
Molecular Simulation of Shale Gas Adsorption and Diffusion in Clay Nanopores

Abstract
:1. Introduction
2 Models and Methodology
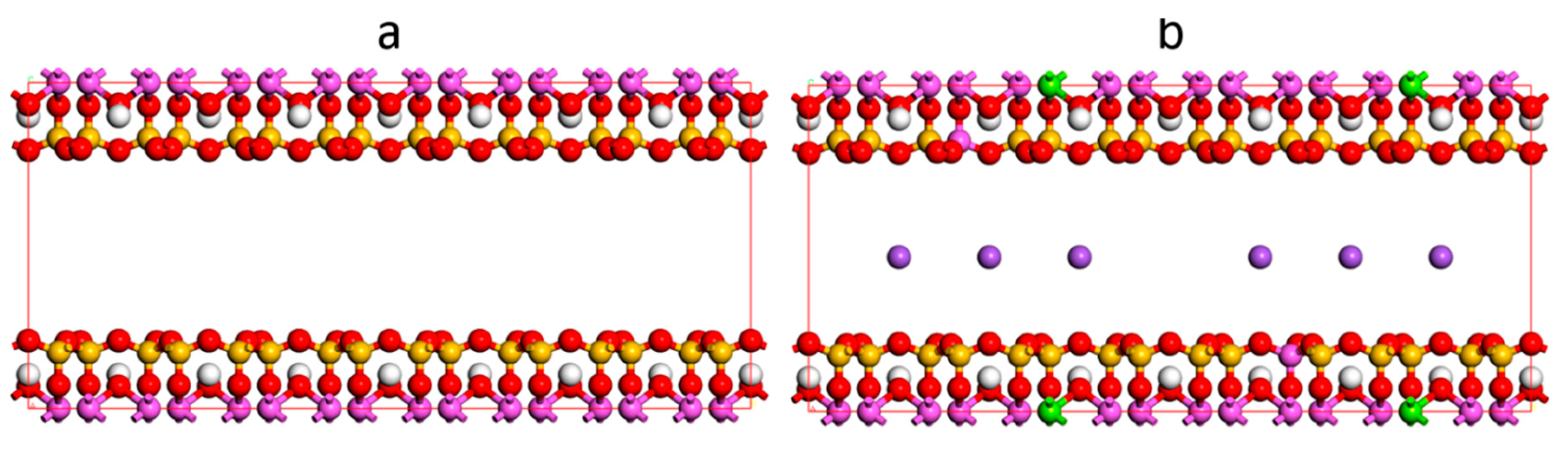
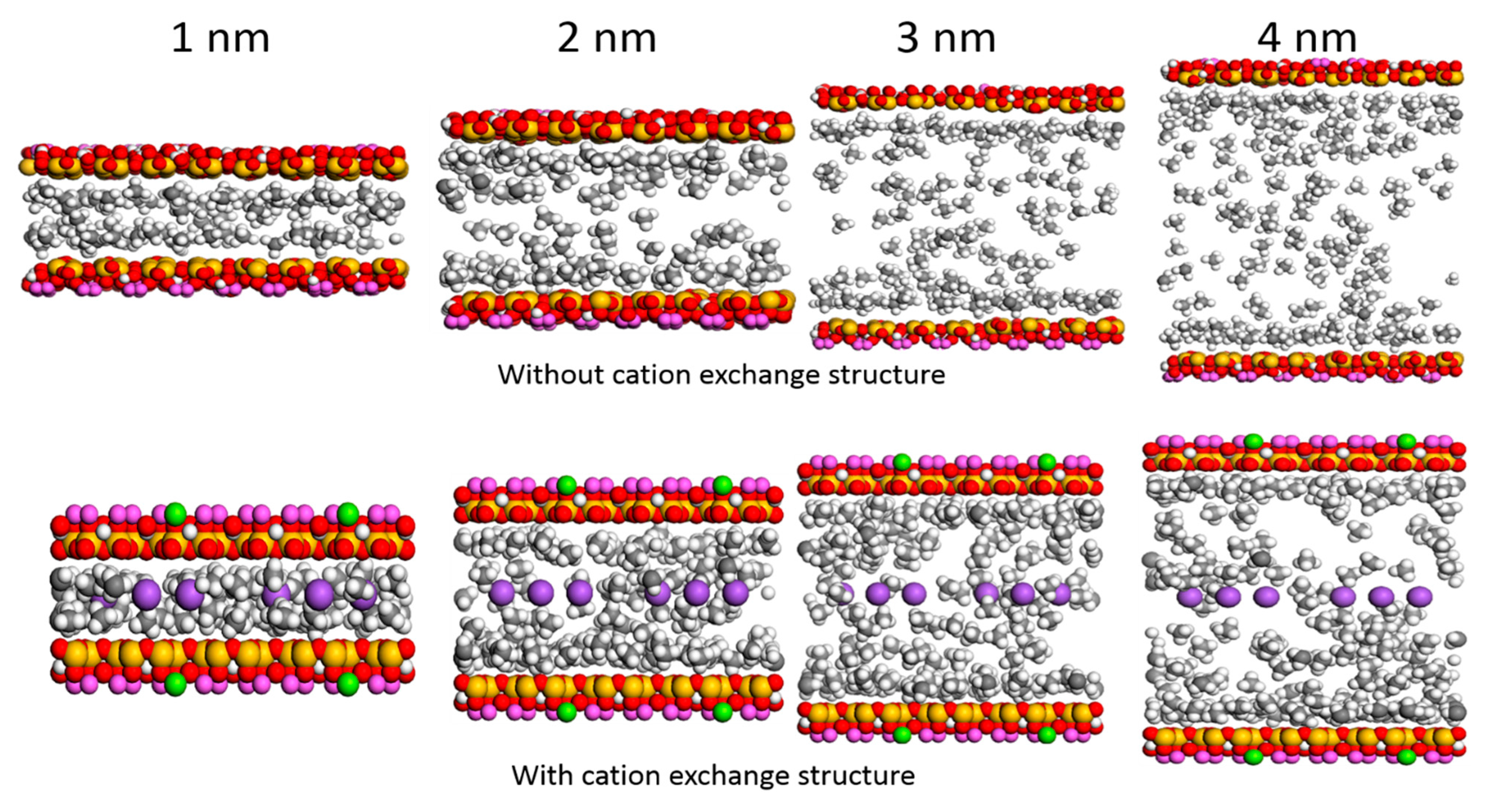
2.1. Model
2.2. Grand Canonical Monte Carlo (GCMC)

2.3. Molecular Dynamic (MD)
3. Results and Discussions
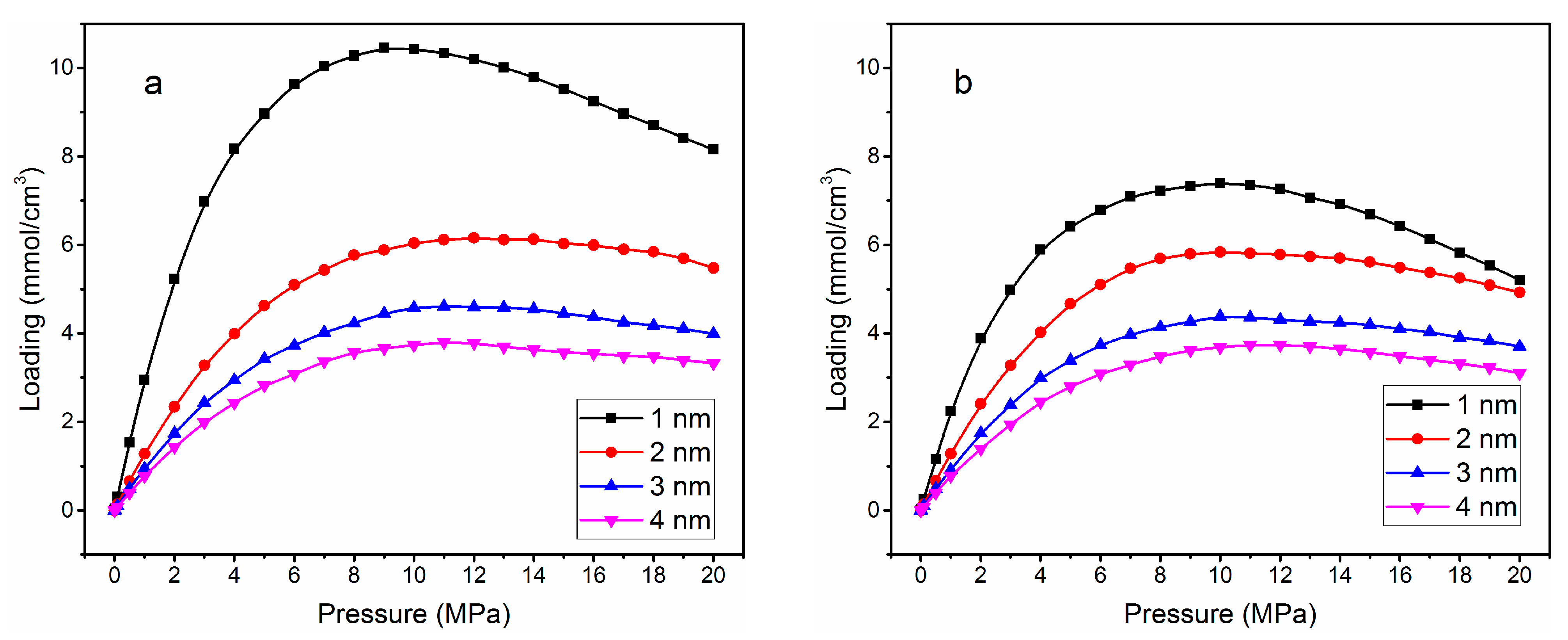
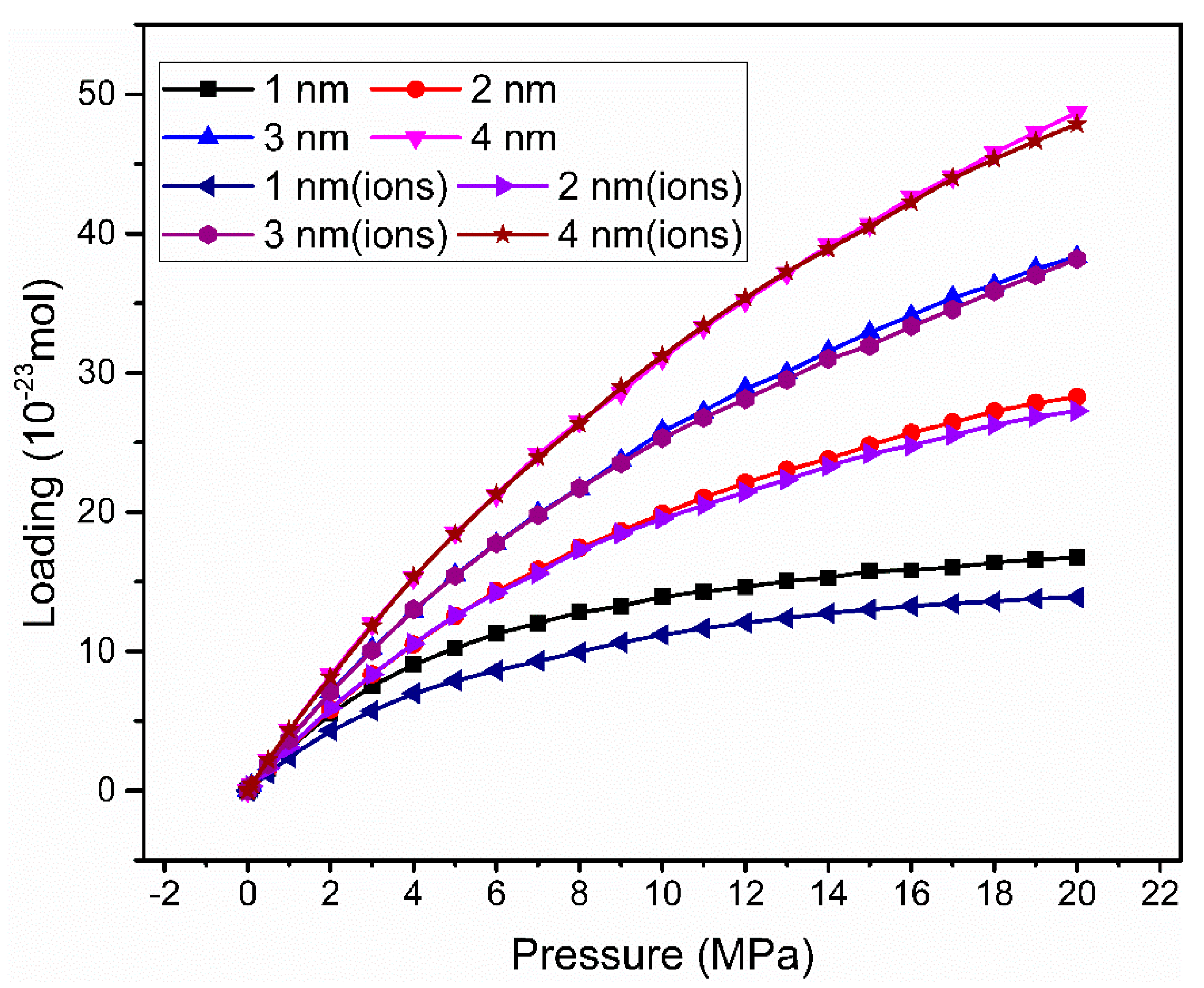
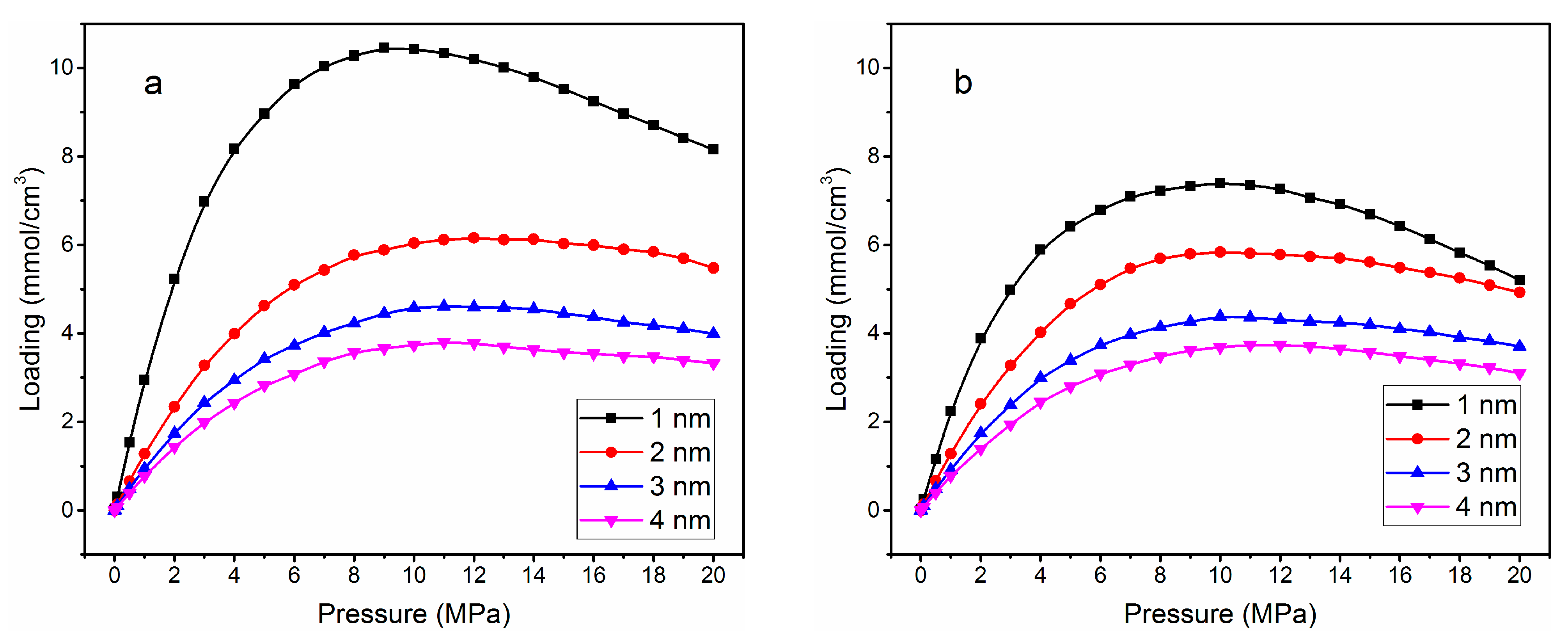
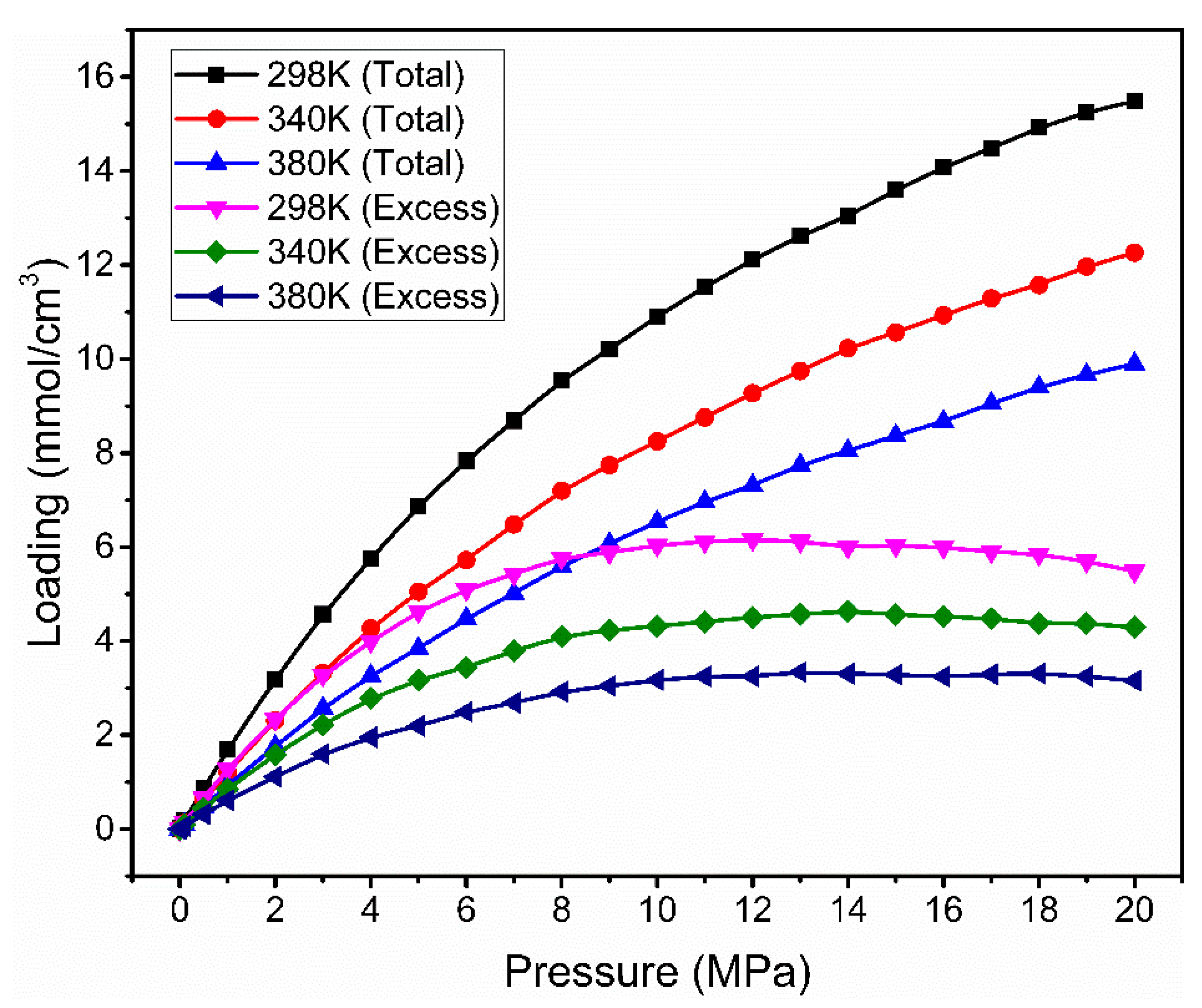
3.1. Adsorption Isotherm
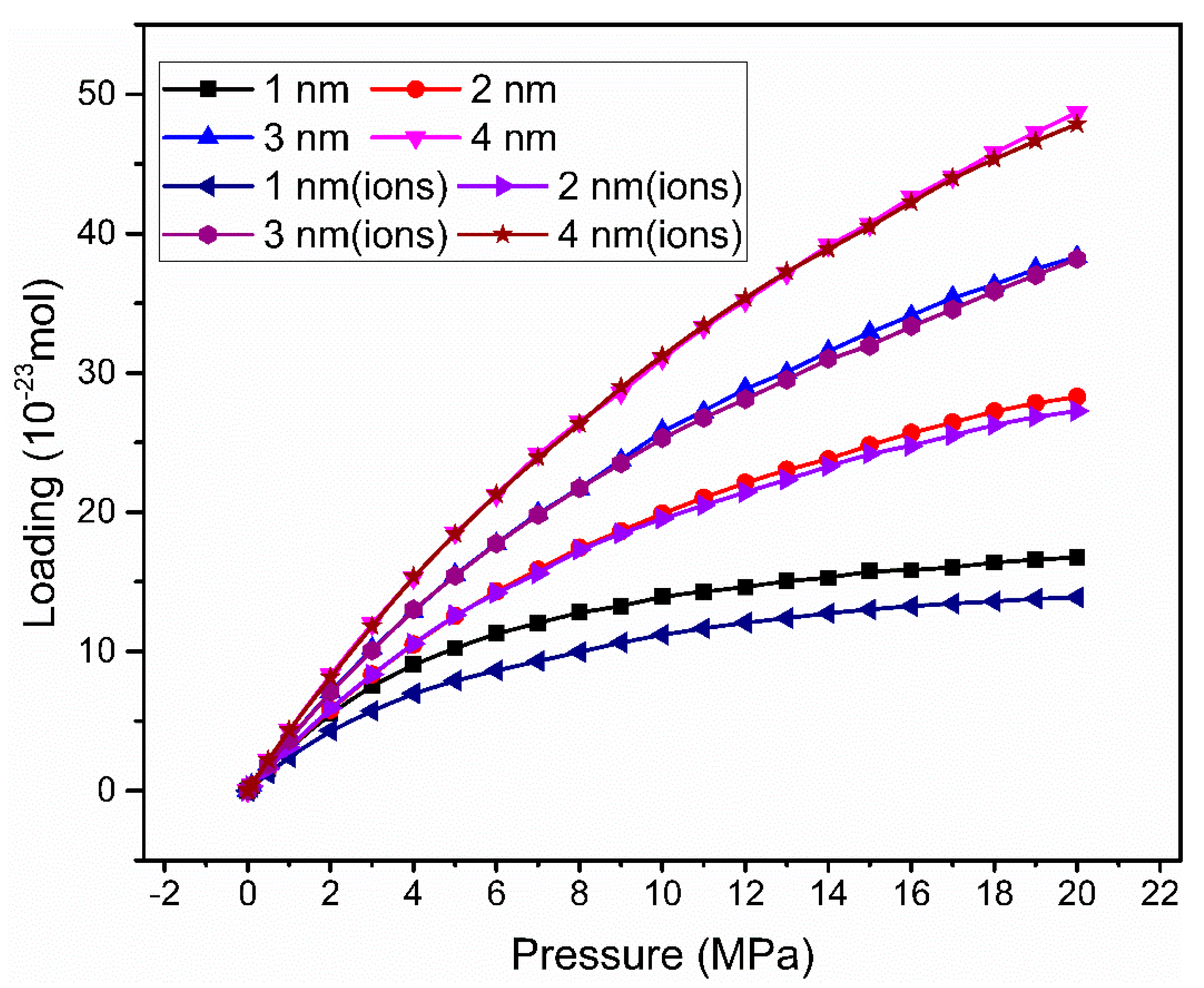

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Without Cation Exchange Structure | With Cation Exchange Structure | |||||||
|---|---|---|---|---|---|---|---|---|
| Pore size (nm) | 1.0 | 2.0 | 3.0 | 4.0 | 1.0 | 2.0 | 3.0 | 4.0 |
| Loading (mmol/cm3) | 10.46 | 6.15 | 4.61 | 3.78 | 7.40 | 5.83 | 4.38 | 3.73 |
| Pressure (MPa) | 8 | 11 | 12 | 13 | 8 | 9 | 10 | 11 |
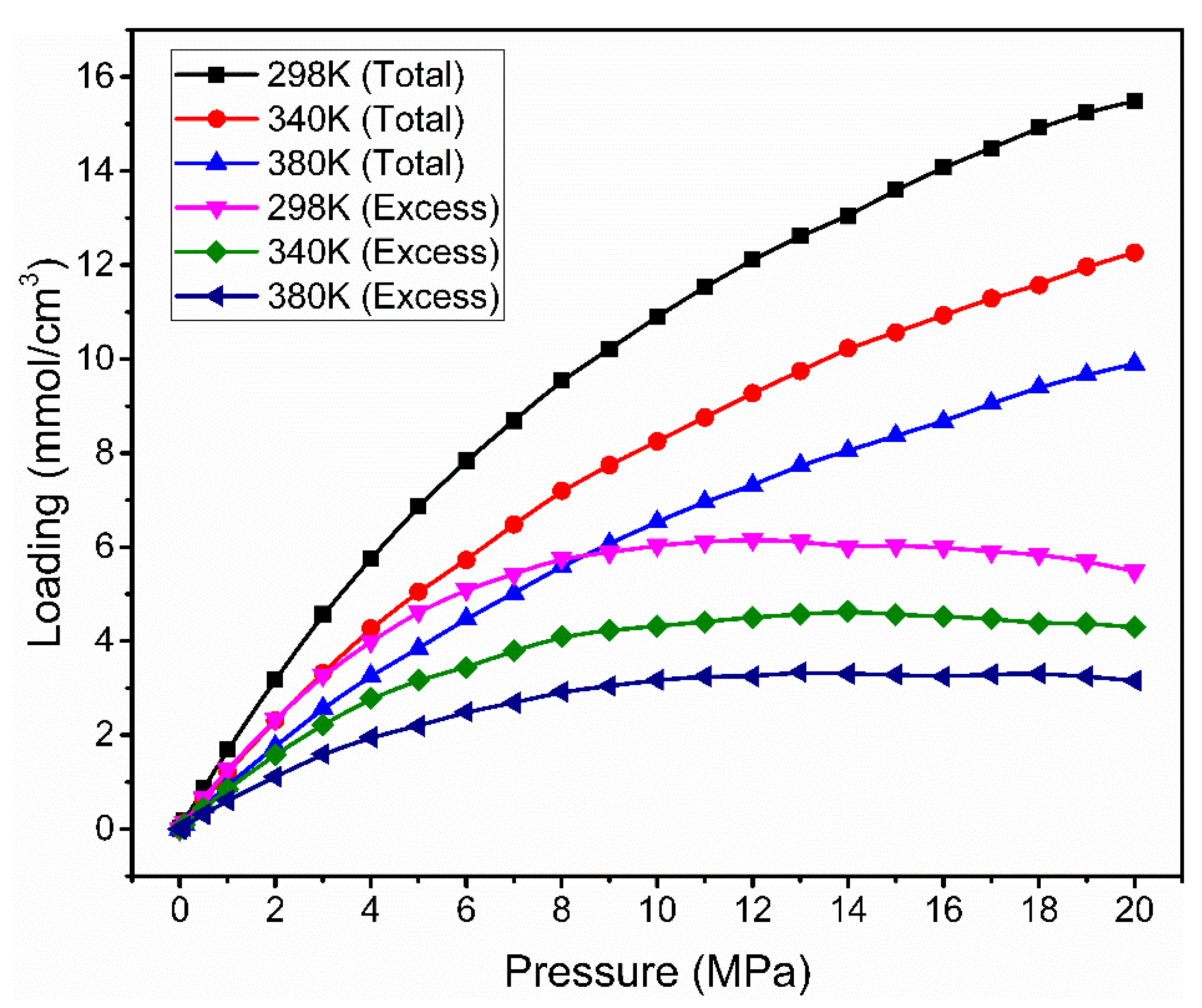
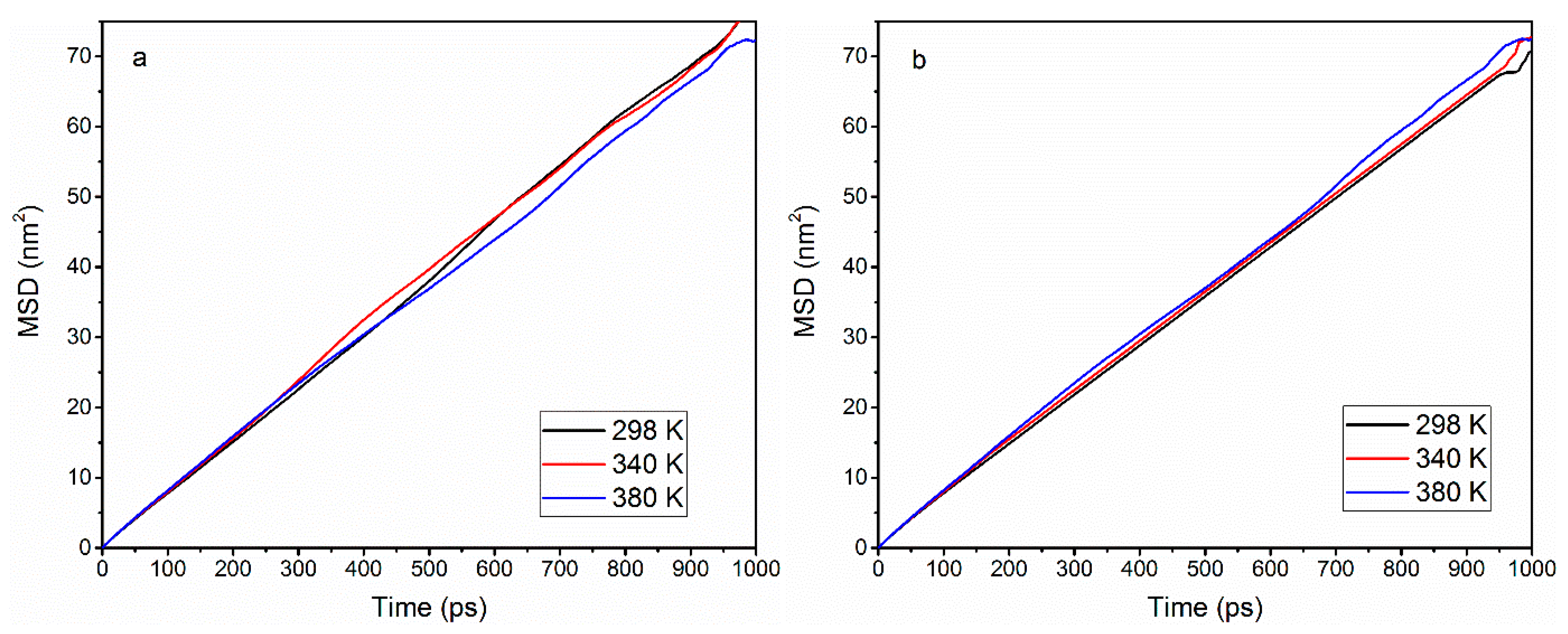
3.2. Effect of Temperature
3.3. Density of Methane in Slit Pores


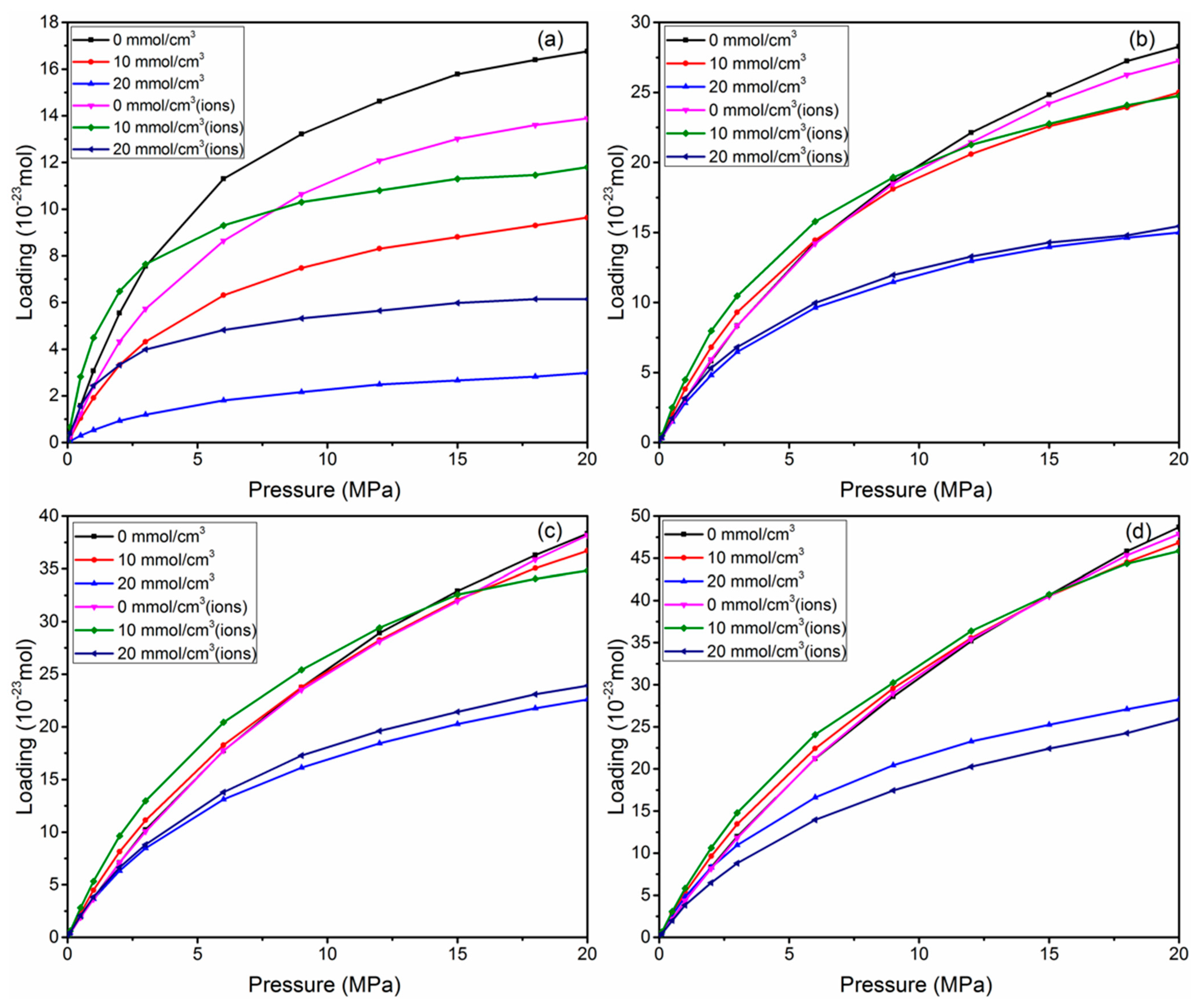
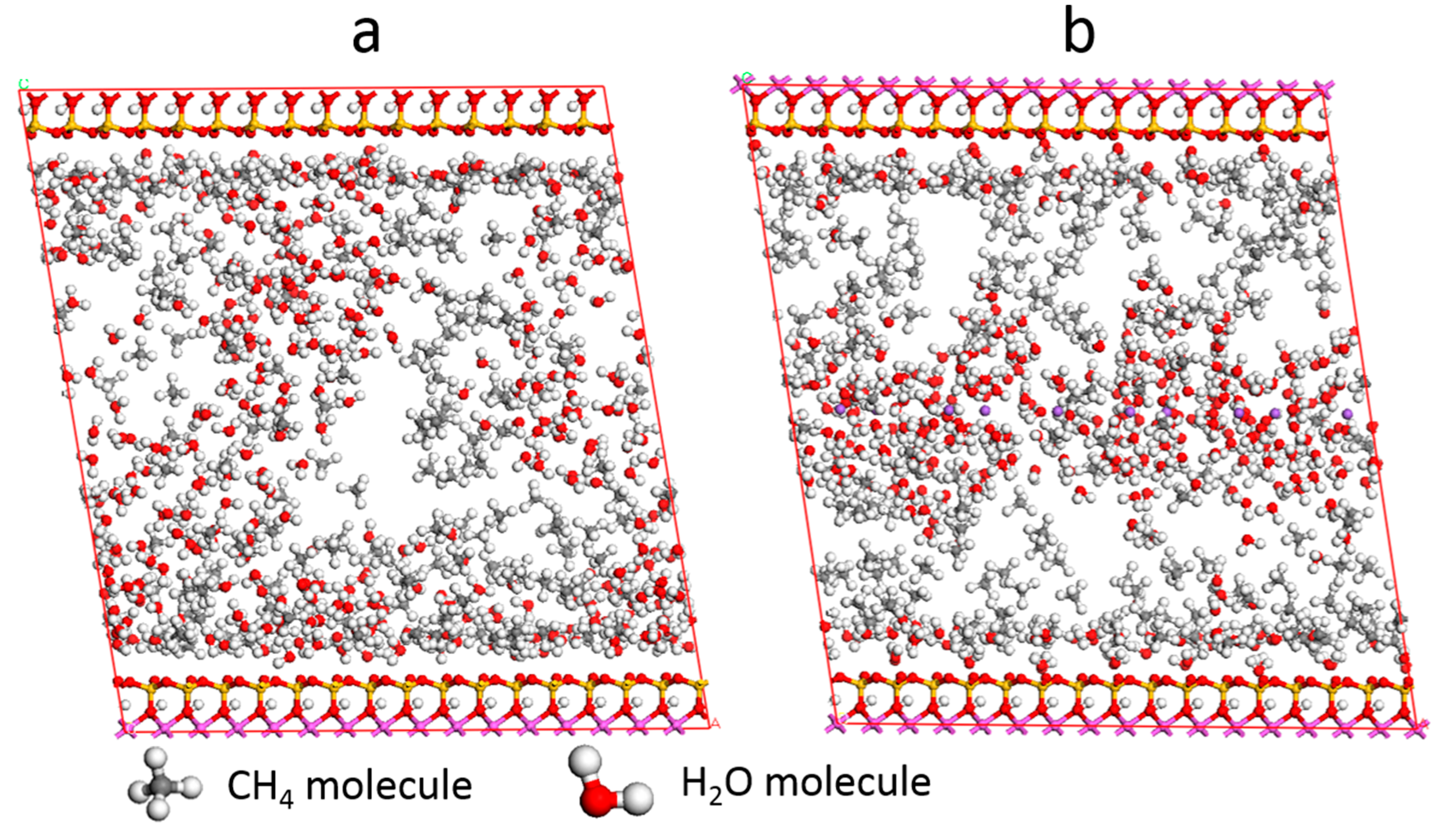
3.4. Effect of Water
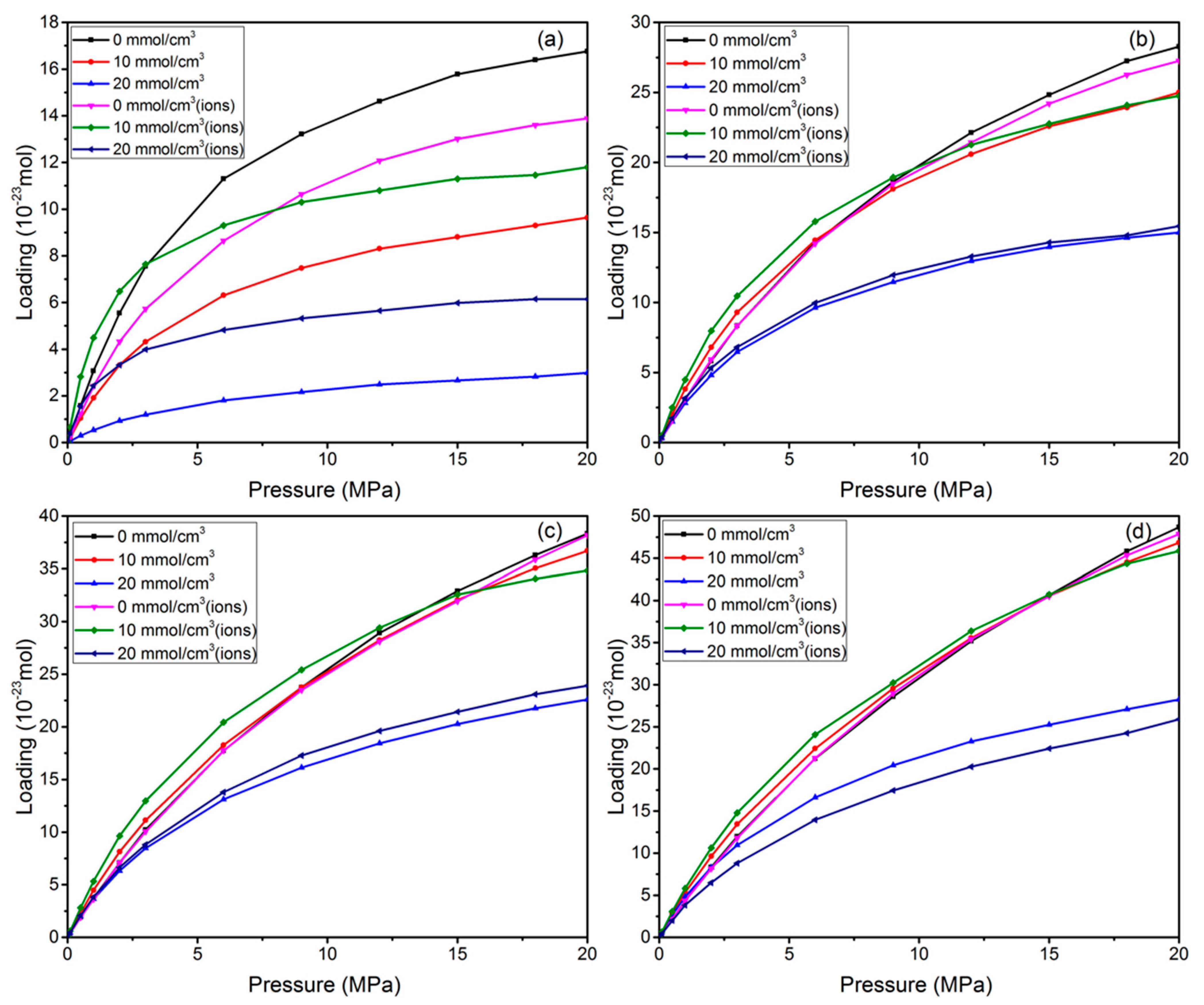
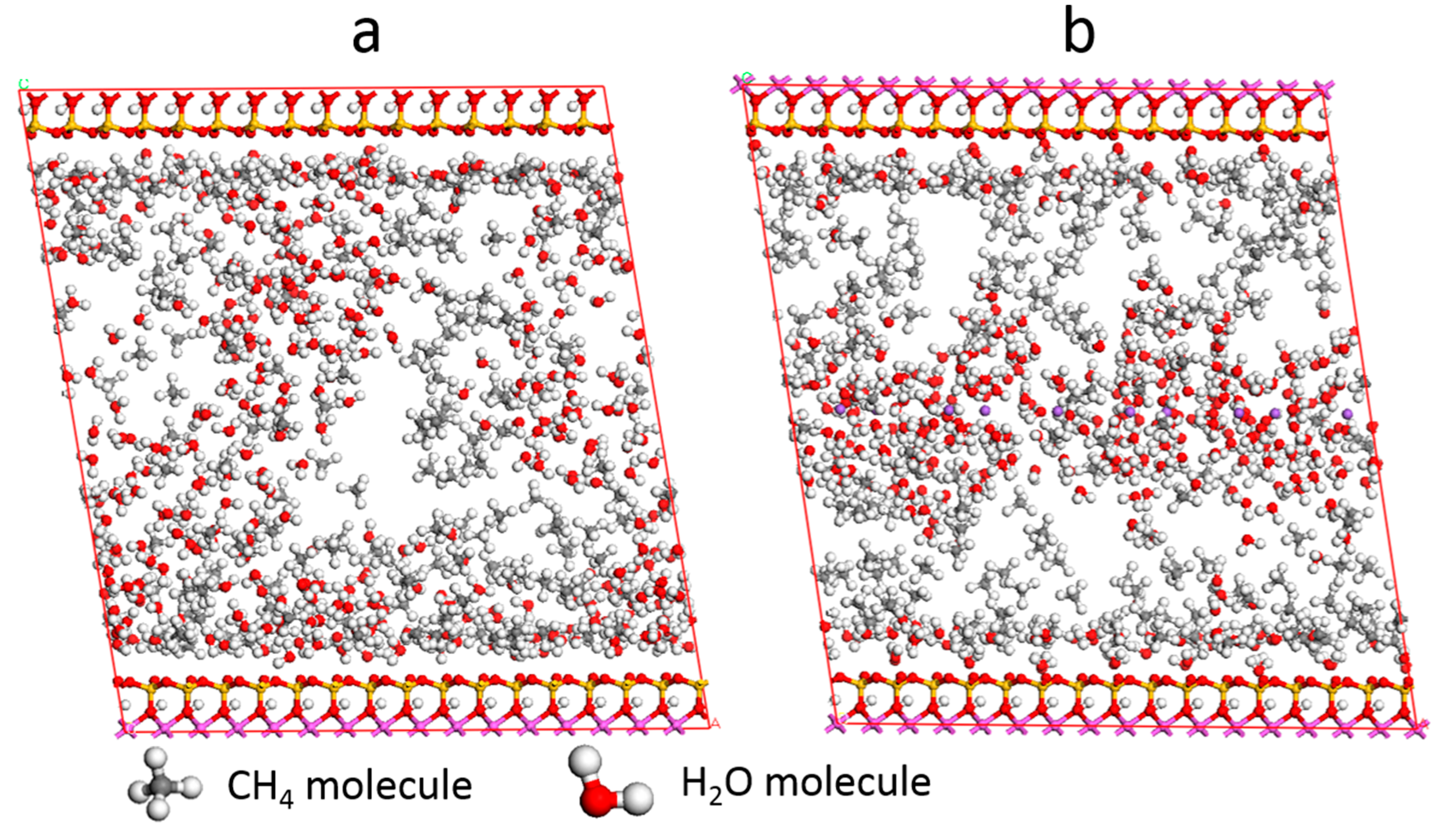
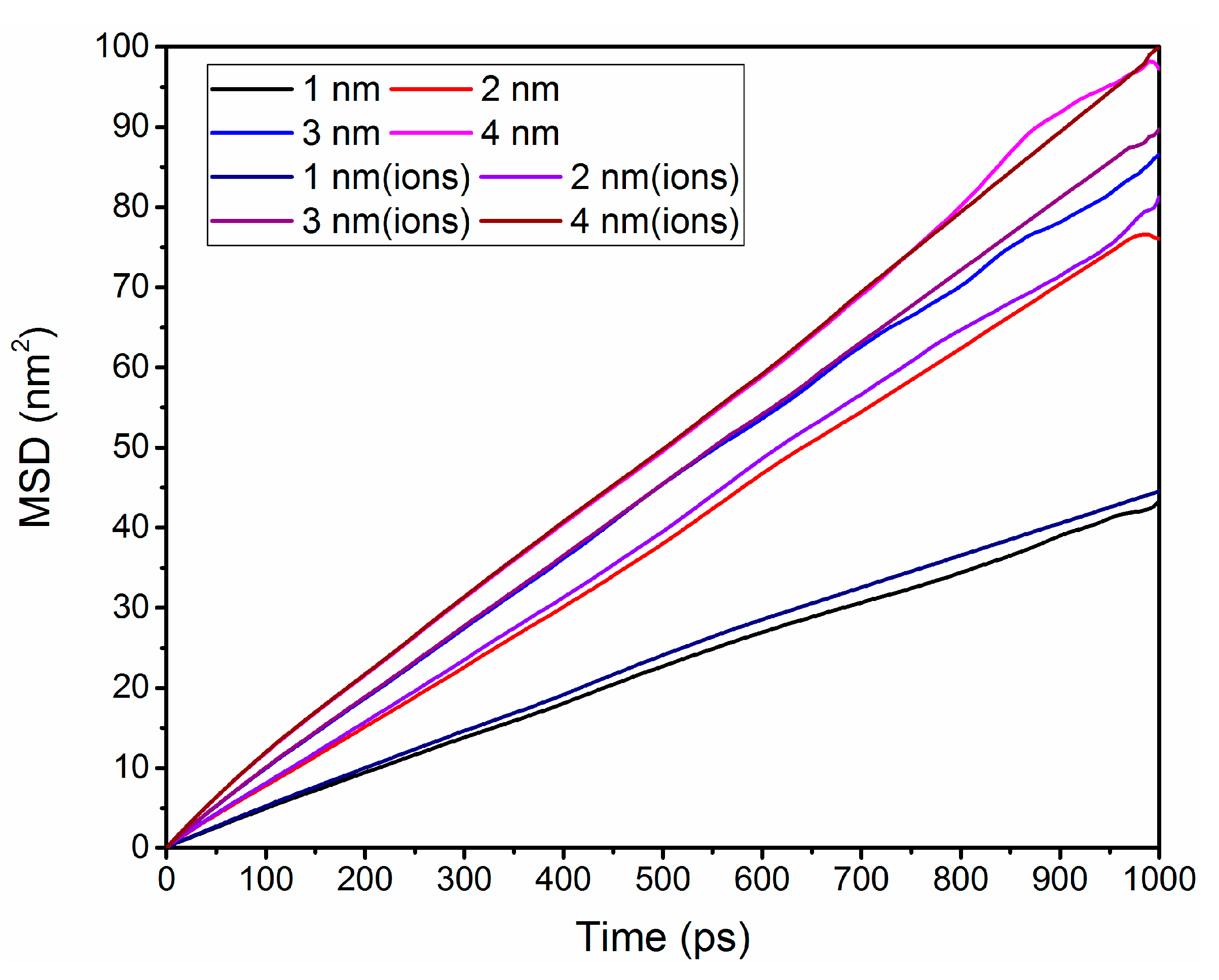
