Exploring the SARS-CoV-2 Proteome in the Search of Potential Inhibitors via Structure-Based Pharmacophore Modeling/Docking Approach

 , , ,
, , , 
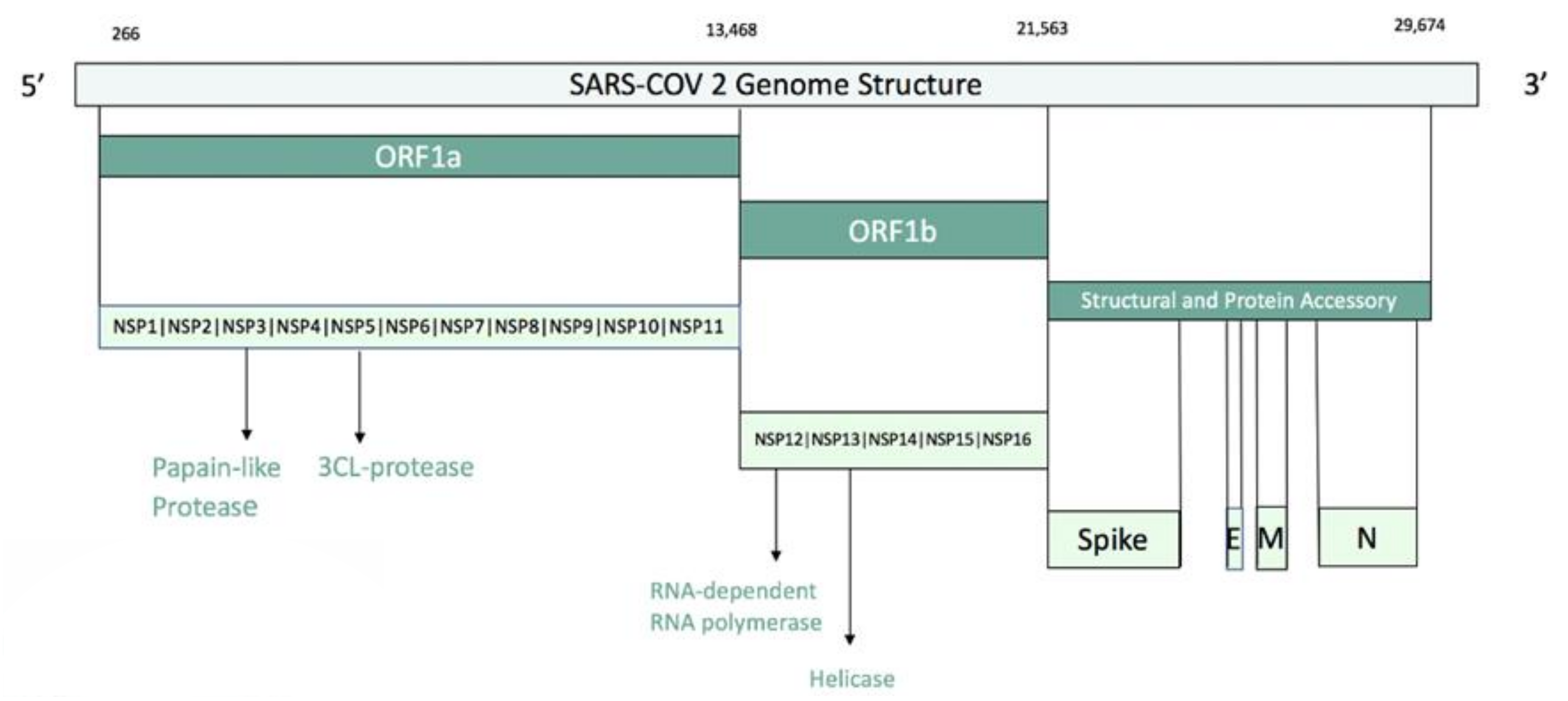{kind=link}
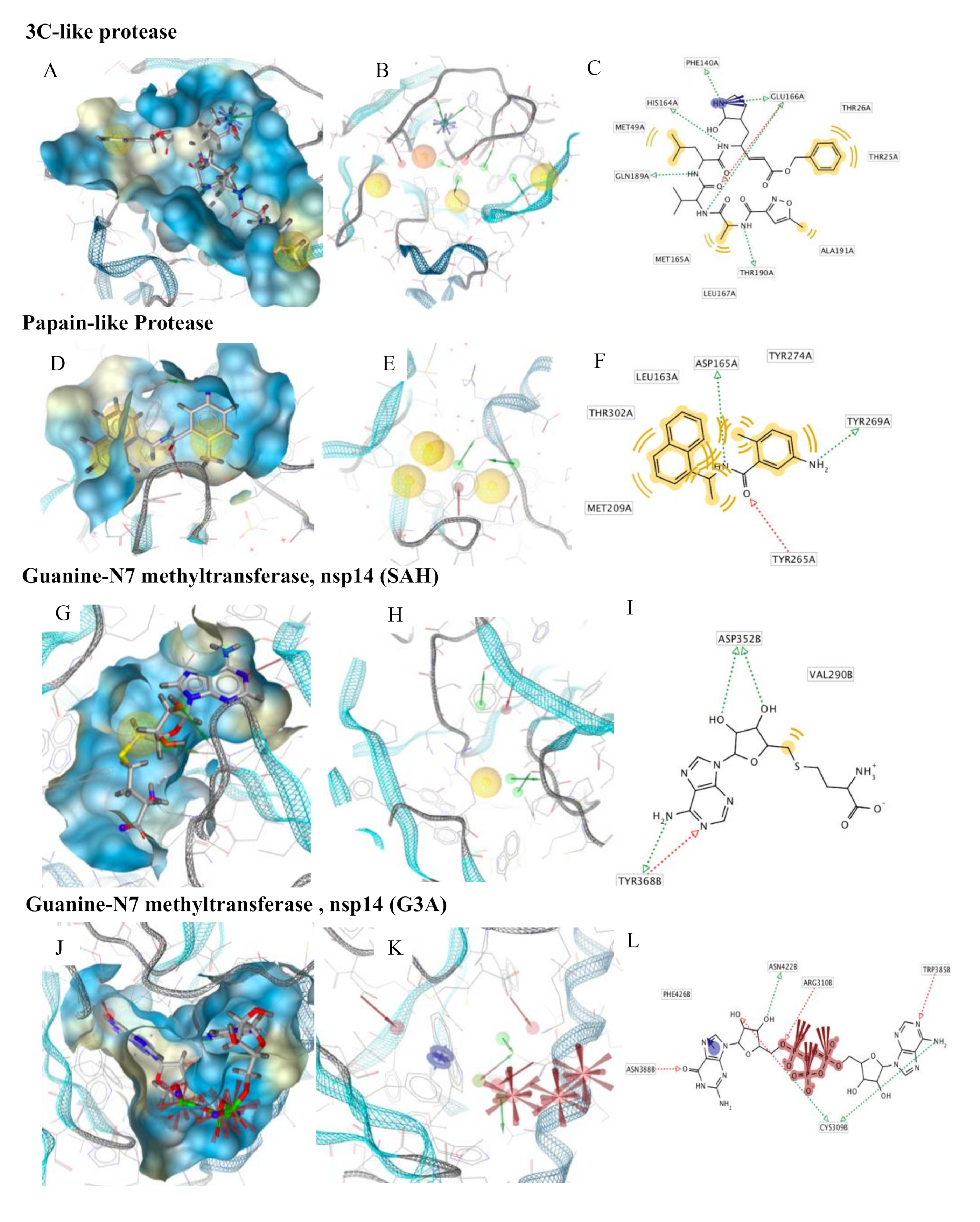{kind=link}
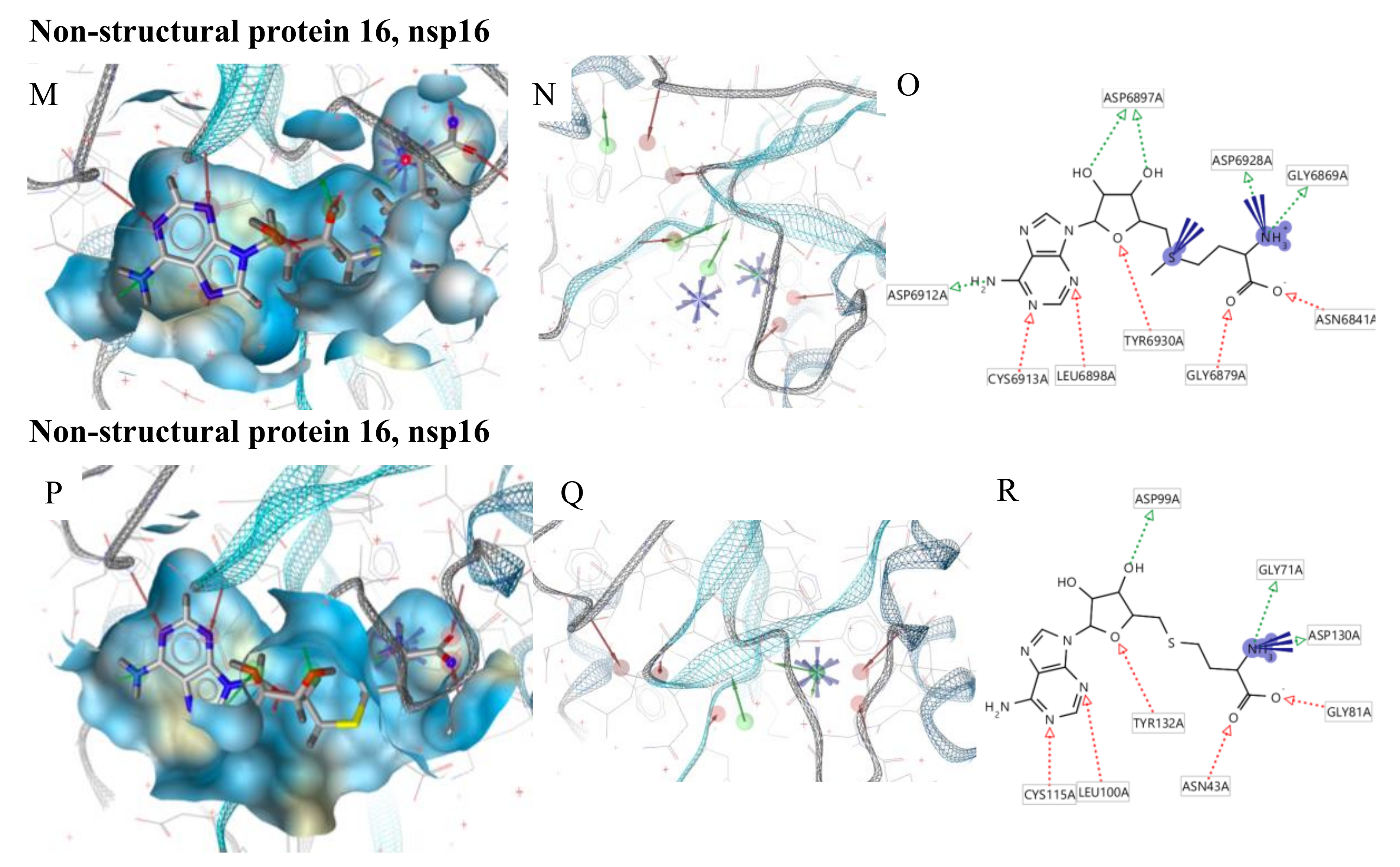{kind=link}
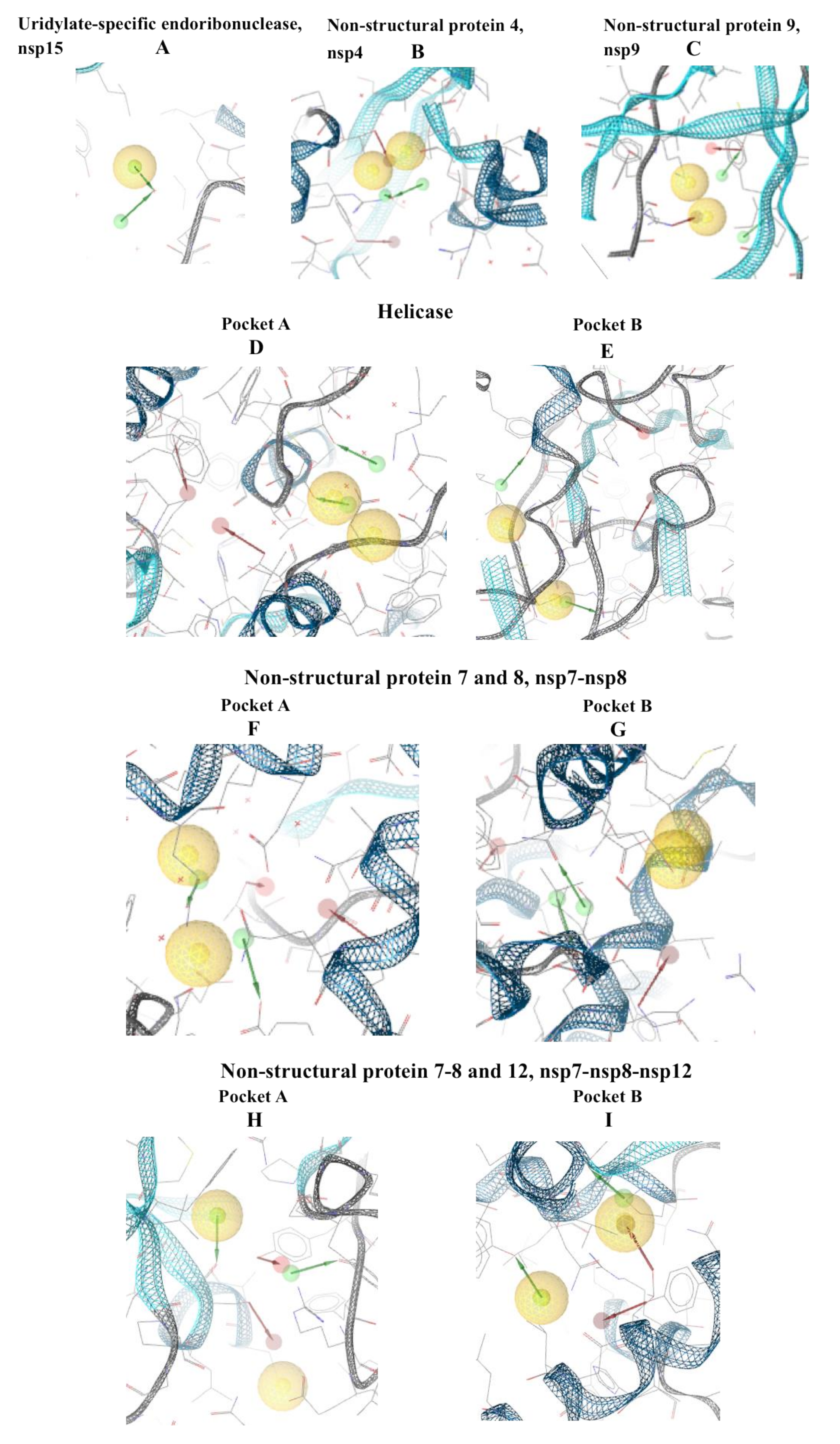{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Library Preparation
2.2. Homology Modeling and Protein Preparation
2.3. Pharmacophore Modeling
2.4. Docking
2.5. Induced-Fit Docking and MM-GBSA
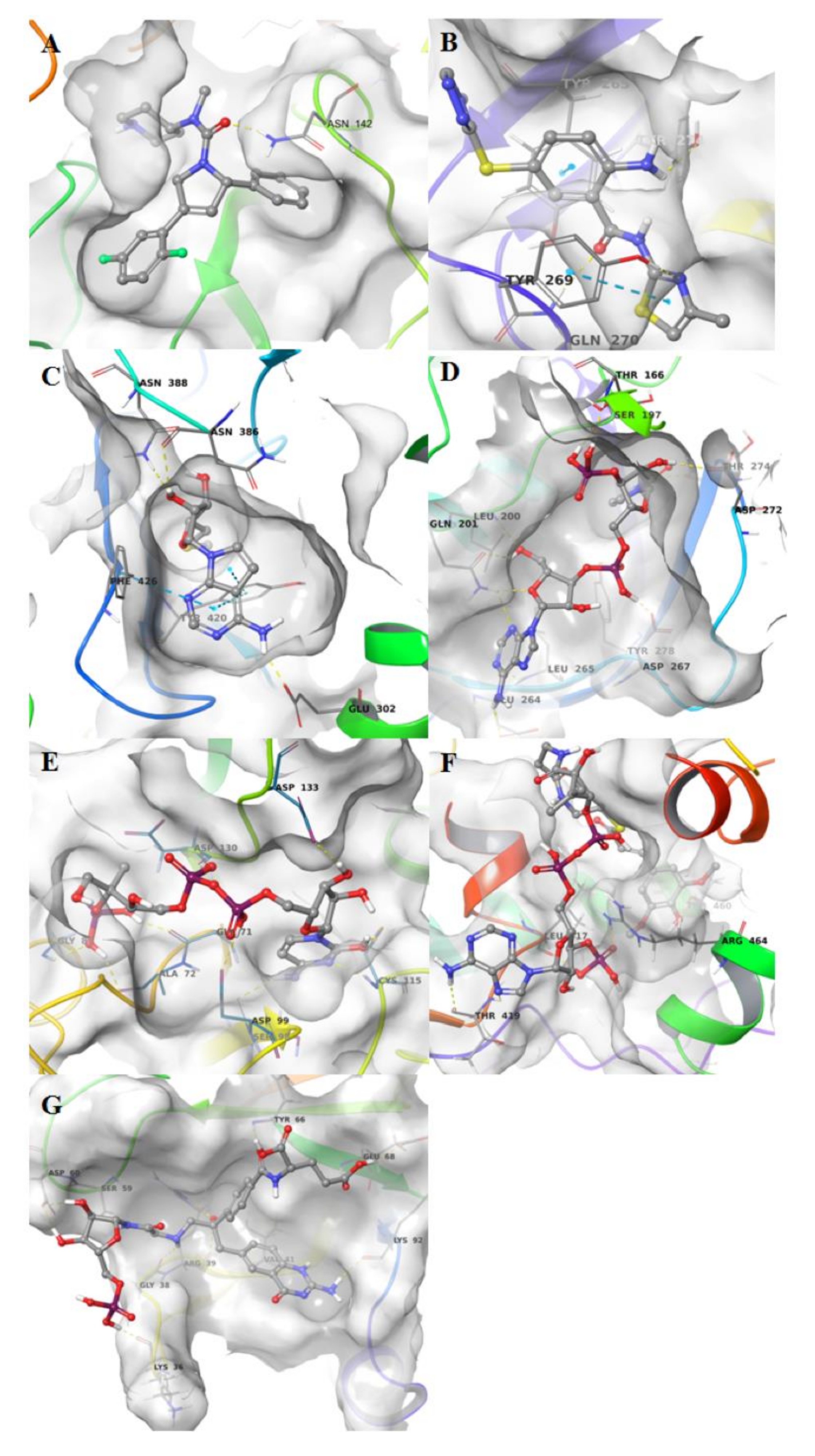
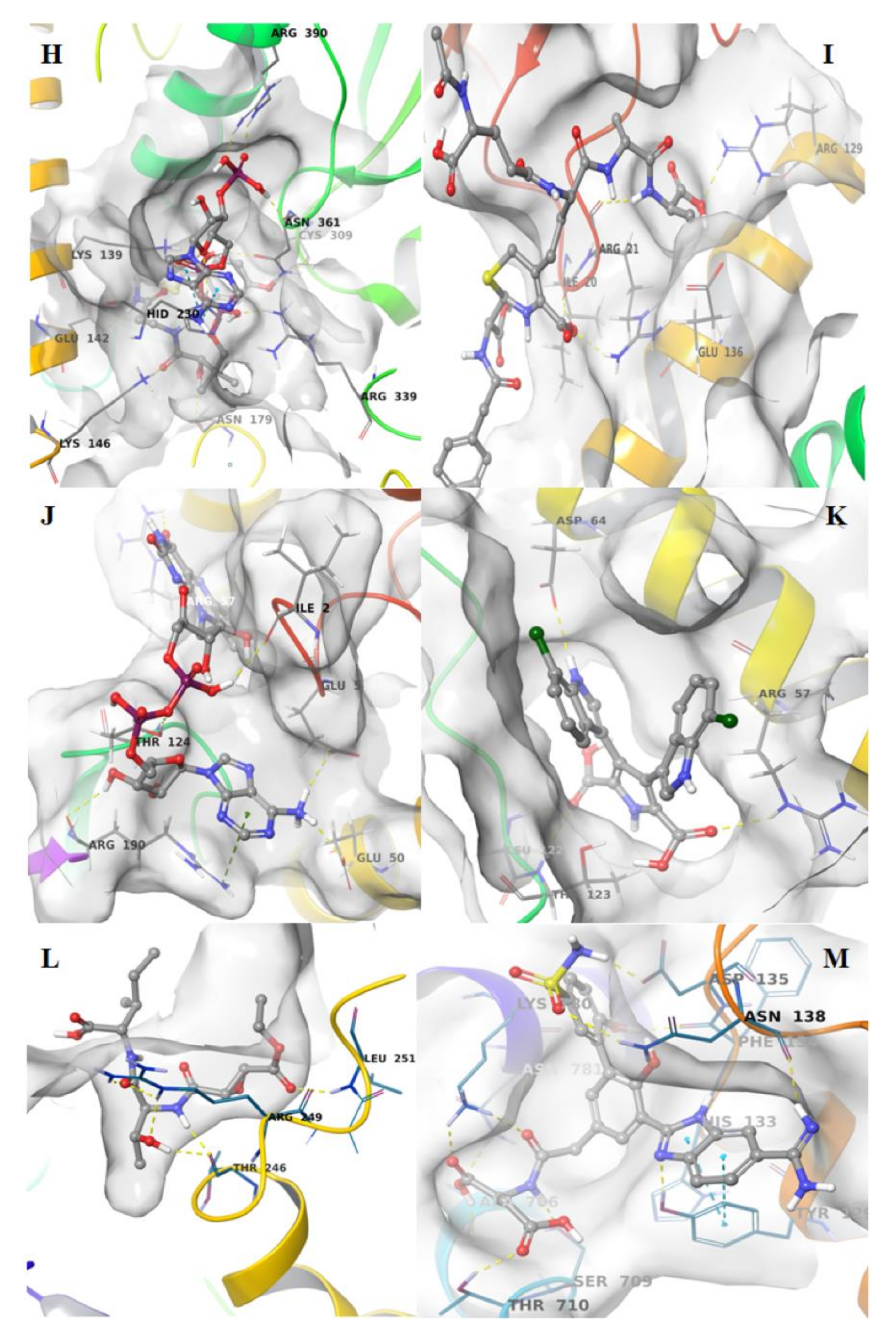
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Chen, L.; Lan, R.; Shen, R.; Li, P. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int. J. Antimicrob. Agents 2020, 2, 3–8. [Google Scholar]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Ciliberto, G.; Cardone, L. Boosting the arsenal against COVID-19 through computational drug repurposing. Drug Discov. Today 2020, 26, 946–948. