Abstract
SLITRK1 is a critical protein involved in neural development and is associated with various neurological disorders, including Tourette Syndrome. This study investigates the structural dynamics, intrinsic disorder propensity, and pharmacological interactions of SLITRK1, with a particular focus on amino acid substitutions and their pathological implications. A comprehensive computational framework was employed, including intrinsic disorder region analysis, transmembrane topology predictions, and stability assessments of SLITRK1 variants. Integrated with reinforcement learning (RL), molecular docking and dynamics simulations were used to evaluate the pharmacotherapeutic potential of drugs commonly prescribed for Tourette Syndrome, such as Pimozide, Aripiprazole, Risperidone, and Haloperidol. Structural analyses revealed that the S656M mutation significantly alters SLITRK1’s 3D conformation, biological functions, and drug binding profiles. Among the tested drugs, Aripiprazole exhibited the highest binding affinity across various SLITRK1 variants, with reinforcement learning highlighting a notable interaction with the S659K mutation. These findings were supported by Ramachandran plot and molecular dynamics analyses, which identified mutation-induced structural and dynamic changes. This study provides an integrative analysis of SLITRK1, offering insights into its role in Tourette Syndrome and laying a foundation for targeted therapeutic strategies to mitigate SLITRK1-related neurological disorders.
1. Introduction
The intricate mechanisms governing neurological function and dysfunction remain a focal point of contemporary scientific inquiry, especially in understanding protein interactions and their implications in various diseases. The SLITRK1(SLIT and NTRK-like family member 1) protein, a pivotal element in neural development and synaptic formation, emerges as a critical subject of study due to its multifaceted roles in the nervous system. This protein’s relevance is further underscored by its involvement in neurological disorders, a domain where molecular insights can significantly advance therapeutic strategies [1,2,3].
In recent years, the emphasis on protein structure and function has led to a deeper appreciation of how proteins, particularly those like SLITRK1, influence neurobiology. Intrinsically disordered proteins (IDPs), characterized by regions of structural flexibility, play a vital role in cellular signaling and regulation [4,5]. Their dynamic nature, as epitomized by SLITRK1, is crucial for adaptive functioning within the complex cellular environments of the nervous system. These aspects of protein dynamics are not just academic curiosities; they hold the key to understanding the underpinnings of synaptic plasticity, neurotransmission, and, by extension, the pathology of numerous neurological disorders [6,7]. In pursuing this understanding, our study comprehensively analyzes the SLITRK1 protein. We delve into its structure at the amino acid level, examining unstructured regions, transmembrane topology, and the implications of stability changes due to amino acid substitutions [8,9,10].
Furthermore, we explore its interactions within complex protein networks, emphasizing the significance of these interactions in the broader context of neural development and synaptic function [11,12]. This exploration is particularly pertinent given the growing recognition of the impact of protein–protein interactions and stability in neurodegenerative diseases. Moreover, the study extends into molecular docking, assessing how mutations, particularly the S656M variant in SLITRK1, influence drug–protein interactions. This aspect is crucial for understanding the pharmacodynamics of drugs used in treating neurological disorders like Tourette Syndrome [13,14,15]. By analyzing changes in drug binding affinities resulting from specific amino acid substitutions, we can glean insights into the implications of genetic variations on drug efficacy. This approach aligns with the burgeoning field of personalized medicine, where understanding individual genetic makeup is critical to optimizing therapeutic interventions. The research also incorporates advanced techniques such as molecular dynamics simulations and Ramachandran plot analysis to assess the structural integrity and dynamic behavior of SLITRK1 [16,17,18,19,20]. These methods provide a window into the conformational changes induced by mutations and their potential impacts on protein function and therapeutic effectiveness.
