be the case due to accuracy or stability limits of the continuum solver, e.g., due to strong gradients in the flow (cf. simulation 2).
To meet sampling requirements, we considered macroscopic cells of size
, making them bigger than the cells used in our prior work [
22]. Our multi-instance sampling approach is an additional ingredient for coping with this challenge.
4.5. Lesson 5: Modular Programming, Testing, Interoperability and Portability Are Essential to Robust Coupling!
Although this is a well-known fact for most complex computational problems in science and engineering [
29], we want to stress that software design, modular programming and (automated) unit and integration testing are essential requirements for long-term simulation software development. Indeed, we were faced with the challenge of fixing a few software bugs that were hard to detect and only became visible in large-scale/long-term runs through minor perturbations in the overall solution. Our software MaMiCo already incorporates several of these kinds of tests, and we are currently working towards extending upon this in the near future.
Similarly, interoperability—in our case: interfacing various MD and continuum solver software packages—is a major challenge, and in this contribution, we outlined how to also include bigger continuum solver software packages with the example of OpenFOAM.
Portability is also important, not only for porting software from laptops to supercomputers, but also for porting between different supercomputing platforms. Amongst others, we currently use the ARM A64FX platform for molecular dynamics simulations, which requires extra instructions (e.g., SVE) to optimally exploit the hardware. Although it is well-known that MD is the most computationally intensive part in a molecular-continuum system, portability aspects are also relevant for the interfacing and coupling software, such as MaMiCo.







{kind=link}
{kind=link}
{kind=link}