
Extracting Conformational Ensembles of Small Molecules from Molecular Dynamics Simulations: Ampicillin as a Test Case


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Methods
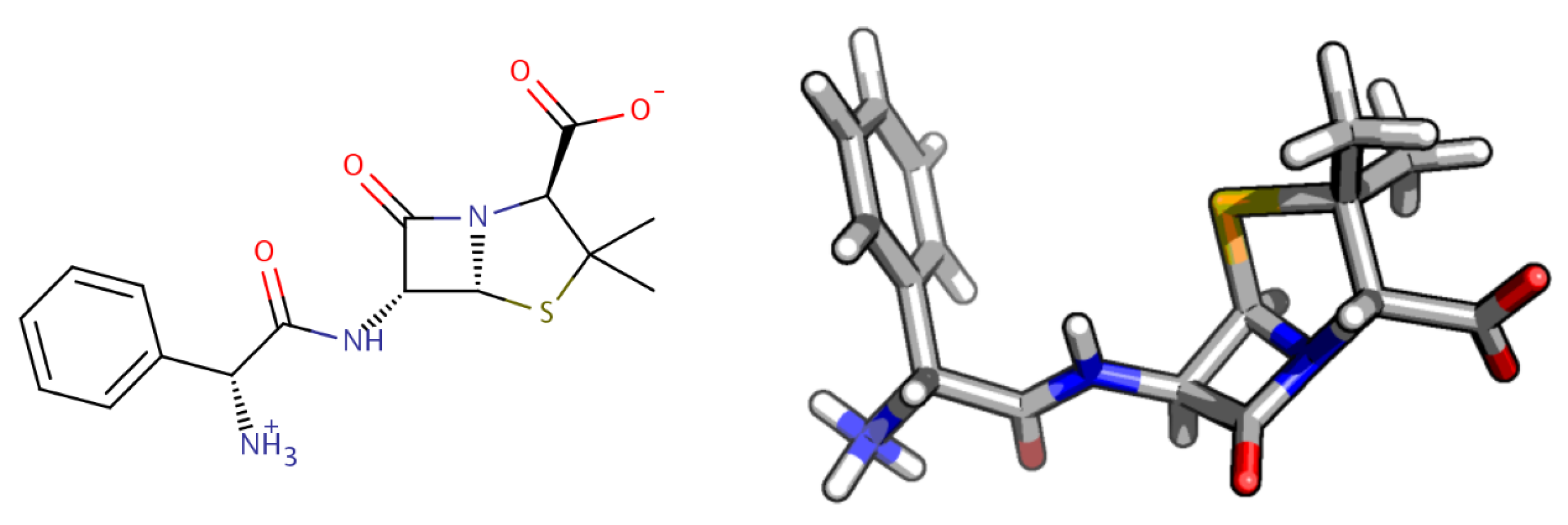
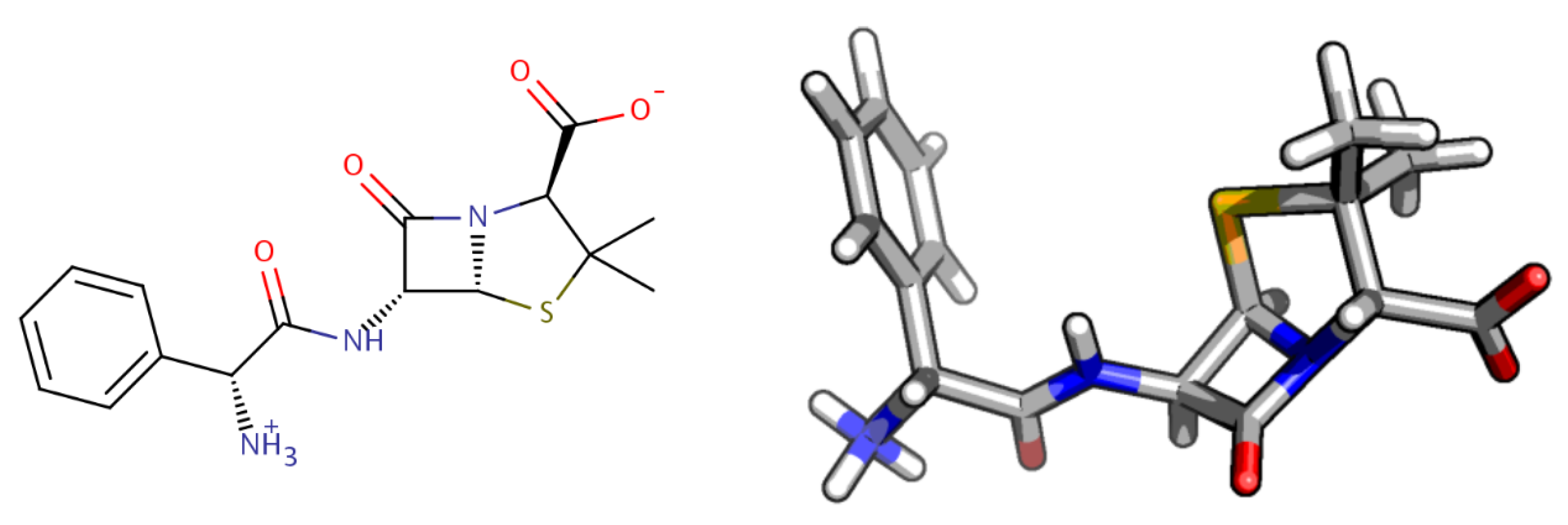
2.1. Molecular Characterization and Quantum-Chemistry Calculations
2.2. MD Simulations and Post-Processing of the MD Trajectories
2.3. REMD Calculations and Conformer Generation Methods Adopted
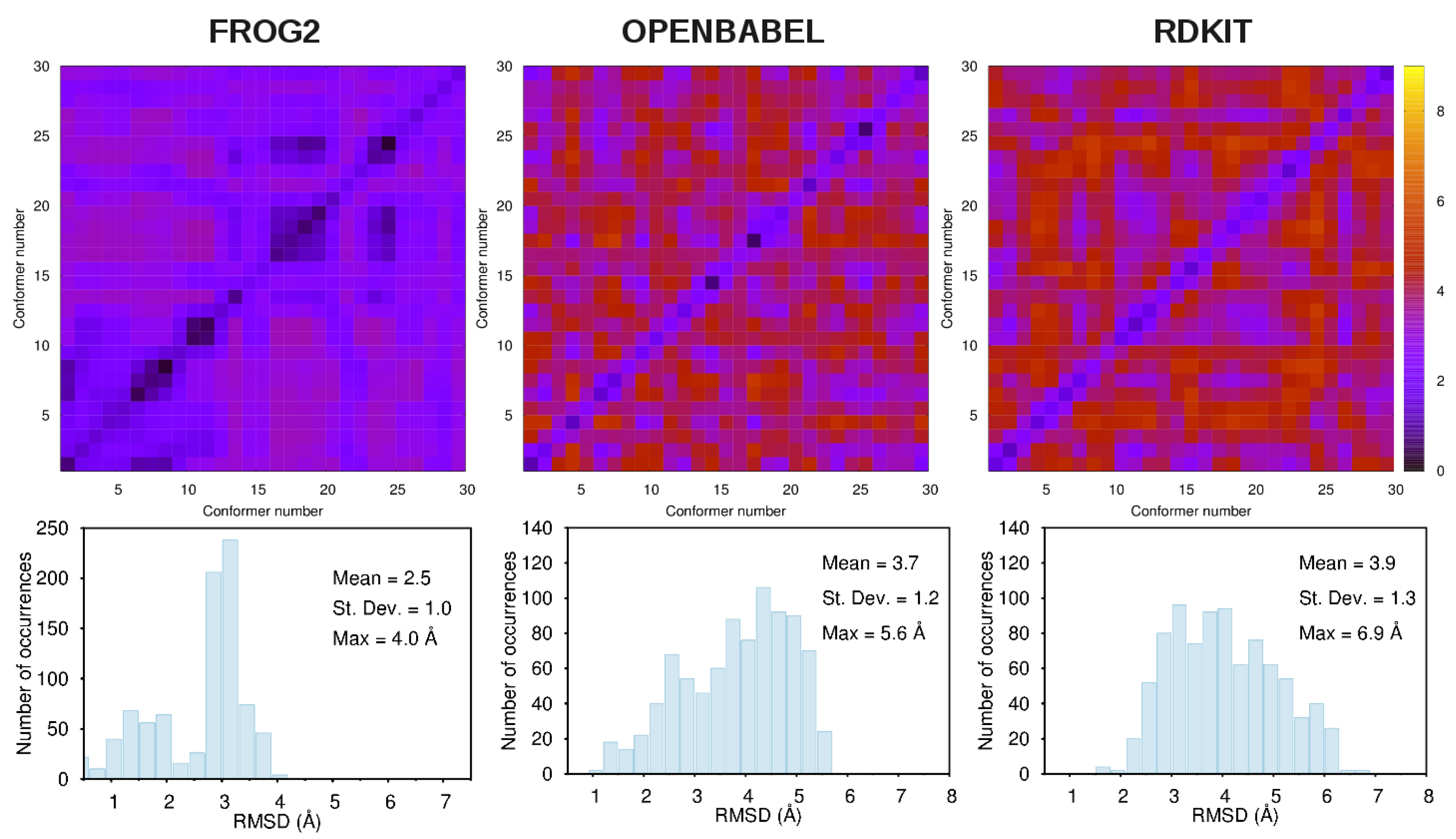
- frog2: We loaded the 3D structure data file CID_6249.sdf taken from PubChem [43] in the web portal and adopted default settings. The server returned an ensemble of diverse conformers generated using a two-stage Monte-Carlo approach in the dihedral space.
- openbabel: We used the same structure CID_6249.sdf as an input to the genetic algorithm code implemented in the program. This is a stochastic conformer generator producing a population of conformers that arrive iteratively at an optimal solution in terms of root-mean-squared diversity (we used a cutoff of 2.0 Å), after a series of generations.
- rdkit: We used the python script provided in the user manual by loading the CID_6249 structure in .mol2format. The script generates the desired number of conformers and, for each of them, performs energy minimizations with the Universal Force Field [67].