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef]
- Lauria, A.; Tutone, M.; Almerico, A.M. Virtual lock-and-key approach: The in silico revival of Fischer model by means of molecular descriptors. Eur. J. Med. Chem. 2011, 46, 4274–4280. [Google Scholar] [CrossRef]
- Oprea, T.I.; Mestres, J. Drug repurposing: Far beyond new targets for old drugs. AAPS J. 2012, 14, 759–763. [Google Scholar] [CrossRef]
- Vanhaelen, Q.; Mamoshina, P.; Aliper, A.M.; Artemov, A.; Lezhnina, K.; Ozerov, I.; Labat, I.; Zhavoronkov, A. Design of efficient computational workflows for in silico drug repurposing. Drug Discov. Today 2017, 22, 210–222. [Google Scholar] [CrossRef]
- March-Vila, E.; Pinzi, L.; Sturm, N.; Tinivella, A.; Engkvist, O.; Chen, H.; Rastelli, G. On the integration of in silico drug design methods for drug repurposing. Front. Pharmacol. 2017, 8, 298. [Google Scholar] [CrossRef]
- Liu, Z.; Fang, H.; Reagan, K.; Xu, X.; Mendrick, D.L.; Slikker, W.; Tong, W. In silico drug repositioning-what we need to know. Drug Discov. Today 2013, 18, 110–115. [Google Scholar] [CrossRef]
- Oprea, T.I.; Bauman, J.E.; Bologa, C.G.; Buranda, T.; Chigaev, A.; Edwards, B.S.; Jarvik, J.W.; Gresham, H.D.; Haynes, M.K.; Hjelle, B.; et al. Drug repurposing from an academic perspective. Drug Discov. Today Ther. Strateg. 2011, 8, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Lauria, A.; Tutone, M.; Barone, G.; Almerico, A.M. Multivariate analysis in the identification of biological targets for designed molecular structures: The BIOTA protocol. Eur. J. Med. Chem. 2014, 75, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Farha, M.A.; Brown, E.D. Drug repurposing for antimicrobial discovery. Nat. Microbiol. 2019, 4, 565–577. [Google Scholar] [CrossRef]