3.5. Self-Diffusion


| Width | Temperature | Dwithout ions (10−8 m2/s) | Dwith ions (10−8 m2/s) |
|---|---|---|---|
| (nm) | (K) | (8 MPa) | (8 MPa) |
| 1.0 | 298 | 0.706 | 0.710 |
| 2.0 | 298 | 1.236 | 1.234 |
| 2.0 | 340 | 1.264 | 1.261 |
| 2.0 | 380 | 1.288 | 1.284 |
| 3.0 | 298 | 1.431 | 1.432 |
| 4.0 | 298 | 1.638 | 1.637 |
4. Conclusion
Supplementary Files
Supplementary File 1Acknowledgement
Author Contributions
Conflicts of Interest
References
- Howarth, R.W.; Ingraffea, A.; Engelder, T. Natural gas: Should fracking stop? Nature 2011, 477, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.D. Energy: A reality check on the shale revolution. Nature 2013, 494, 307–308. [Google Scholar] [CrossRef] [PubMed]
- McGlade, C.; Speirs, J.; Sorrell, S. Unconventional gas—A review of regional and global resource estimates. Energy 2013, 55, 571–584. [Google Scholar] [CrossRef]
- Gouth, F.; Collell, J.; Galliero, G.; Wang, J. Molecular Simulation to Determine Key Shale Gas Parameters, and Their Use in a Commercial Simulator for Production Forecasting; Society of Petroleum Engineers: Richardson, TX, USA, 2013. [Google Scholar]
- Chen, S.; Zhu, Y.; Wang, H.; Liu, H.; Wei, W.; Fang, J. Shale gas reservoir characterisation: A typical case in the southern sichuan basin of china. Energy 2011, 36, 6609–6616. [Google Scholar] [CrossRef]