3. Results and Discussion
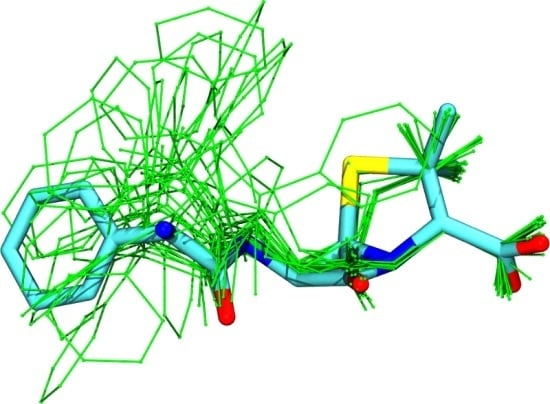
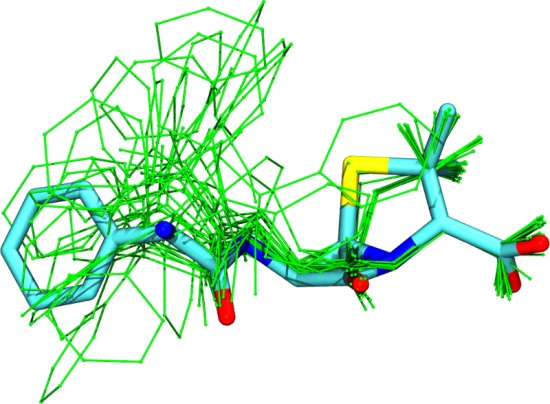
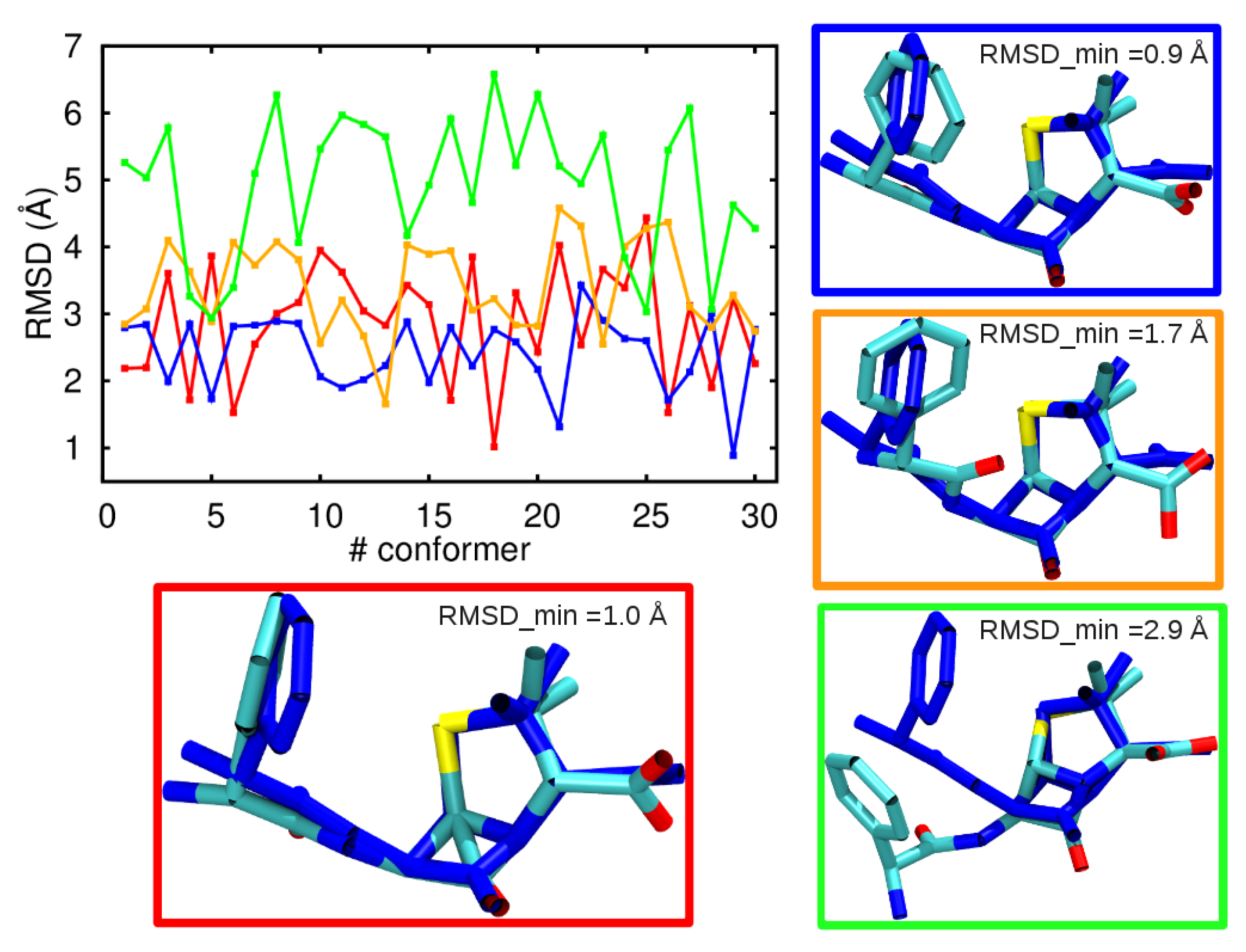
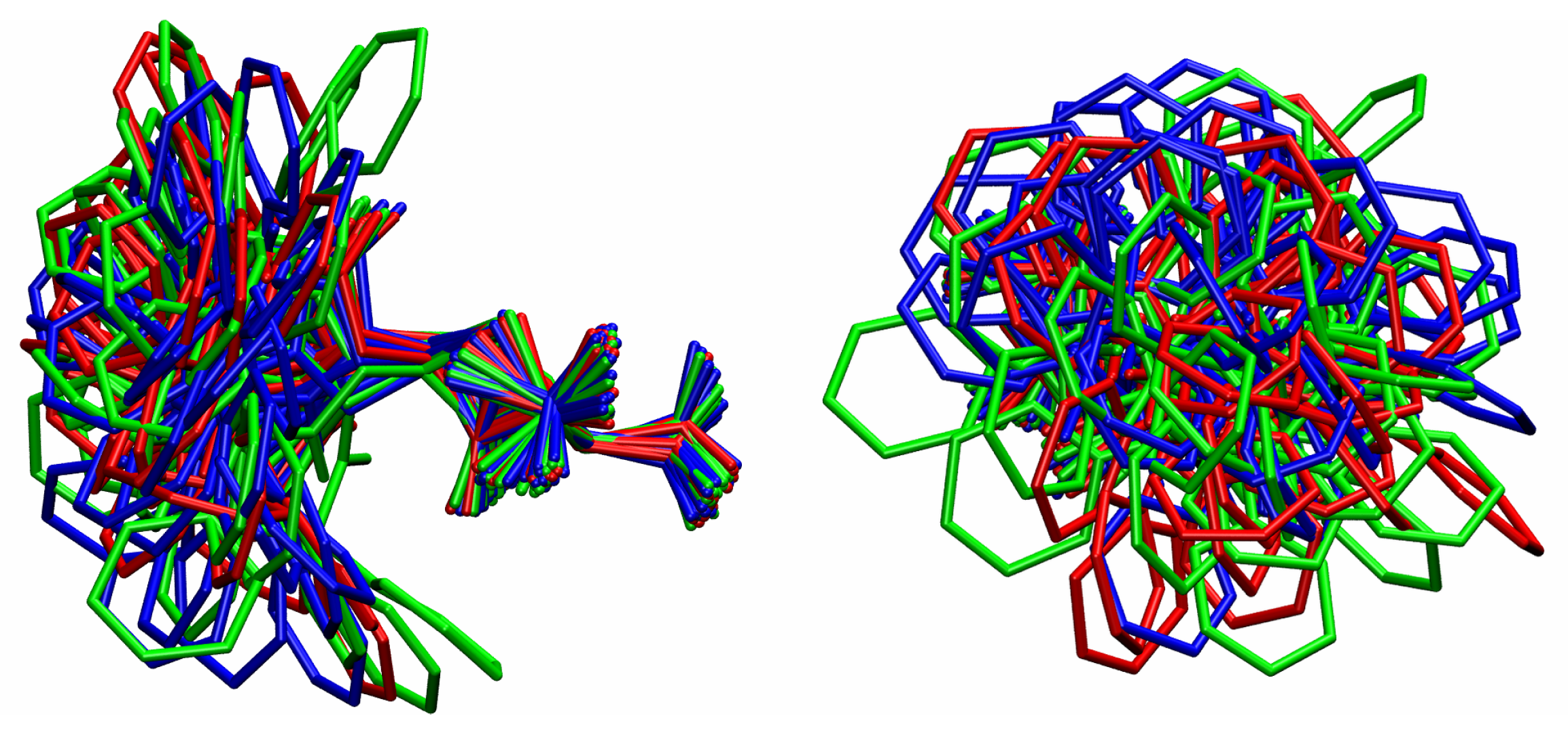
3.1. Comparison among Different Conformational Ensembles
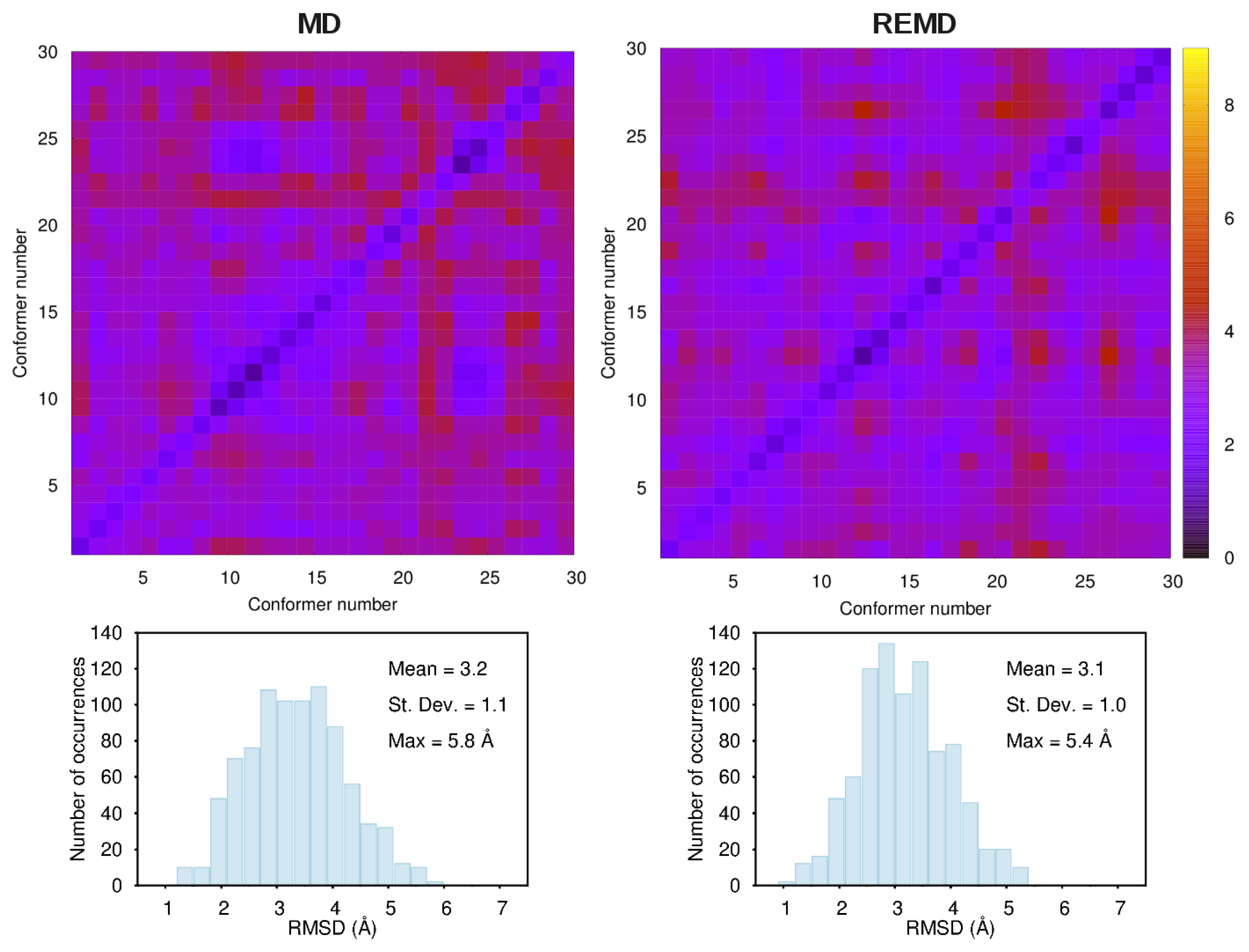
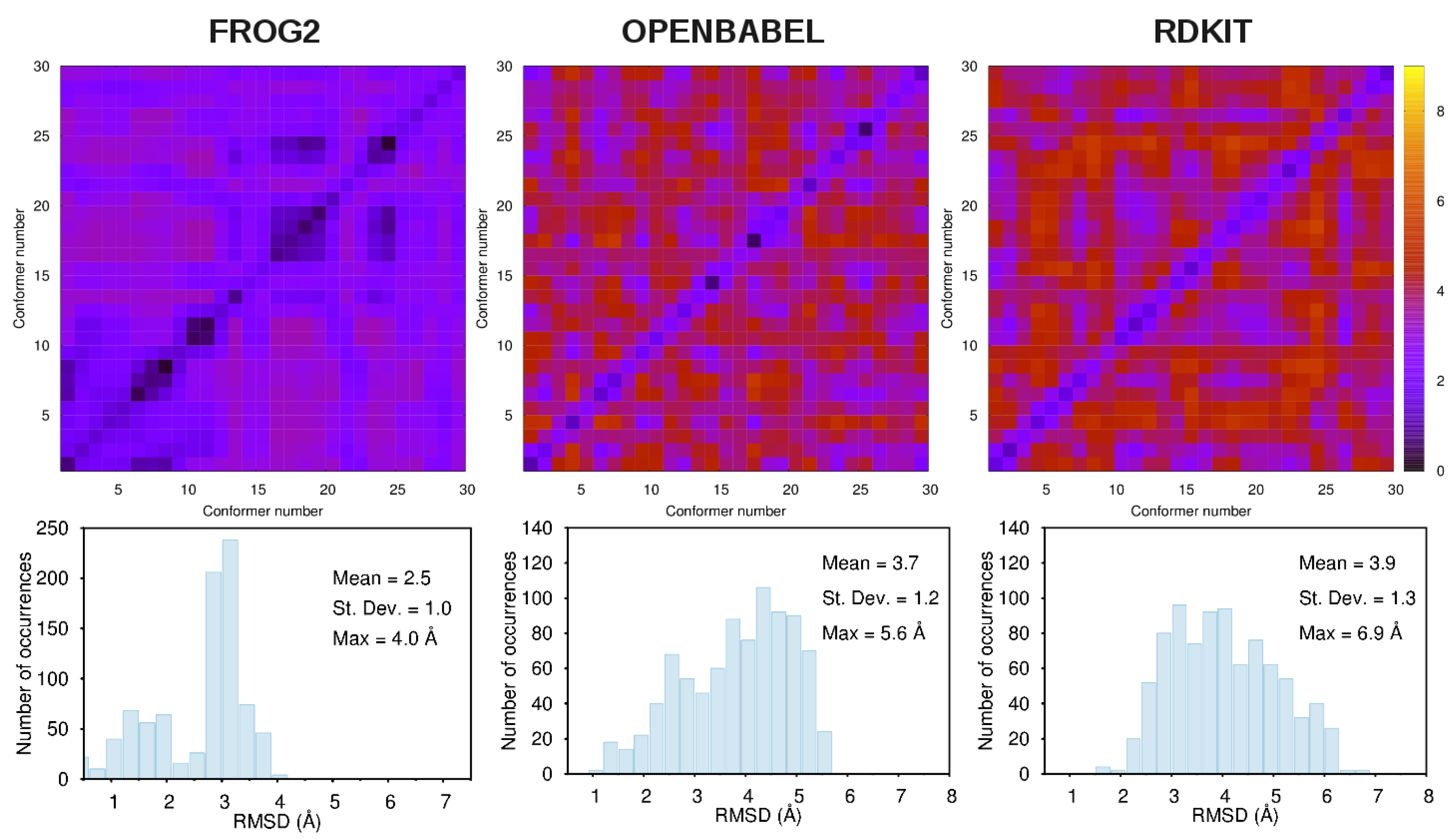
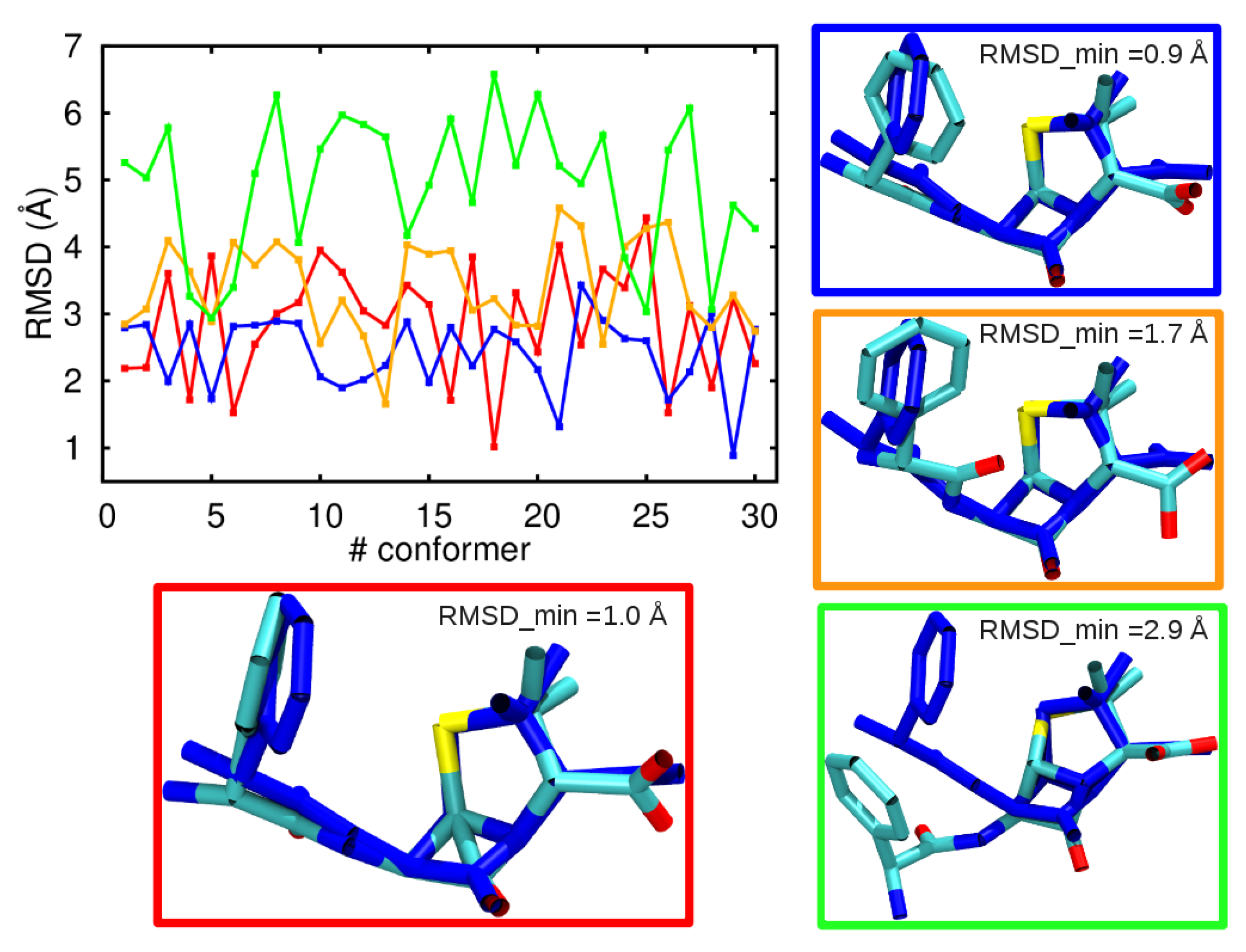
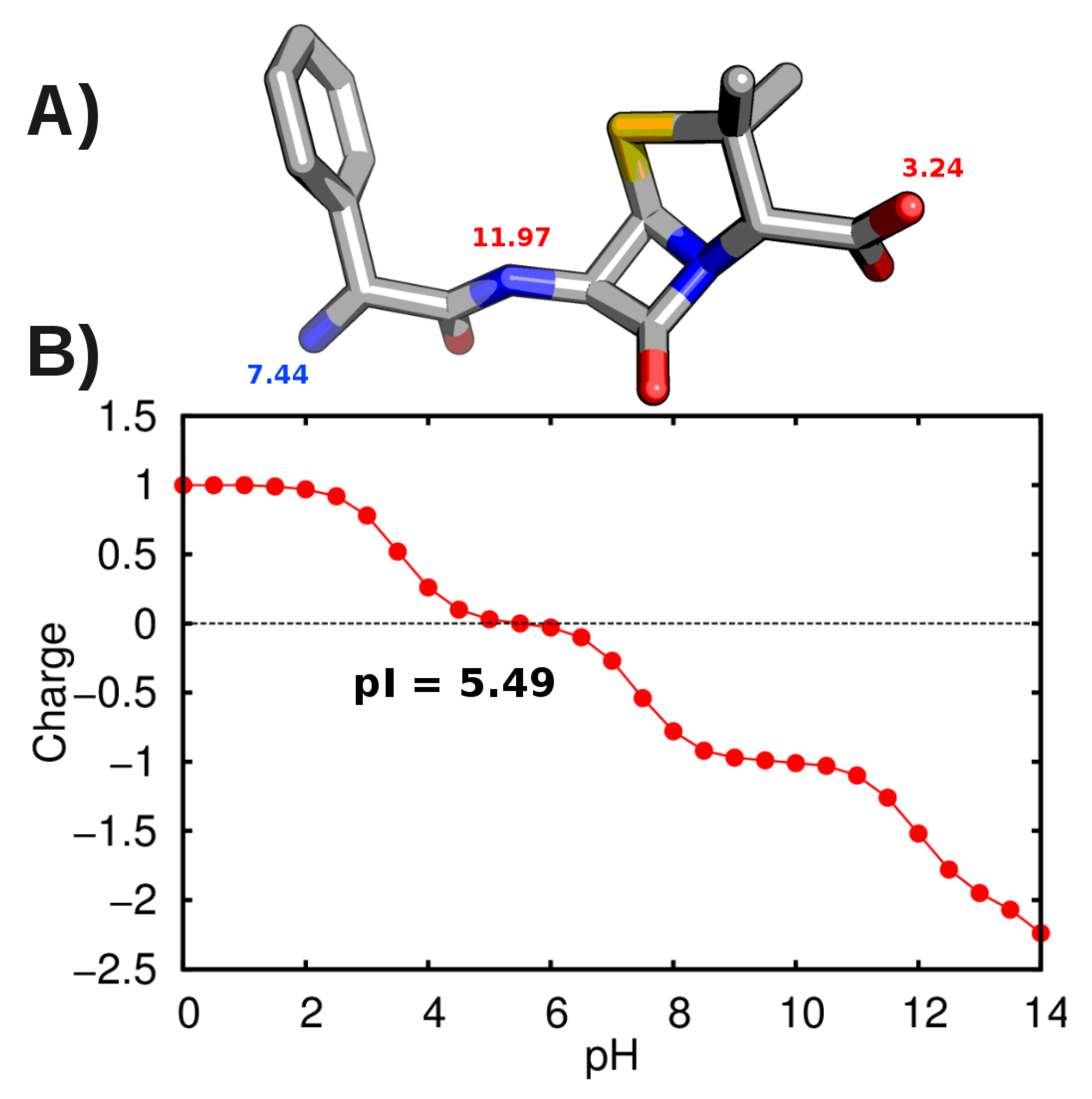
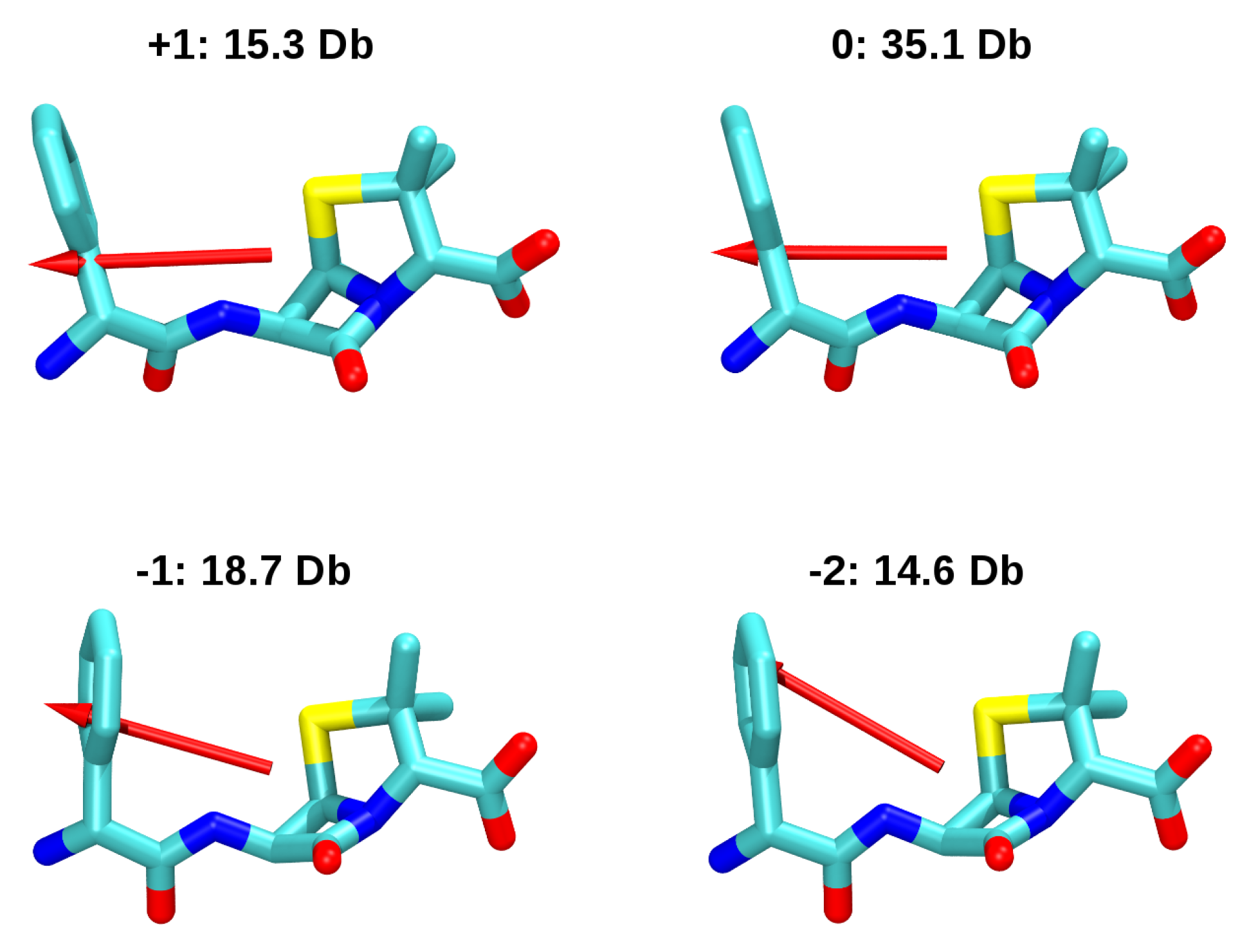
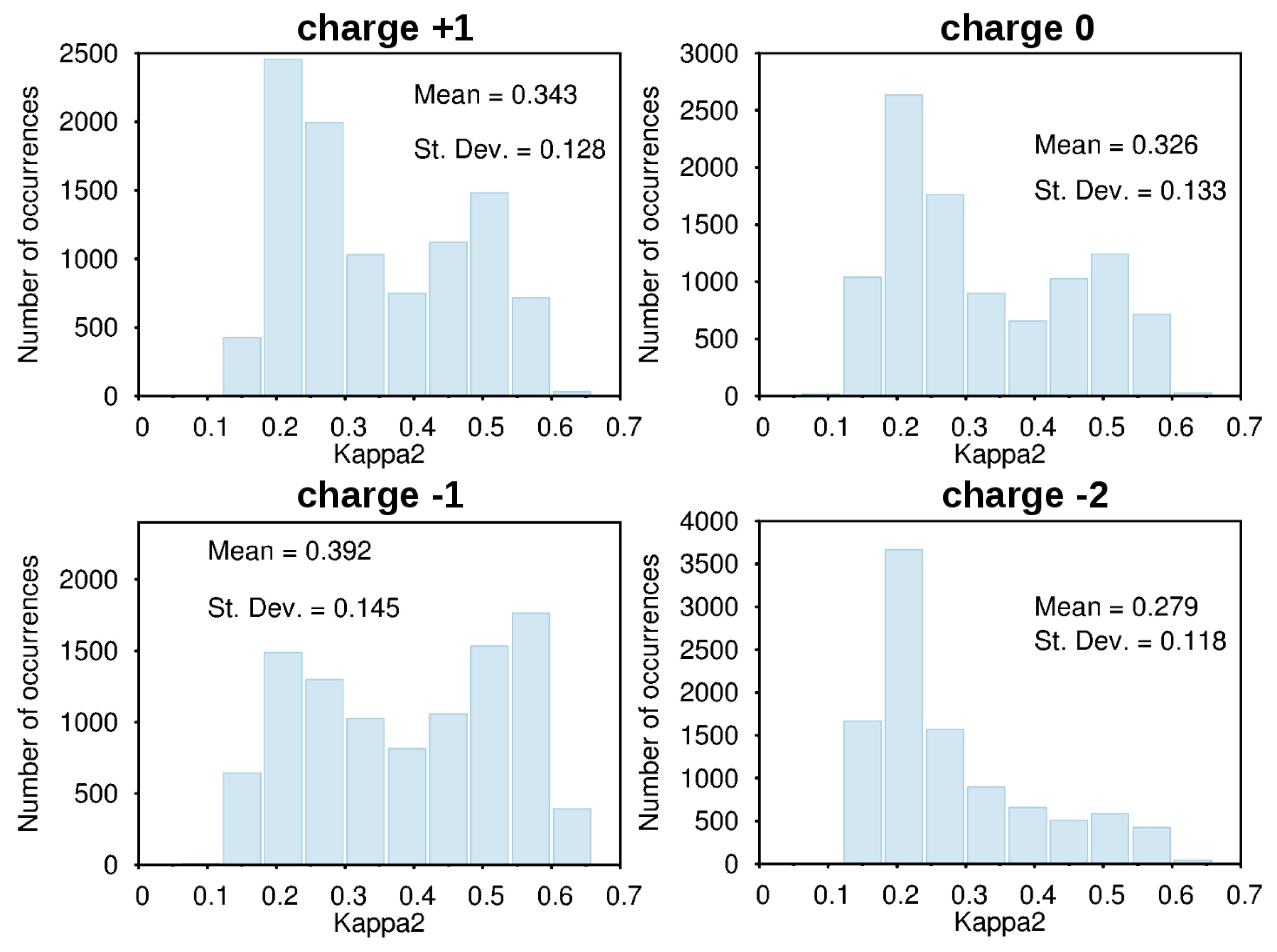
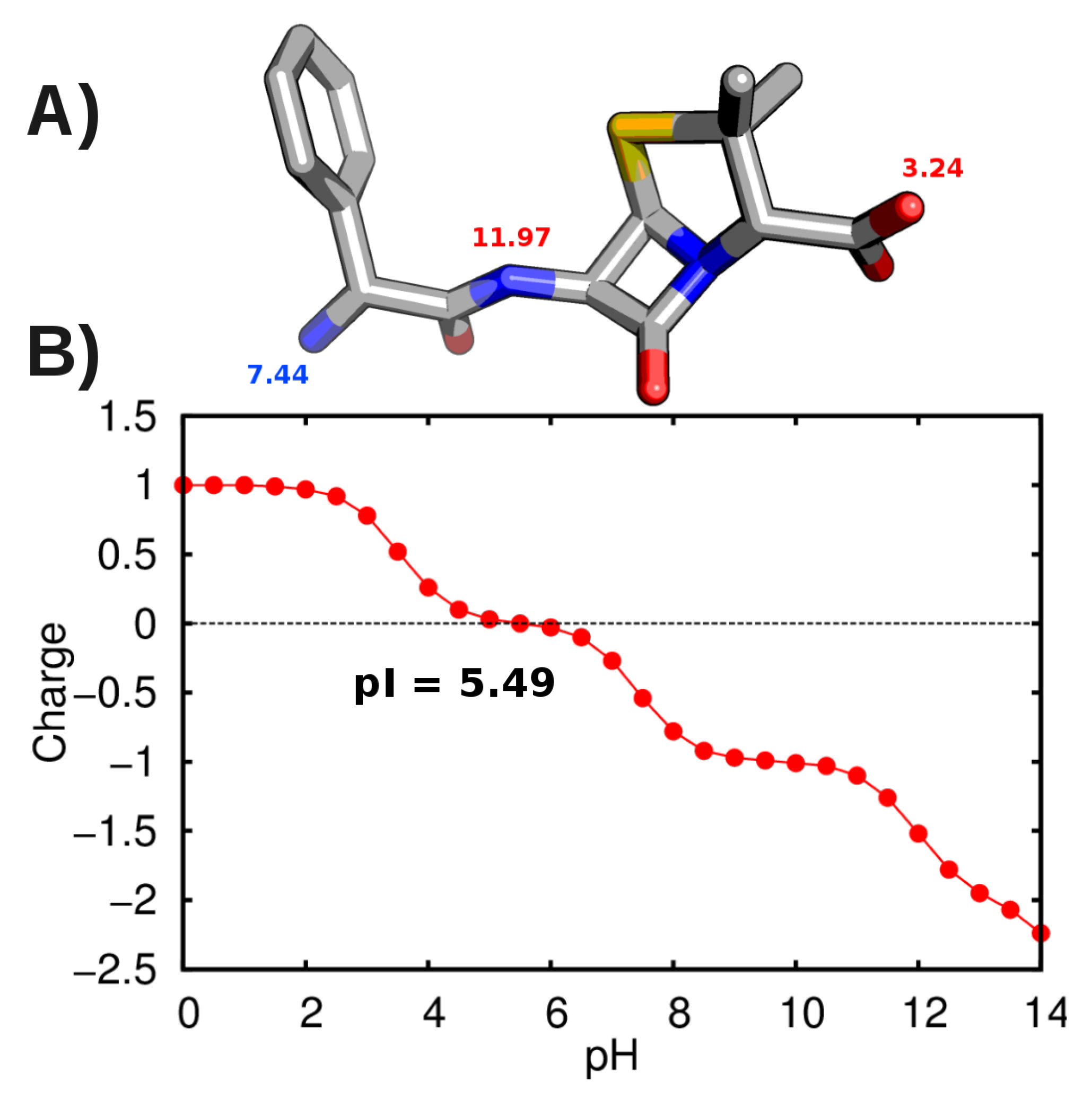
3.2. Force-Field Parameters and Molecular Properties of Different Microspecies
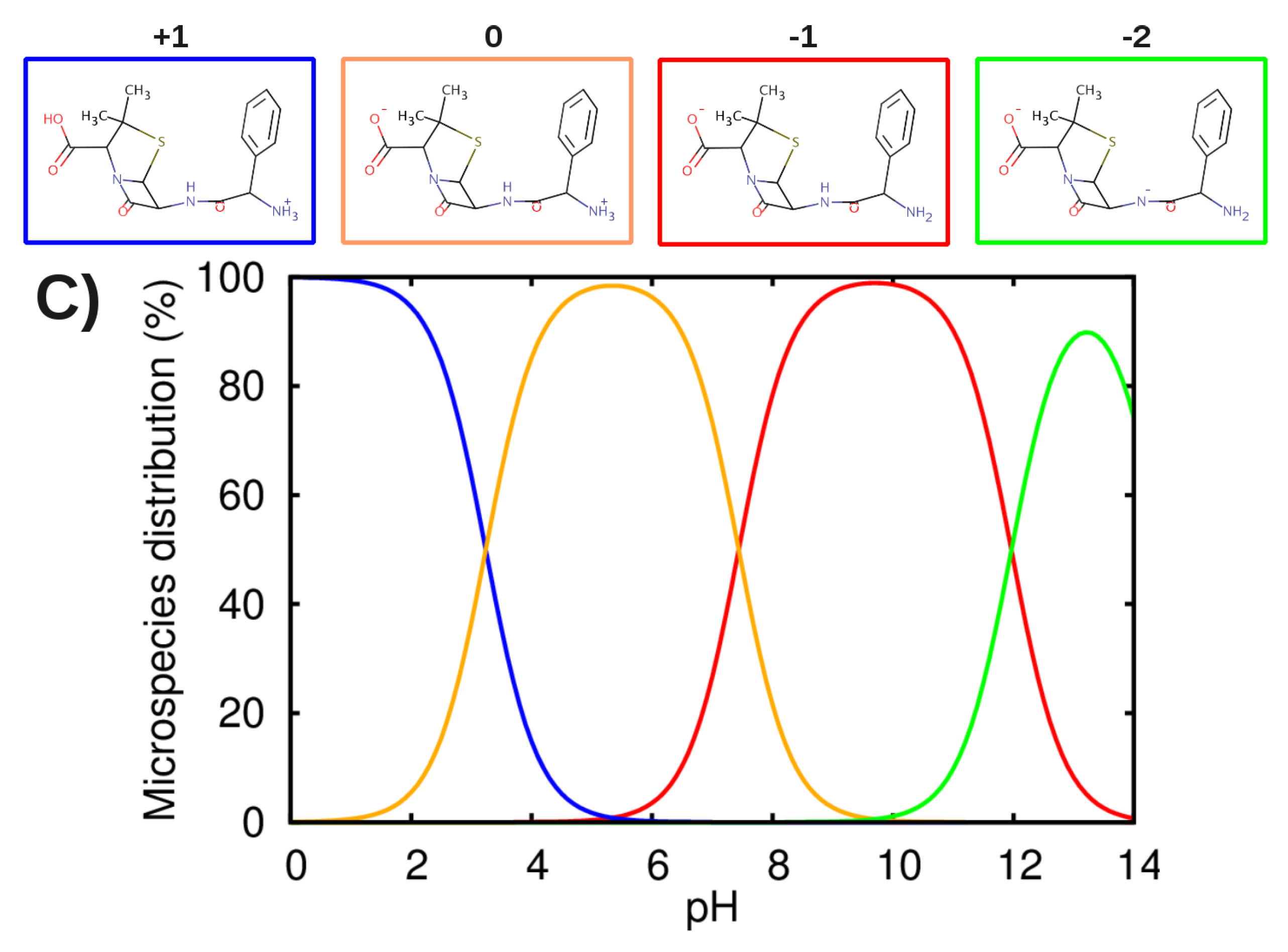
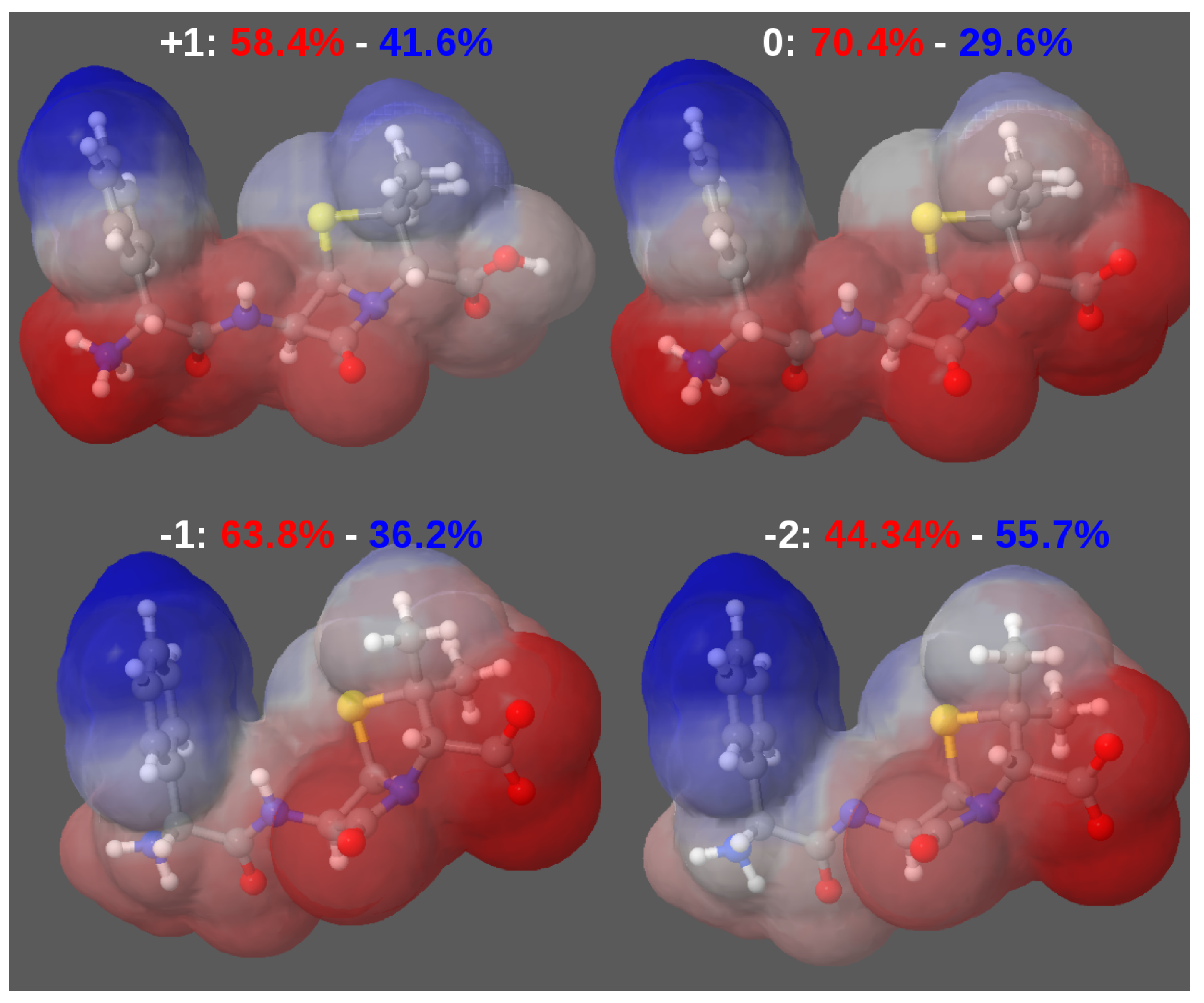
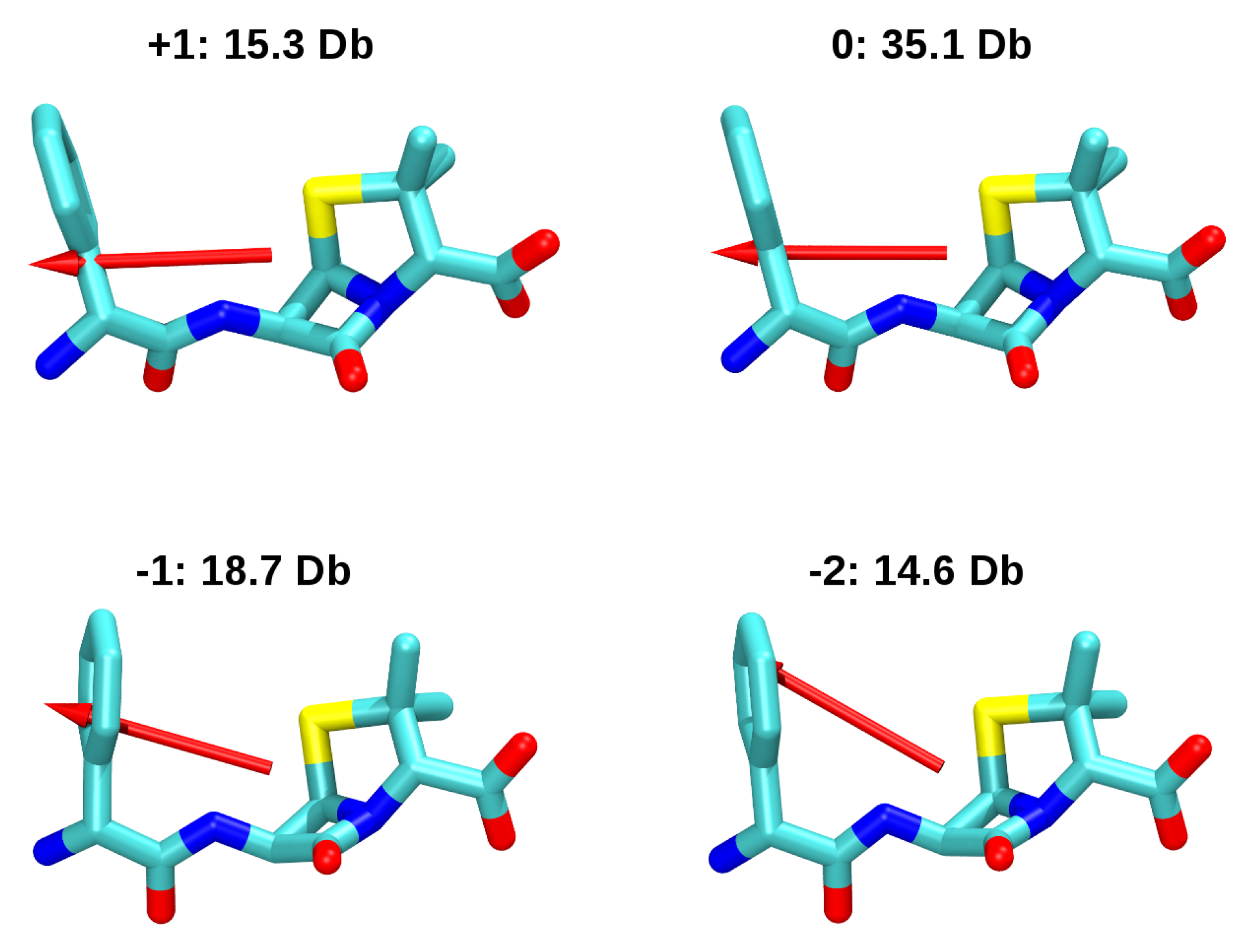
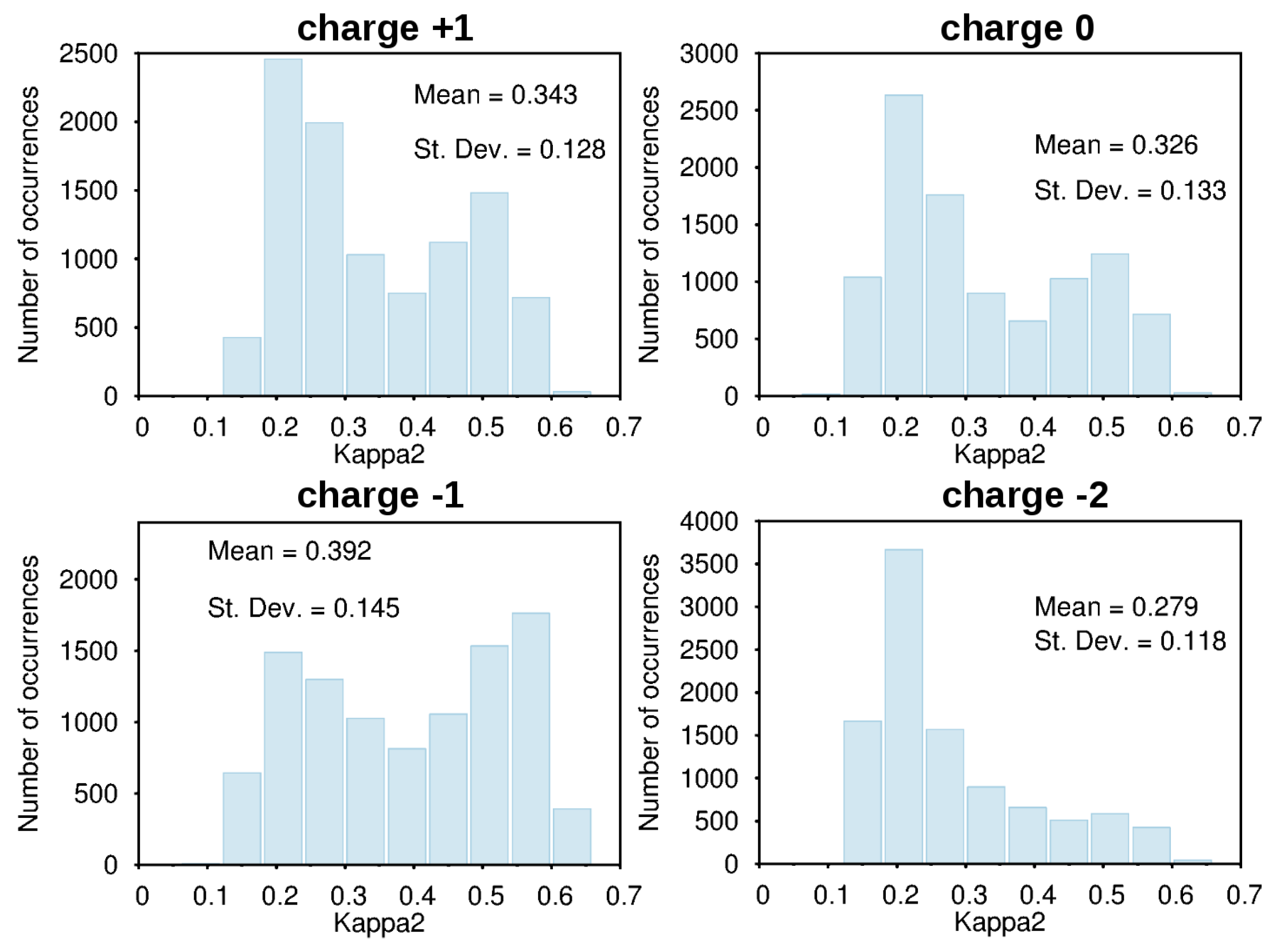
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
Appendix
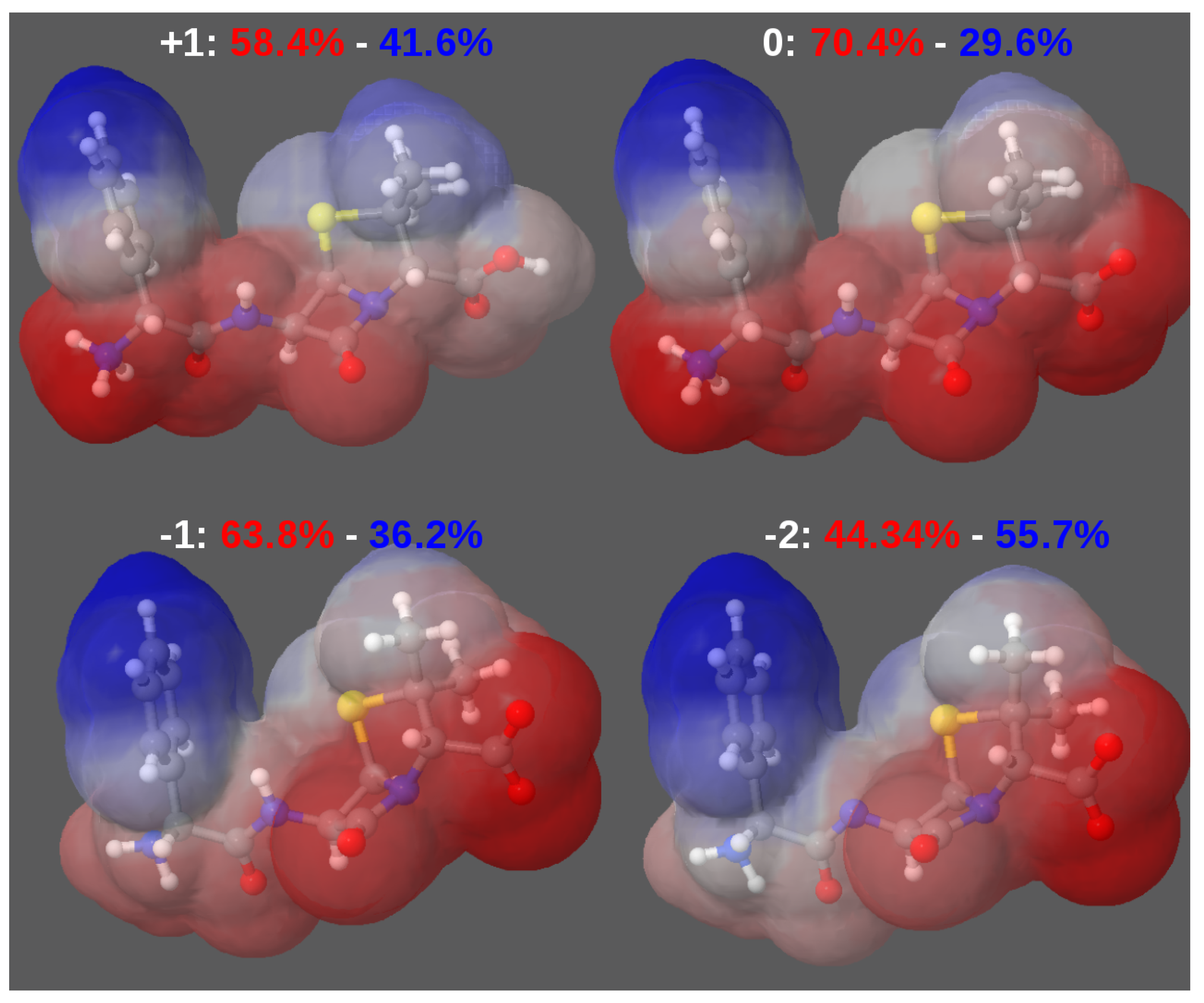
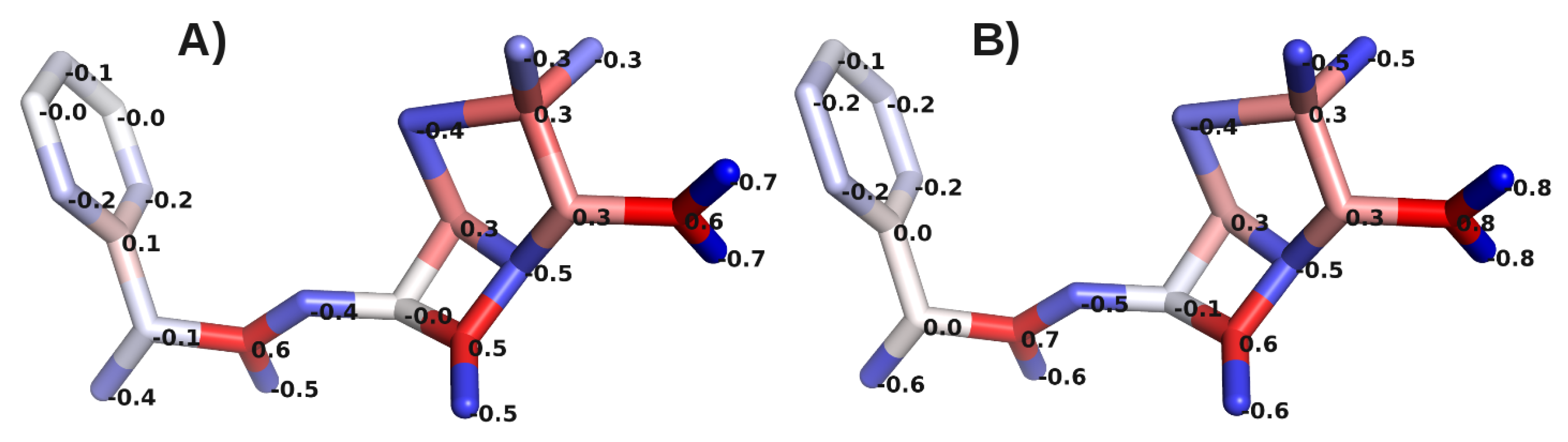
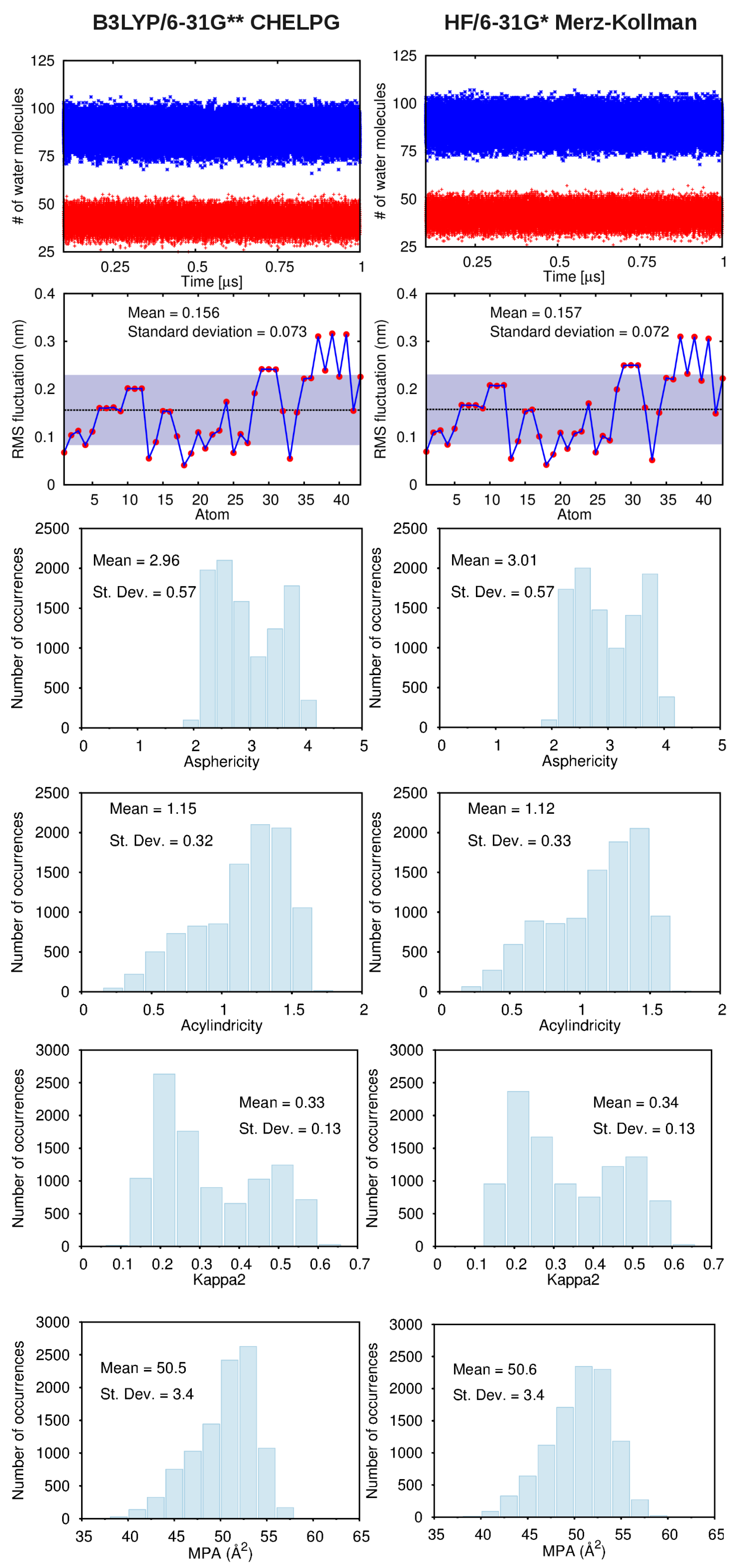
A. Impact of Atomic Partial Charges
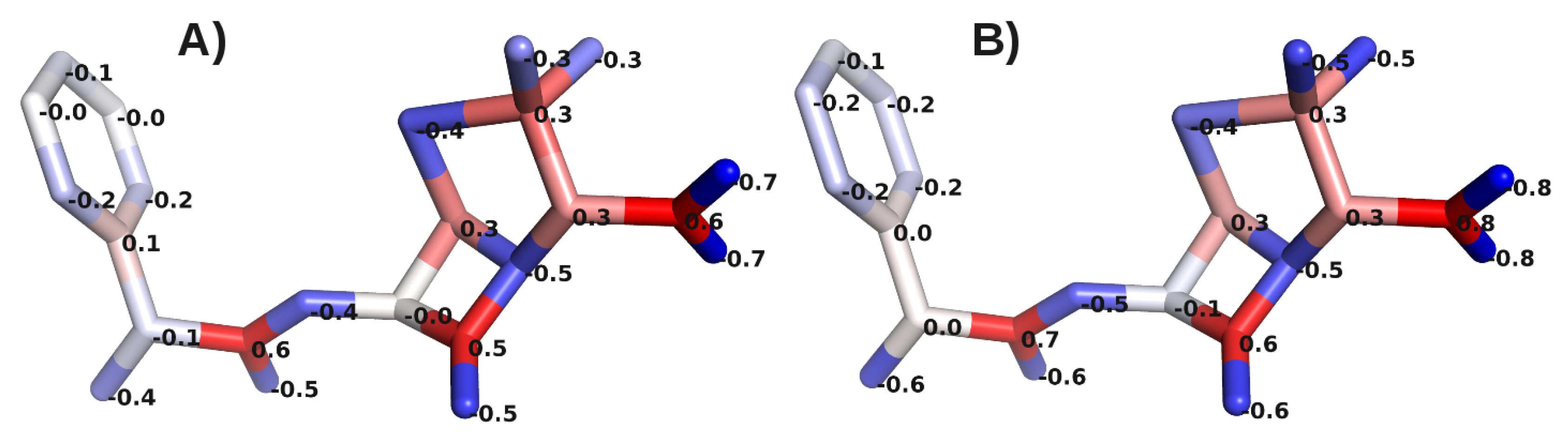
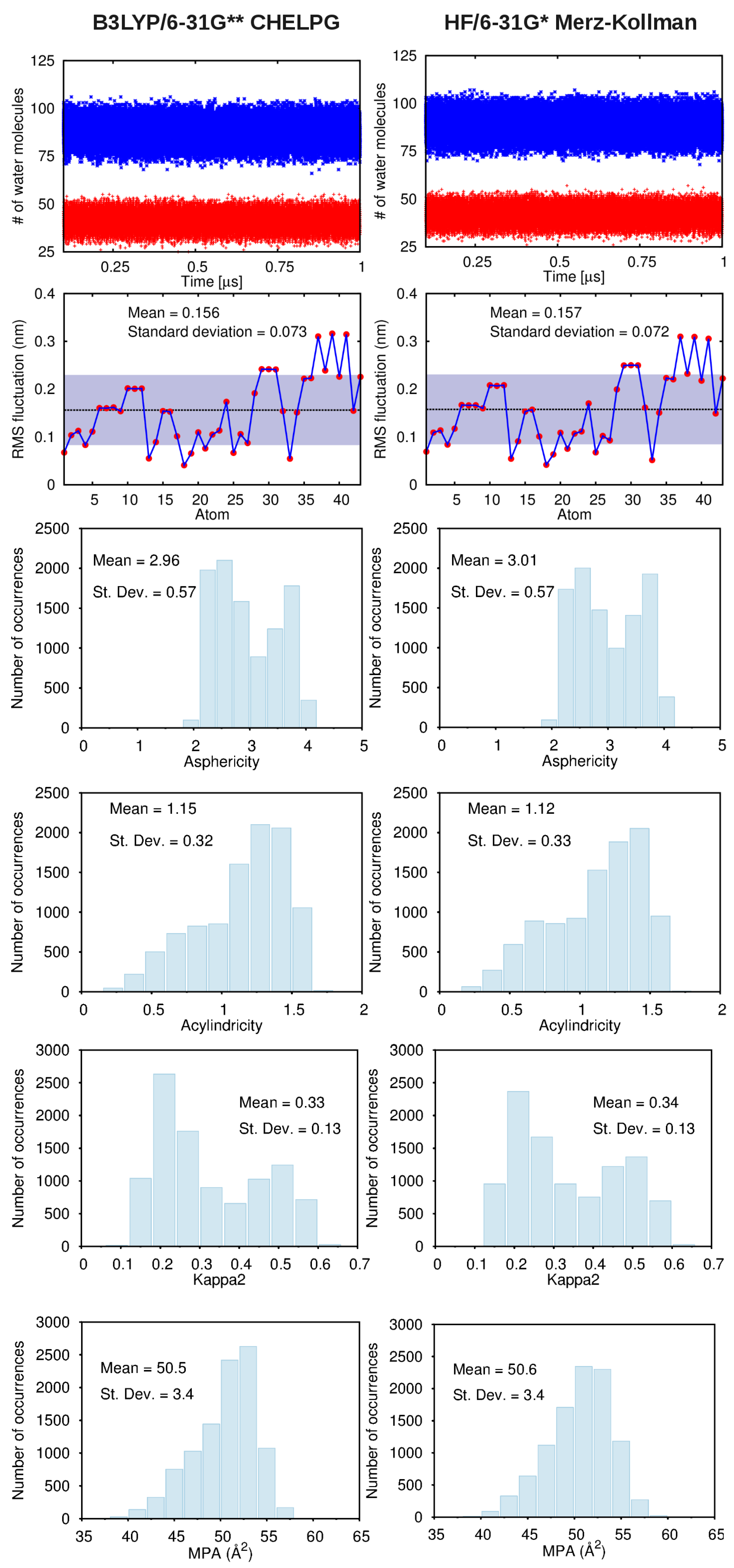
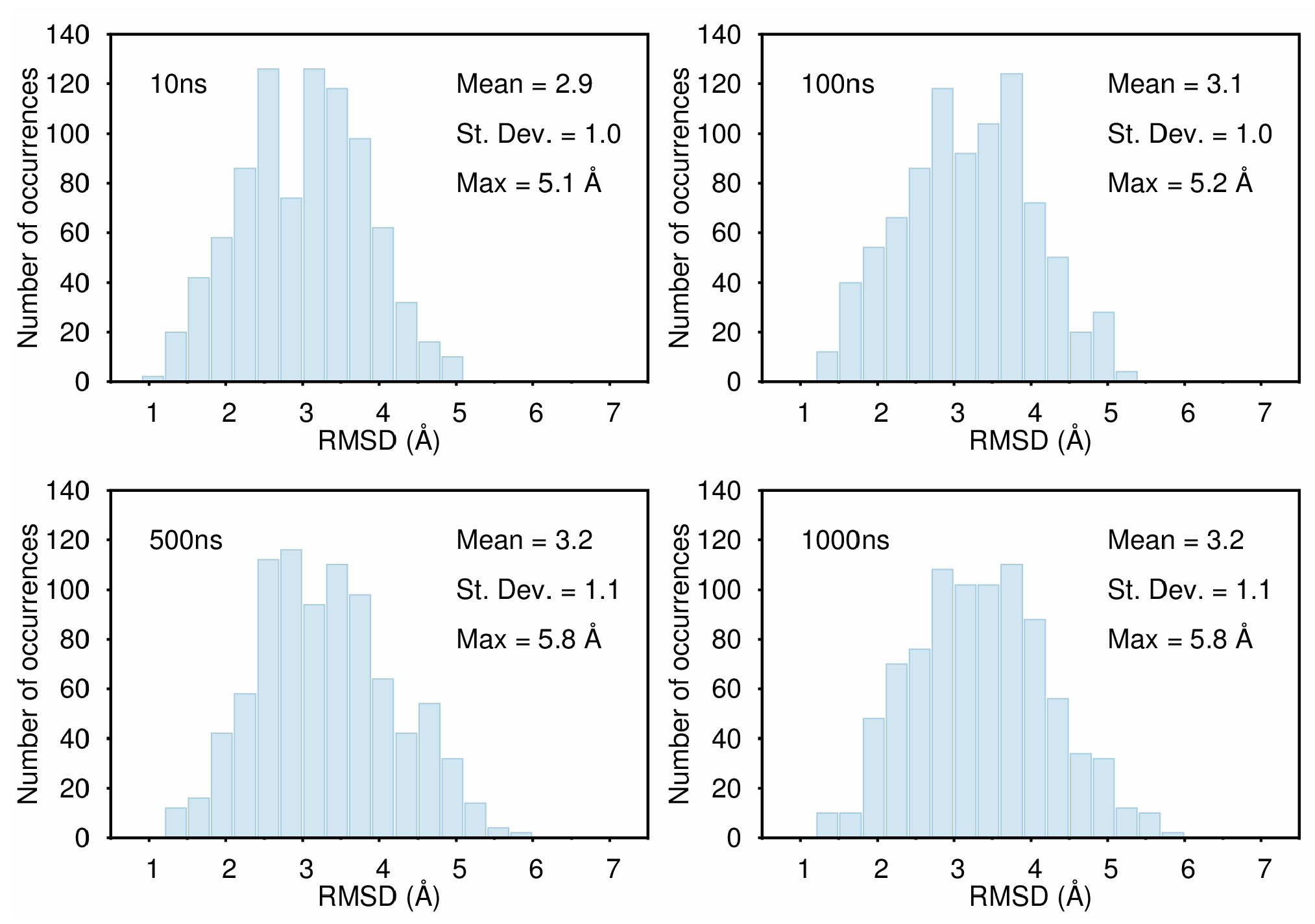
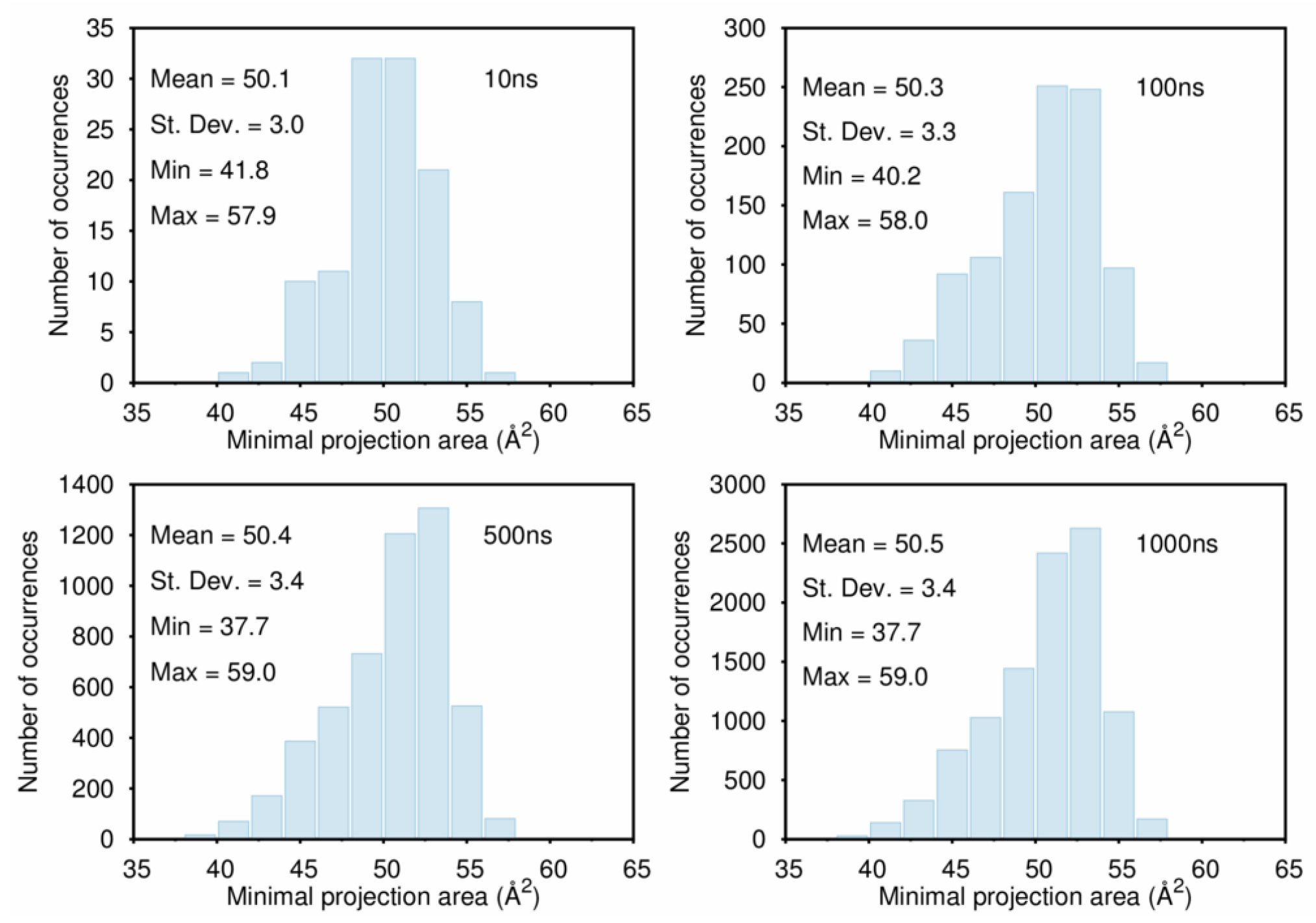
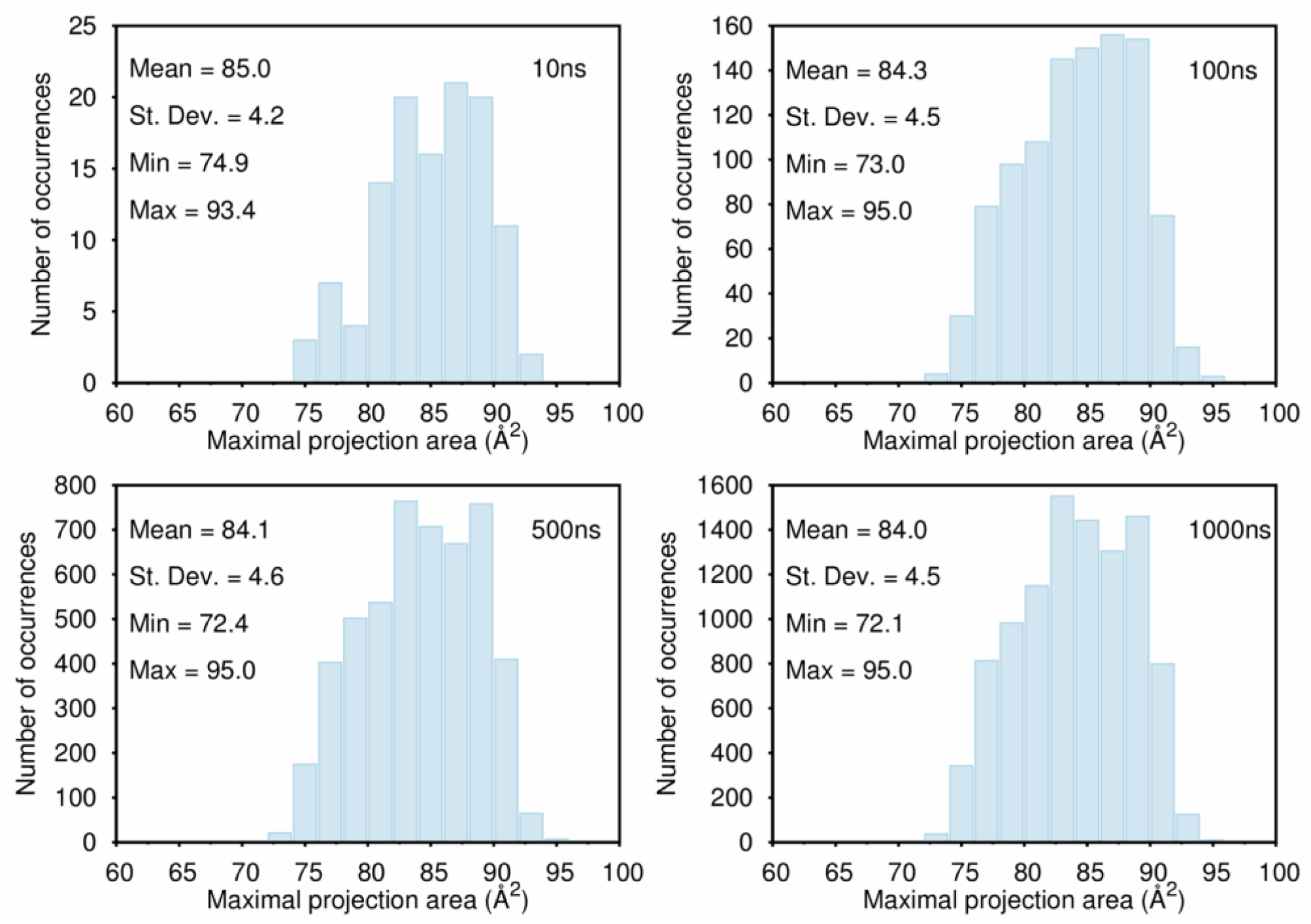
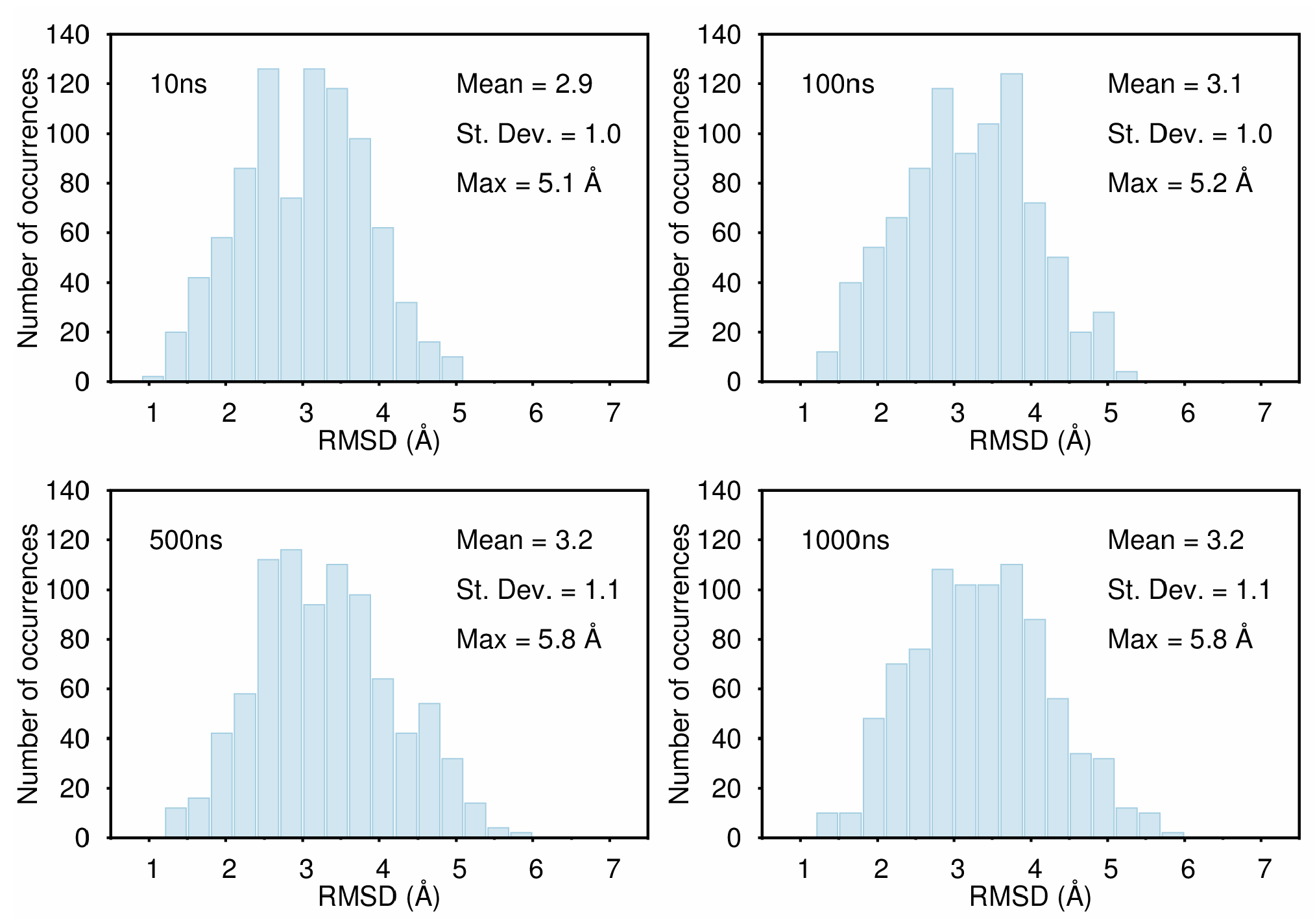
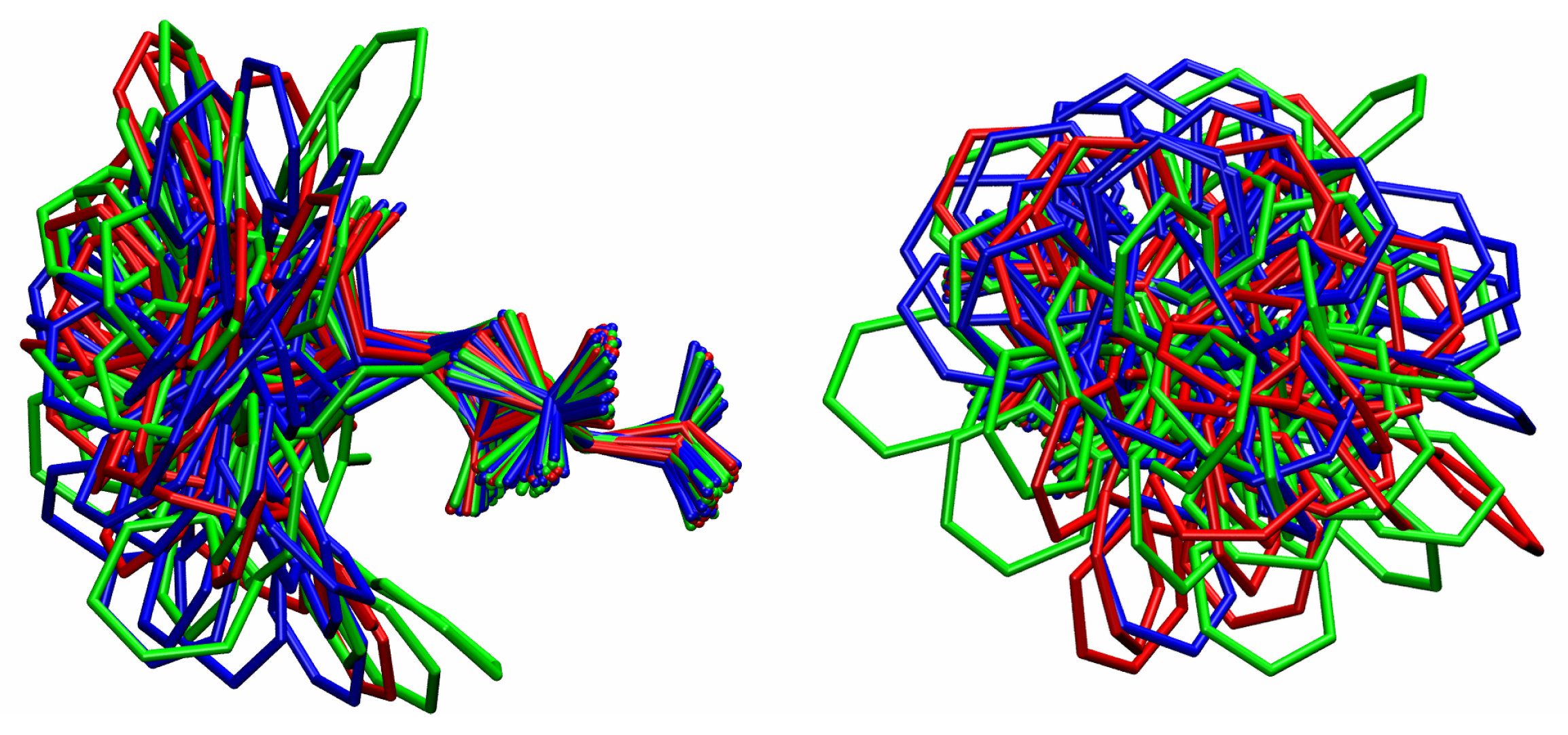
B. Assessment of the Convergence of Structural Properties vs. Simulation Time
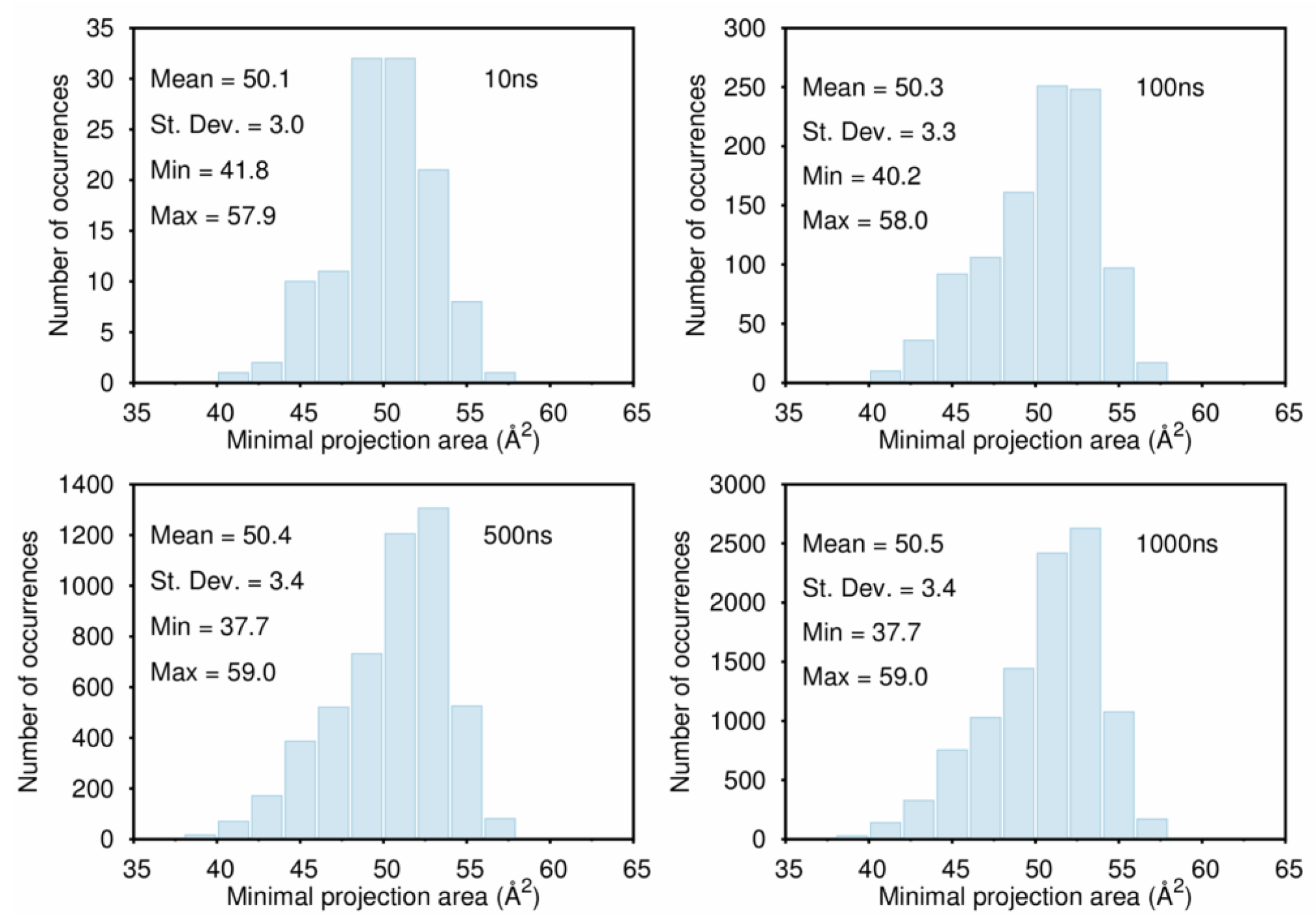
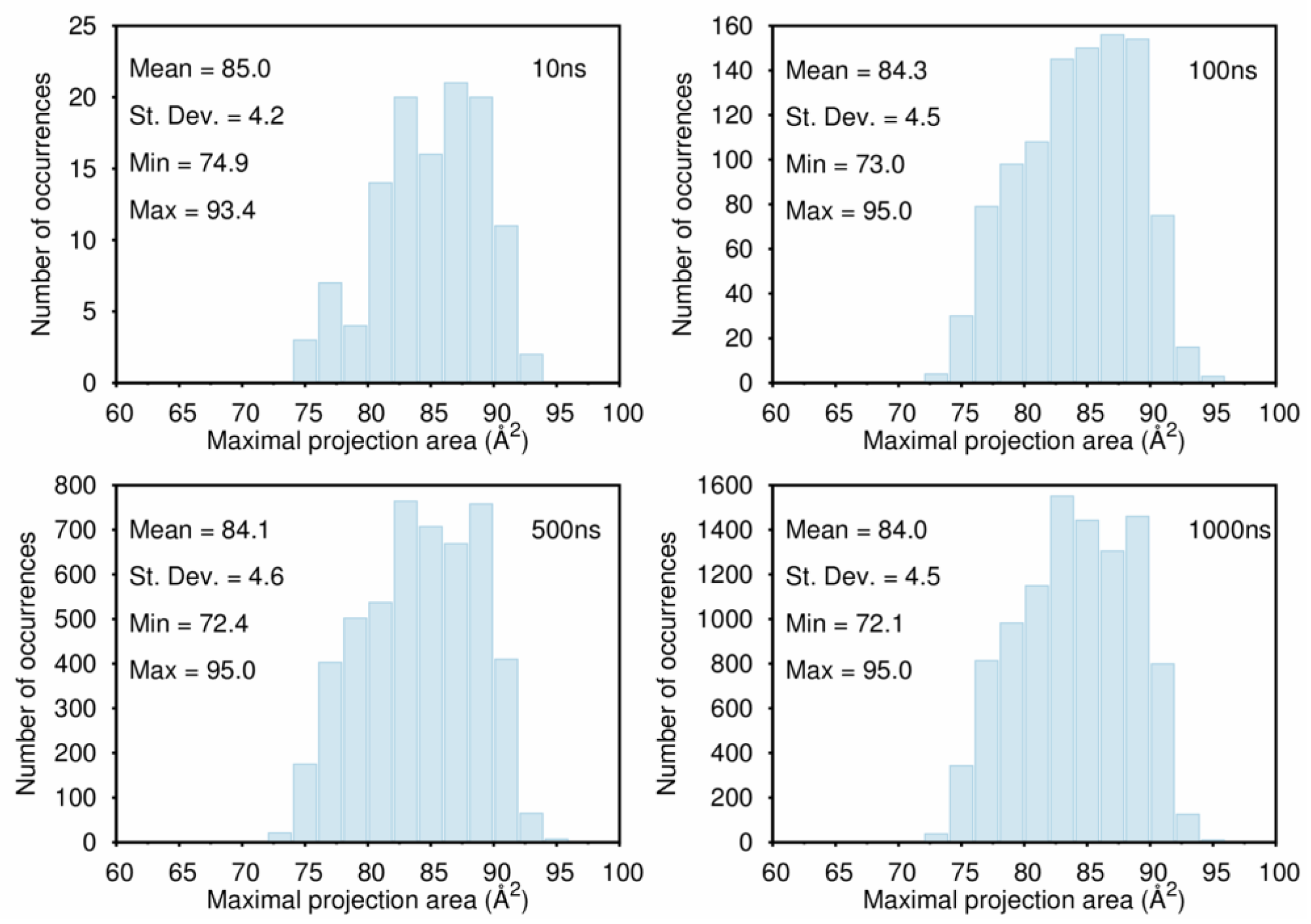
References
- Csermely, P.; Palotai, R.; Nussinov, R. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem. Sci. 2010, 35, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; McCammon, J.A. Molecular Recognition and Ligand Association. Ann. Rev. Phys. Chem. 2013, 64, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Pisani, P.; Piro, P.; Decherchi, S.; Bottegoni, G.; Sona, D.; Murino, V.; Rocchia, W.; Cavalli, A. Describing the Conformational Landscape of Small Organic Molecules through Gaussian Mixtures in Dihedral Space. J. Chem. Theory Comput. 