- Sleire, L.; Førde-Tislevoll, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.Ø. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef]
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug repurposing from the perspective of pharmaceutical companies. Br. J. Pharmacol. 2018, 175, 168–180. [Google Scholar] [CrossRef]
- Tutone, M.; Perricone, U.; Almerico, A.M. Conf-VLKA: A structure-based revisitation of the Virtual Lock-and-key Approach. J. Mol. Graph. Model. 2017, 71, 50–57. [Google Scholar] [CrossRef]
- Tutone, M.; Almerico, A.M. The In Silico Fischer Lock-and-Key Model: The Combined Use of Molecular Descriptors and Docking Poses for the Repurposing of Old Drugs. Targeting Enzymes for Pharmaceutical Development. Methods Mol. Biol. 2020, 2089, 29–39. [Google Scholar]
- Gao, J.; Zhang, L.; Liu, X.; Li, F.; Ma, R.; Zhu, Z.; Zhang, J.; Wu, J.; Shi, Y.; Pan, Y.; et al. Repurposing Low-Molecular-Weight Drugs Against the Main Protease of Severe Acute Respiratory Syndrome Coronavirus 2. J. Phys. Chem. Lett. 2020. [Google Scholar] [CrossRef]
- Meyer-Almes, F.J. Repurposing approved drugs as potential inhibitors of 3CL-protease of SARS-CoV-2: Virtual screening and structure based drug design. Comput. Biol. Chem. 2020, 88. [Google Scholar] [CrossRef]
- Touret, F.; Gilles, M.; Barral, K.; Nougairède, A.; van Helden, J.; Decroly, E.; de Lamballerie, X.; Coutard, B. In vitro screening of a FDA approved chemical library reveals potential inhibitors of SARS-CoV-2 replication. Sci. Rep. 2020, 10, 13093. [Google Scholar] [CrossRef] [PubMed]
- Shyr, Z.A.; Gorshkov, K.; Chen, C.Z.; Zheng, W. Drug discovery strategies for SARS- CoV-2. J. Pharmacol. Exp. Ther. 2020, 374. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.; Di Filippo, J. In silico Drug Repurposing for COVID-19: Targeting SARS-CoV-2 Proteins through Docking and Consensus Ranking. Mol. Inform. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef]