- Bustin, A.M.M.; Bustin, R.M. Importance of rock properties on the producibility of gas shales. Int. J. Coal Geol. 2012, 103, 132–147. [Google Scholar] [CrossRef]
- Zhang, T.; Ellis, G.S.; Ruppel, S.C.; Milliken, K.; Yang, R. Effect of organic-matter type and thermal maturity on methane adsorption in shale-gas systems. Org. Geochem. 2012, 47, 120–131. [Google Scholar] [CrossRef]
- Sondergeld, C.H.; Ambrose, R.J.; Rai, C.S.; Moncrieff, J. Micro-Structural Studies of Gas Shales; Society of Petroleum Engineers: Richardson, TX, USA, 2010. [Google Scholar]
- Ross, D.J.K.; Marc Bustin, R. The importance of shale composition and pore structure upon gas storage potential of shale gas reservoirs. Mar. Pet. Geol. 2009, 26, 916–927. [Google Scholar] [CrossRef]
- Ross, D.J.; Bustin, R.M. Characterizing the shale gas resource potential of devonian–mississippian strata in the western canada sedimentary basin: Application of an integrated formation evaluation. AAPG Bull. 2008, 92, 87–125. [Google Scholar] [CrossRef]
- Weniger, P.; Kalkreuth, W.; Busch, A.; Krooss, B.M. High-pressure methane and carbon dioxide sorption on coal and shale samples from the paraná basin, Brazil. Int. J. Coal Geol. 2010, 84, 190–205. [Google Scholar] [CrossRef]