2014, 10, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Van Gunsteren, W.F.; Bakowies, D.; Baron, R.; Chandrasekhar, I.; Christen, M.; Daura, X.; Gee, P.; Geerke, D.P.; Glaettli, A.; Huenenberger, P.H.; et al. Biomolecular modeling: Goals, problems, perspectives. Angew. Chem. Int. Ed. 2006, 45, 4064–4092. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Zhou, H.X. Calculation of Protein-Ligand Binding Affinities. Ann. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, P.; Brooks, B.R.; Sansom, M.S.P. Multiscale methods for macromolecular simulations. Curr. Opin. Struc. Biol. 2008, 18, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Dodson, G.G.; Lane, D.P.; Verma, C.S. Molecular simulations of protein dynamics: New windows on mechanisms in biology. EMBO Rep. 2008, 9, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Hsin, J.; Sotomayor, M.; Comellas, G.; Schulten, K. Discovery Through the Computational Microscope. Structure 2009, 17, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Ann. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; McCammon, J.A. Molecular Recognition and Ligand Association. Ann. Rev. Phys. Chem. 2013, 64, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Cheatham, T.E.; Case, D.A. Twenty-Five Years of Nucleic Acid Simulations. Biopolymers 2013, 99, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; Lavery, R. Significance of Molecular Dynamics Simulations for Life Sciences. Isr. J. Chem. 2014, 54, 1042–1051. [Google Scholar] [CrossRef]