In addition, this study utilized Reinforcement Learning (RL) to analyze the pharmacological interactions of SLITRK1 variants with FDA-approved drugs. RL, a machine learning (ML) branch where an agent learns decision-making by performing actions and receiving rewards, offers significant advantages over traditional methods [21,22,23]. Our study employed the Q-learning algorithm to optimize drug–variant interactions based on reward values derived from docking scores [24]. RL’s ability to balance exploration and exploitation ensured comprehensive analysis by identifying both well-known and less obvious high-reward interactions. The integration of RL represents a notable advancement in computational drug discovery. Traditional molecular docking methods often rely on static models and may be limited by biological system complexities. Conversely, RL’s dynamic learning process allows continuous improvement and adaptation, leading to more accurate predictions. This approach aligns well with the intricate nature of protein-drug interactions, where factors like conformational flexibility and allosteric effects play a critical role. We efficiently simulated and evaluated numerous drug–variant interactions by applying RL, accelerating the drug discovery. This method enhances our understanding of SLITRK1’s pharmacological profiles and demonstrates RL’s potential in broader bioinformatics applications, paving the way for future research and development in personalized medicine and targeted therapies(Figure 1). This study investigates a single protein and a foray into the broader landscape of neurobiology and protein dynamics. By elucidating the various roles of SLITRK1 and its interactions within the neural context, we aim to contribute to the ongoing efforts in developing targeted therapies for neurological disorders, thereby enhancing our understanding of the complex interplay between protein structure, function, and disease [25,26,27,28,29].

Figure 1.
This research extensively computes the SLITRK1 protein, emphasizing its structural behavior, tendency toward disorder, and interactions with various drugs. Beginning with the acquisition of the protein sequence and related drug data, the study explores the prediction of disordered regions, evaluates the effects of amino acid substitutions on protein stability, and maps its network of protein–protein interactions. The study also includes predictions of transmembrane topology, identifies evolutionarily conserved regions, and assesses the physicochemical properties of the protein. Additionally, 3D structural modeling and validation are performed, followed by molecular docking to examine drug binding affinities. The study leverages reinforcement learning to optimize drug–protein interactions and concludes with molecular dynamics simulations to explore structural changes caused by mutations. These combined analyses offer deeper insights into SLITRK1’s role in neurological disorders and pave the way for developing targeted therapeutic approaches.
2. Materials and Methods
2.1. Protein Sequence Data Retrieval and Drug Accession Retrieval
The entire amino acid sequence of the SLITRK1 protein (NP_001268432.1) was retrieved from the NCBI (National Center for Biotechnology Information) database [30]. This sequence was thoroughly analyzed to predict disordered regions using various bioinformatic tools. This analysis identified specific amino acid positions, as mentioned in the results section. The potential impact of these disordered regions on drug interactions was investigated. Therefore, the drugs Pimozide (CID:16362), Aripiprazole (CID:60795), Risperidone (CID:5073), and Haloperidol (CID:3559), which are used in the treatment of Tourette Syndrome (a condition in which SLITRK1 plays a role), were obtained from PubChem [31,32,33,34]. The consistency of the results was further validated by docking and molecular dynamics simulations using the FASTA version of this protein, both in its wild-type form and in variants containing changes in the identified disordered regions, with these drugs.
2.2. Prediction of Unregulated/Unstructured and Disorder Regions for SLITRK1 Protein
It is critical to highlight areas of potential structural disorder within proteins, as this is essential for understanding their functional dynamics and interaction capabilities [5,10,35]. Numerous methods for predicting intrinsically disordered proteins (IDPs) and regions have been published [36,37]. However, the large variety of available predictors can make it difficult to compare their performance, which may confound biologists seeking to make informed choices [35,37]. We used the Critical Assessment of Protein Intrinsic Disorder (CAID) to address this issue. CAID provides extensive documentation that explains the meaning of different CAID statistics and briefly describes all methods. It also offers results from many bioinformatic tools [37]. Consequently, multiple bioinformatic prediction tools were employed to predict unregulated/unstructured and disordered regions in the SLITRK1 protein. Using various bioinformatics tools to predict possible disorder regions enhances the scientific rigor of the study [36,37,38,39,40,41]. This approach yields more scientifically reliable outcomes than analyzing individual amino acid positions. All obtained results were downloaded as a ‘tsv’ file, and a heatmap of all bioinformatic tools was created using Python. We adhered to this heatmap and based on its average, the most likely disordered regions were predicted. The amino acid positions identified in this step were used in all other bioinformatic analyses.