- Ji, L.; Zhang, T.; Milliken, K.L.; Qu, J.; Zhang, X. Experimental investigation of main controls to methane adsorption in clay-rich rocks. Appl. Geochem. 2012, 27, 2533–2545. [Google Scholar] [CrossRef]
- Wang, C.C.; Juang, L.C.; Lee, C.K.; Hsu, T.C.; Lee, J.F.; Chao, H.P. Effects of exchanged surfactant cations on the pore structure and adsorption characteristics of montmorillonite. J. Colloid Interface Sci. 2004, 280, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Aringhieri, R. Nanoporosity characteristics of some natural clay minerals and soils. Clays Clay Miner. 2004, 52, 700–704. [Google Scholar] [CrossRef]
- Volzone, C.; Thompson, J.G.; Melnitchenko, A.; Ortiga, J.; Palethorpe, S.R. Selective gas adsorption by amorphous clay-mineral derivatives. Clays Clay Miner. 1999, 47, 647–657. [Google Scholar] [CrossRef]
- Lu, X.C.; Li, F.C.; Watson, A.T. Adsorption studies of natural gas storage in devonian shales. SPE Form. Evaluation 1995, 10, 109–113. [Google Scholar] [CrossRef]
- Schettler, P., Jr.; Parmely, C. Contributions to total storage capacity in devonian shales. In Proceedings of the SPE Eastern Regional Meeting, Lexington, KY, USA, 22–25 October 1991; Society of Petroleum Engineers: Richardson, TX, USA, 1991. [Google Scholar]