- Mortier, J.; Rakers, C.; Bermudez, M.; Murgueitio, M.S.; Riniker, S.; Wolber, G. The impact of molecular dynamics on drug design: Applications for the characterization of ligand-macromolecule complexes. Drug Discov. Today 2015, 20, 686–702. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.H.; Kandt, C. LAMBADA and InflateGRO2: Efficient Membrane Alignment and Insertion of Membrane Proteins for Molecular Dynamics Simulations. J. Chem. Inf. Mod. 2012, 52, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Piana, S.; Klepeis, J.L.; Shaw, D.E. Assessing the accuracy of physical models used in protein-folding simulations: quantitative evidence from long molecular dynamics simulations. Curr. Opin. Struc. Biol. 2014, 24, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Šponer, J.; Banáš, P.; Jurečka, P.; Zgarbová, M.; Kührová, P.; Havrila, M.; Krepl, M.; Stadlbauer, P.; Otyepka, M. Molecular Dynamics Simulations of Nucleic Acids. From Tetranucleotides to the Ribosome. J. Phys. Chem. Lett. 2014, 5, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Graen, T.; Hoefling, M.; Grubmueller, H. AMBER-DYES: Characterization of Charge Fluctuations and Force Field Parameterization of Fluorescent Dyes for Molecular Dynamics Simulations. J. Chem. Theory Comput. 2014, 10, 5505–5512. [Google Scholar] [CrossRef] [PubMed]
- Dupradeau, F.Y.; Cézard, C.; Lelong, R.; Stanislawiak, É.; Pêcher, J.; Delepine, J.C.; Cieplak, P. R.E. DD. B.: A database for RESP and ESP atomic charges, and force field libraries. Nucleic Acids Res. 2008, 36, D360–D367. [Google Scholar] [CrossRef] [PubMed]
- Dupradeau, F.Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef] [PubMed]
- AMBER Parameter Database. Available online: http://www.pharmacy.manchester.ac.uk/bryce/amber/ (accessed on 22 January 2016).