- Ferraz, W.R.; Gomes, R.A.; S Novaes, A.L.; Goulart Trossini, G.H. Ligand and structure- based virtual screening applied to the SARS-CoV-2 main protease: An in silico repurposing study. Future Med. Chem. 2020. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell. Discov. 2020, 6, 14. [Google Scholar] [CrossRef]
- Mirza, M.U.; Froeyen, M. Structural elucidation of SARS-CoV-2 vital proteins: Computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J. Pharm. Anal. 2020. [Google Scholar] [CrossRef]
- Harrison, C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020, 38, 379–381. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Battisti, V.; Wieder, O.; Garon, A.; Seidel, T.; Urban, E.; Langer, T. A Computational Approach to Identify Potential Novel Inhibitors against the Coronavirus SARS-CoV-2. Mol. Inform. 2020. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic. Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Epik; Schrödinger: München, Germany, 2018.
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic. Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Battistuz, T.; Bhat, T.N. The protein data bank. Acta Crystallogr. Sect. D. Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Protein Preparation Wizard; Schrödinger: München, Germany, 2018.
- Schaller, D.; Šribar, D.; Noonan, T.; Lihua Deng, L.; Nguyen, T.N.; Pach, S.; Machalz, D.; Bermudez, M.; Wolber, G. Next generation 3D pharmacophore modeling. WIREsComput. Mol. Sci. 2020, 10, e1468. [Google Scholar] [CrossRef]
- Schrödinger Glide; Schrödinger: München, Germany, 2018.
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Schrödinger LLC Prime; Version 3; Schrödinger: New York, NY, USA, 2018.