- Busch, A.; Alles, S.; Gensterblum, Y.; Prinz, D.; Dewhurst, D.N.; Raven, M.D.; Stanjek, H.; Krooss, B.M. Carbon dioxide storage potential of shales. Int. J. Greenh. Gas Control 2008, 2, 297–308. [Google Scholar] [CrossRef]
- Cheng, A.L.; Huang, W.L. Selective adsorption of hydrocarbon gases on clays and organic matter. Org. Geochem. 2004, 35, 413–423. [Google Scholar] [CrossRef]
- Jin, Z.; Firoozabadi, A. Methane and carbon dioxide adsorption in clay-like slit pores by monte carlo simulations. Fluid Phase Equilibria 2013, 360, 456–465. [Google Scholar] [CrossRef]
- Jin, Z.; Firoozabadi, A. Effect of water on methane and carbon dioxide sorption in clay minerals by monte carlo simulations. Fluid Phase Equilibria 2014, 382, 10–20. [Google Scholar] [CrossRef]
- Fathi, E.; Akkutlu, I.Y. Nonlinear Sorption Kinetics and Surface Diffusion Effects on Gas Transport in Low-Permeability Formations; Society of Petroleum Engineers: Richardson, TX, USA, 2009. [Google Scholar]
- Darabi, H.; Ettehad, A.; Javadpour, F.; Sepehrnoori, K. Gas flow in ultra-tight shale strata. J. Fluid Mech. 2012, 710, 641–658. [Google Scholar] [CrossRef]
- Krishna, R.; Wesselingh, J.A. The maxwell-stefan approach to mass transfer. Chem. Eng. Sci. 1997, 52, 861–911. [Google Scholar] [CrossRef]