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Mod. 2012, 52, 3144–3154. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Mod. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Stavenger, R.A.; Winterhalter, M. TRANSLOCATION Project: How to Get Good Drugs into Bad Bugs. Sci. Transl. Med. 2014, 6, 228ed7. [Google Scholar] [CrossRef] [PubMed]
- Malloci, G.; Vargiu, A.V.; Serra, G.; Bosin, A.; Ruggerone, P.; Ceccarelli, M. A Database of Force-Field Parameters, Dynamics, and Properties of Antimicrobial Compounds. Molecules 2015, 20, 13997–14021. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Hajjar, E.; Ruggerone, P.; Ceccarelli, M. Molecular simulations reveal the mechanism and the determinants for ampicillin translocation through OmpF. J. Phys. Chem. B 2010, 114, 9608–9616. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, E.; Bessonov, A.; Molitor, A.; Kumar, A.; Mahendran, K.R.; Winterhalter, M.; Pagès, J.M.; Ruggerone, P.; Ceccarelli, M. Toward screening for antibiotics with enhanced permeation properties through bacterial porins. Biochemistry 2010, 49, 6928–6935. [Google Scholar] [CrossRef] [PubMed]
- Collu, F.; Vargiu, A.V.; Dreier, J.; Cascella, M.; Ruggerone, P. Recognition of Imipenem and Meropenem by the RND-Transporter MexB Studied by Computer Simulations. J. Am. Chem. Soc. 2012, 134, 19146–19158. [Google Scholar] [CrossRef] [PubMed]
- Vargiu, A.V.; Nikaido, H. Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2012, 109, 20637–20642. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Forli, S. Charting a Path to Success in Virtual Screening. Molecules 2015, 20, 18732–18758. [Google Scholar] [CrossRef] [PubMed]