- Perricone, U.; Wieder, M.; Seidel, T.; Langer, T.; Padova, A.; Almerico, A.M.; Tutone, M. A molecular dynamics–shared pharmacophore approach to boost early enrichment virtual screening. A case study on PPAR alpha. ChemMedChem 2017, 12, 1399–1407. [Google Scholar] [CrossRef]
- Almerico, A.M.; Tutone, M.; Lauria, A. Receptor-guided 3D-QSAR approach for the discovery of c-kit tyrosine kinase inhibitors. J. Mol. Model. 2012, 18, 2885–2895. [Google Scholar] [CrossRef][Green Version]
- Almerico, A.M.; Tutone, M.; Pantano, L.; Lauria, A. Molecular dynamics studies on Mdm2 complexes: An analysis of the inhibitor influence. Biochem. Biophys. Res. Commun. 2012, 424, 341–347. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Tutone, M.; Pibiri, I.; Lentini, L.; Pace, A.; Almerico, A.M. Deciphering the Nonsense Readthrough Mechanism of Action of Ataluren: An in Silico Compared Study. ACS Med. Chem. Lett. 2019, 10, 522–527. [Google Scholar] [CrossRef]
- Tutone, M.; Virzì, A.; Almerico, A.M. Reverse screening on indicaxanthin from Opuntia ficus-indica as natural chemoactive and chemopreventive agent. J. Theor. Biol. 2018, 455, 147–160. [Google Scholar] [CrossRef]
- Hou, T.; Yu, R. Molecular dynamics and free energy studies on the wild-type and double mutant HIV-1 protease complexed with amprenavir and two amprenavir-related inhibitors: Mechanism for binding and drug resistance. J. Med. Chem. 2007, 50, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in Reproducing the Binding Free Energies to HIV-1 RT of TIBO Derivatives and Predicting the Binding Mode to HIV-1 RT of Efavirenz by Docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Zia, K.; Ashraf, S.; Uddin, R.; Ul-Haq, Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.; Das, A.; Prashar, V.; Kumar, M. Potential inhibitors against papain-like protease of novel coronavirus (SARS-CoV-2) from FDA approved drugs. Chemrxiv. Org. 2020. [Google Scholar] [CrossRef]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.A.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug targets and potential treatments. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Xu, X.; Lou, Z.; Ma, Y.; Chen, X.; Yang, Z.; Tong, X.; Zhao, Q.; Xu, Y.; Deng, H.; Bartlam, M.; et al. Crystal Structure of the C-Terminal Cytoplasmic Domain of Non-Structural Protein 4 from Mouse Hepatitis Virus A59. PLoS ONE 2009, 4, e6217. [Google Scholar] [CrossRef]
- Sutton, G.; Fry, E.; Carter, L.; Sainsbury, S.; Walter, T.; Nettleship, J.; Berrow, N.; Owens, R.; Gilbert, R.; Davidson, A.; et al. The nsp9 Replicase Protein of SARS-Coronavirus, Structure and Functional Insights. Structure 2004, 12, 341–353. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Culletta, G.; Gulotta, M.R.; Perricone, U.; Zappalà, M.; Almerico, A.M.; Tutone, M. Exploring the SARS-CoV-2 Proteome in the Search of Potential Inhibitors via Structure-Based Pharmacophore Modeling/Docking Approach. Computation 2020, 8, 77. https://doi.org/10.3390/computation8030077
Culletta G, Gulotta MR, Perricone U, Zappalà M, Almerico AM, Tutone M. Exploring the SARS-CoV-2 Proteome in the Search of Potential Inhibitors via Structure-Based Pharmacophore Modeling/Docking Approach. Computation. 2020; 8(3):77. https://doi.org/10.3390/computation8030077
Chicago/Turabian StyleCulletta, Giulia, Maria Rita Gulotta, Ugo Perricone, Maria Zappalà, Anna Maria Almerico, and Marco Tutone. 2020. "Exploring the SARS-CoV-2 Proteome in the Search of Potential Inhibitors via Structure-Based Pharmacophore Modeling/Docking Approach" Computation 8, no. 3: 77. https://doi.org/10.3390/computation8030077
APA StyleCulletta, G., Gulotta, M. R., Perricone, U., Zappalà, M., Almerico, A. M., & Tutone, M. (2020). Exploring the SARS-CoV-2 Proteome in the Search of Potential Inhibitors via Structure-Based Pharmacophore Modeling/Docking Approach. Computation, 8(3), 77. https://doi.org/10.3390/computation8030077