- Yuan, W.; Pan, Z.; Li, X.; Yang, Y.; Zhao, C.; Connell, L.D.; Li, S.; He, J. Experimental study and modelling of methane adsorption and diffusion in shale. Fuel 2014, 117, 509–519. [Google Scholar] [CrossRef]
- Cracknell, R.F.; Nicholson, D.; Gubbins, K.E. Molecular dynamics study of the self-diffusion of supercritical methane in slit-shaped graphitic micropores. J. Chem. Soc. Faraday Trans. 1995, 91, 1377–1383. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, C. Structure and diffusion behavior of dense carbon dioxide fluid in clay-like slit pores by molecular dynamics simulation. Chem. Phys. Lett. 2005, 407, 427–432. [Google Scholar] [CrossRef]
- Botan, A.; Rotenberg, B.; Marry, V.; Turq, P.; Noetinger, B. Carbon dioxide in montmorillonite clay hydrates: Thermodynamics, structure, and transport from molecular simulation. J. Phys. Chem. C 2010, 114, 14962–14969. [Google Scholar] [CrossRef]
- Sharma, A.; Namsani, S.; Singh, J.K. Molecular simulation of shale gas adsorption and diffusion in inorganic nanopores. Mol. Simul. 2015, 41, 414–422. [Google Scholar] [CrossRef]
- Lu, X.C.; Li, F.C.; Watson, A.T. Adsorption measurements in devonian shales. Fuel 1995, 74, 599–603. [Google Scholar] [CrossRef]