- Sinko, W.; Lindert, S.; McCammon, J.A. Accounting for Receptor Flexibility and Enhanced Sampling Methods in Computer-Aided Drug Design. Chem. Biol. Drug Des. 2013, 81, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Amaro, R.E.; Baron, R.; McCammon, J.A. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J. Comput.-Aided Mol. Des. 2008, 22, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, M. Protein-protein docking with a reduced protein model accounting for side-chain flexibility. Protein Sci. 2003, 12, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Acred, P.; Brown, D.M.; Turner, D.H.; Wilson, M.J. Pharmacology and Chemotherapy of Ampicillin. A New Broad-Spectrum Penicillin. Brit. J. Pharm. Chemother. 1962, 18, 356–369. [Google Scholar] [CrossRef]
- Ebejer, J.P.; Morris, G.M.; Deane, C.M. Freely Available Conformer Generation Methods: How Good Are They? J. Chem. Inf. Mod. 2012, 52, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Bolton, E.E.; Wangand, Y.; Thiessenand, P.A.; Bryant, S.H. PubChem: Integrated Platform of Small Molecules and Biological Activities. Annu. Rep. Comput. Chem. 2008, 4, 217–241. [Google Scholar]
- Marvin 14.8.25.0. ChemAxon 2014. Available online: http://www.chemaxon.com (accessed on 22 January 2016).
- Calculator Plugins. ChemAxon 2014. Available online: https://www.chemaxon.com/products/calculator-plugins/ (accessed on 22 January 2016).
- Kohn, W. Nobel Lecture: Electronic structure of matter-wave functions and density functionals. Rev. Mod. Phys. 1999, 71, 1253–1266. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09 Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Kim, K.; Jordan, K.D. Comparison of Density Functional and MP2 Calculations on the Water Monomer and Dimer. J. Phys. Chem. 1994, 98, 10089–10094. [Google Scholar] [CrossRef]