2.3. Prediction of SLITRK1 Protein Stability Changes
After predicting the unregulated/unstructured and disordered regions of the SLITRK1 protein, I-Mutant 2.0 was used to estimate the effects of potential single-point mutations on the stability of the SLITRK1 protein. The temperature was set to 37 °C, and the pH was adjusted to 7.4 [42].
2.4. Evolution of the SLITRK1 Protein and SNP Prediction Associated
The regions predicted through the combined utilization of CIAD and Python-based algorithms were subsequently analyzed using the Protein Analysis Through Evolutionary Relationships (PANTHER) classification system [43]. This analysis evaluated the evolutionary and functional aspects of the SLITRK1 protein regions. Additionally, the Prediction of Harmful Single Nucleotide Polymorphism (PhD-SNP) tool was employed to assess the potential impact of point mutations within these regions [44].
2.5. Prediction of Wild-Type and Mutant-Type SLITRK1 Amino Acid Properties
The regions predicted through the combined utilization of CIAD and Python-based algorithms were examined using HOPE (Have Our Protein Explained) to predict the changes that point mutations can cause in the structure and function of the SLITRK1 protein [45].
2.6. Prediction of Interaction SLITRK1’s Protein with Possible Proteins
The STRING tool is essential because it provides a comprehensive and systematic resource for predicting, analyzing, and interpreting protein–protein interactions [46]. It aids in hypothesis generation, functional annotation, network analysis, and comparative genomics, thereby advancing our understanding of the complex interactions and regulatory mechanisms within biological systems [46,47]. Consequently, we used this tool to predict which proteins may interact with the SLITRK1 protein. Both the normal FASTA sequences and those containing mutations of the SLITRK1 protein were separately uploaded into the tool.
2.7. Prediction of the Transmembrane Topology of the SLITRK1 Protein
Understanding the transmembrane topology of the SLITRK1 protein is essential for predicting its interactions with drugs [48]. his knowledge enables us to comprehend how drugs may interact with the protein at the cellular level, influencing its function and potential therapeutic applications. Therefore, we used Phobius to predict the transmembrane topology and signal peptides from the SLITRK1 amino acid sequence [49,50]. The topology of a transmembrane protein refers to the positions of the N- and C-termini of the membrane-spanning polypeptide chain relative to the inner or outer sides of the biological membrane occupied by the protein [50].
2.8. Prediction of the Evolutionary Pattern of Amino Acids on SLITRK1 Protein
The ConSurf tool was used to predict the conservation of amino acids at the evolutionary level [27]. The results from this tool are based on a set of homologous sequences gathered using hidden Markov model-based search tools recently integrated into the pipeline [51].
2.9. Prediction of Physicochemical Parameters of SLITRK1
Expasy ProtParam was used to predict the physical and chemical properties of the SLITRK1 protein [52]. Predicting these physicochemical parameters enhances our understanding of SLITRK1’s structure, function, and behavior, thereby enabling more informed investigations into its role in biological processes and facilitating targeted drug discovery and protein engineering efforts [53].
2.10. Structural Modeling and Verifying
To obtain the 3D structure of the SLITRK1 protein, we utilized both the Phyre2 and trRosetta servers [54,55]. First, we employed Phyre2 to generate a predicted 3D structure for the SLITRK1 protein with high coverage and at least 80% confidence [54,56]. Since trRosetta requires an initial 3D structure to refine and generate more accurate models, we uploaded the SLITRK1 protein structure file obtained from Phyre2 in the appropriate PDB format [54,55]. After completing the trRosetta analysis, we received a list of predicted models and their corresponding confidence scores. We selected the model with the highest confidence score as the best model. To visualize the selected model, we used Chimera 1.15 software [57]. The Ramachandran plot was then employed to assess the correct folding, stability, and functionality of the protein, providing insights into how different regions of the protein fold and aiding in the design of new protein structures [58,59]. By investigating the structures of both the wild-type and mutant forms of the SLITRK1 protein (from Phyre2 and trRosetta) using the Ramachandran plot, we determined whether the potential mutation altered the amino acids in the structure.