- Gasparik, M.; Ghanizadeh, A.; Bertier, P.; Gensterblum, Y.; Bouw, S.; Krooss, B.M. High-pressure methane sorption isotherms of black shales from the netherlands. Energy Fuels 2012, 26, 4995–5004. [Google Scholar] [CrossRef]
- Ambrose, R.J.; Hartman, R.C.; Diaz-Campos, M.; Akkutlu, I.Y.; Sondergeld, C.H. Shale gas-in-place calculations part i: New pore-scale considerations. SPE J. 2012, 17, 219–229. [Google Scholar] [CrossRef]
- Chávez-Páez, M.; van Workum, K.; de Pablo, L.; de Pablo, J.J. Monte carlo simulations of wyoming sodium montmorillonite hydrates. J. Chem. Phys. 2001, 114, 1405–1413. [Google Scholar] [CrossRef]
- Skipper, N.; Chang, F.R.C.; Sposito, G. Monte carlo simulation of interlayer molecular structure in swelling clay minerals. I: Methodology. Clays Clay Miner. 1995, 43, 285–293. [Google Scholar] [CrossRef]
- Cygan, R.T.; Romanov, V.N.; Myshakin, E.M. Molecular simulation of carbon dioxide capture by montmorillonite using an accurate and flexible force field. J. Phys. Chem. C 2012, 116, 13079–13091. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed-phase applications overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Smirnov, K.S.; Bougeard, D. A Molecular dynamics study of structure and short-time dynamics of water in kaolinite. J. Phys. Chem. B 1999, 103, 5266–5273. [Google Scholar] [CrossRef]