- Pople, J.A. Quantum Chemical Models (Nobel Lecture). Angew. Chem. Int. Ed. 1999, 38, 1894–1902. [Google Scholar] [CrossRef]
- Malloci, G.; Joblin, C.; Mulas, G. On-line database of the spectral properties of polycyclic aromatic hydrocarbons. Chem. Phys. 2007, 332, 353–359. [Google Scholar] [CrossRef]
- Malloci, G.; Cappellini, G.; Mulas, G.; Mattoni, A. Electronic and optical properties of families of polycyclic aromatic hydrocarbons: A systematic (time-dependent) density functional theory study. Chem. Phys. 2011, 384, 19–27. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of Octanol-Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Pyrkov, T.V.; Chugunov, A.O.; Krylov, N.A.; Nolde, D.E.; Efremov, R.G. PLATINUM: a web tool for analysis of hydrophobic/hydrophilic organization of biomolecular complexes. Bioinformatics 2009, 25, 1201–1202. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Modell. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Case, D.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham, T.; Darden, T.; Duke, R.; Gohlke, H.; et al. Amber 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef]
- Theodorou, D.N.; Suter, U.W. Shape of unperturbed linear polymers: polypropylene. Macromolecules 1985, 18, 1206–1214. [Google Scholar] [CrossRef]
- Miteva, M.A.; Guyon, F.; Tufféry, P. Frog2: Efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Res. 2010, 38, W622–W627. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.; Banck, M.; James, C.; Morley, C.; Vandermeersch, T.; Hutchison, G. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- RDKit: Open-Source Cheminformatics Software. Available online: http://www.rdkit.org (accessed on 22 January 2015).
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.G.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Schrodinger LLC (2010) The PyMOL Molecular Graphics System, version 1.3r1. Available online: http://www.pymol.org (accessed on 25 January 2016).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Gnuplot. Available online: http://www.gnuplot.info (accessed on 22 January 2016).
- Ziervogel, B.; Roux, B. The Binding of Antibiotics in OmpF Porin. Structure 2013, 21, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.M.; Ullman, J.L.; Teel, A.L.; Watts, R.J. pH and temperature effects on the hydrolysis of three β-lactam antibiotics: Ampicillin, cefalotin and cefoxitin. Sci. Total Environ. 2014, 466–467, 547–555. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malloci, G.; Serra, G.; Bosin, A.; Vargiu, A.V. Extracting Conformational Ensembles of Small Molecules from Molecular Dynamics Simulations: Ampicillin as a Test Case. Computation 2016, 4, 5. https://doi.org/10.3390/computation4010005
Malloci G, Serra G, Bosin A, Vargiu AV. Extracting Conformational Ensembles of Small Molecules from Molecular Dynamics Simulations: Ampicillin as a Test Case. Computation. 2016; 4(1):5. https://doi.org/10.3390/computation4010005
Chicago/Turabian StyleMalloci, Giuliano, Giovanni Serra, Andrea Bosin, and Attilio Vittorio Vargiu. 2016. "Extracting Conformational Ensembles of Small Molecules from Molecular Dynamics Simulations: Ampicillin as a Test Case" Computation 4, no. 1: 5. https://doi.org/10.3390/computation4010005
APA StyleMalloci, G., Serra, G., Bosin, A., & Vargiu, A. V. (2016). Extracting Conformational Ensembles of Small Molecules from Molecular Dynamics Simulations: Ampicillin as a Test Case. Computation, 4(1), 5. https://doi.org/10.3390/computation4010005