- Cygan, R.T.; Liang, J.J.; Kalinichev, A.G. Molecular models of hydroxide, oxyhydroxide, and clay phases and the development of a general force field. J. Phys. Chem. B 2004, 108, 1255–1266. [Google Scholar] [CrossRef]
- Reid, R.C.; Prausnitz, J.M.; Poling, B.E. The Properties of Gases and Liquids; McGraw-Hill Book Company: New York, NY, USA, 1987. [Google Scholar]
- Brunauer, S.; Deming, L.S.; Deming, W.E.; Teller, E. On a theory of the van der waals adsorption of gases. J. Am. Chem. Soc. 1940, 62, 1723–1732. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sui, H.; Yao, J.; Zhang, L. Molecular Simulation of Shale Gas Adsorption and Diffusion in Clay Nanopores. Computation 2015, 3, 687-700. https://doi.org/10.3390/computation3040687
Sui H, Yao J, Zhang L. Molecular Simulation of Shale Gas Adsorption and Diffusion in Clay Nanopores. Computation. 2015; 3(4):687-700. https://doi.org/10.3390/computation3040687
Chicago/Turabian StyleSui, Hongguang, Jun Yao, and Lei Zhang. 2015. "Molecular Simulation of Shale Gas Adsorption and Diffusion in Clay Nanopores" Computation 3, no. 4: 687-700. https://doi.org/10.3390/computation3040687
APA StyleSui, H., Yao, J., & Zhang, L. (2015). Molecular Simulation of Shale Gas Adsorption and Diffusion in Clay Nanopores. Computation, 3(4), 687-700. https://doi.org/10.3390/computation3040687