2.11. Small Tertiary Structures Are Predicted, and Structures Visualized
The small tertiary structures of the drugs Pimozide (CID:16362), Aripiprazole (CID:60795), Risperidone (CID:5073), and Haloperidol (CID:3559) were obtained and visualized using Chimera 1.15 [57]. These structures were used for both docking and molecular dynamics simulations. Prior to these analyses, energy minimization was performed using Chimera 1.15 [57].
2.12. Molecular Docking Between Drugs and Our Proteins
Both the HDOCK and PyRx servers were used to predict the docking scores between the drugs Pimozide (CID:16362), Aripiprazole (CID:60795), Risperidone (CID:5073), and Haloperidol (CID:3559), and the SLITRK1 protein (including its variants) for receptor interactions [60]. After utilizing HDOCK, we conducted additional molecular docking studies using PyRx to increase the probability of obtaining accurate results. PyRx was employed for both single and multiple docking studies [61]. We then continued our study based on the results obtained from both tools, as it is essential to have multiple bioinformatics tools corroborate each other’s findings [60,61]. The affinity scores of the best complexes were selected for Molecular Dynamics simulations, using an affinity scoring function, ΔG [U total in kcal/mol], to rank the candidates [60,62].
2.13. Evaluation of Drug Interactions for SLITRK1 Protein Variants Using Reinforcement Learning
This study employed reinforcement learning (RL) algorithms to evaluate the interactions of SLITRK1 protein variants with different drugs [63,64]. Initially, data files were loaded using the Pandas library, followed by data cleaning steps. First, the presence of NaN values and data types were checked. Non-numeric columns were converted to numeric values, and rows containing NaN values were removed from the dataset. The RL algorithms were then applied to the cleaned dataset [24]. In the RL algorithm, the discount factor for future rewards (gamma) was set at 0.95, the learning rate (alpha) at 0.8, and the exploration rate (epsilon) at 0.1. The Q-table was initialized as a matrix of zeros, with dimensions corresponding to the number of variants in the cleaned dataset and four different drugs (4 columns) [22]. The reward function was designed to convert negative energy values into positive rewards. An epsilon-greedy policy was applied during the training process to randomly select or choose the best drug for each variant. The reward value for the selected drug was obtained, and the Q-table was updated using the Q-learning update rule. This process was repeated for 1000 episodes, with rewards obtained in each episode recorded [65,66]. The best drug selection for each variant was determined upon completion of the training, and performance metrics were generated. These metrics were presented as a data frame containing each variant’s cumulative reward and the best drug information. Additionally, the average episode reward was plotted against the number of episodes. This method demonstrates the applicability of RL algorithms for evaluating drug interactions of SLITRK1 protein variants and identifying the most effective treatment options.
2.14. Molecular Dynamics Simulation of SLITRK1 Protein with Drugs
In this study, molecular dynamics simulations were conducted using GROMACS 2024.4 software (http://www.mdtutorials.com/gmx/complex/index.html, (accessed on 1 February 2024)), with the AMBER ff99SB force field applied for precise energy calculations and system interactions [67,68]. The protein–ligand complexes were solvated in an SPC (Simple Point Charge) water model within a triclinic box, ensuring ample space for molecular movement and minimizing boundary effects. The system was neutralized using NaCl to achieve a physiological salt concentration of 0.15 M, which was used to explore its impact on system stability. Energy minimization was performed using the Steepest Descent method over 5000 steps, ensuring the system reached a local energy minimum and was adequately prepared for subsequent simulation stages. Equilibration of the system was carried out using both NVT (constant Number of particles, Volume, and Temperature) and NPT (constant Number of particles, Pressure, and Temperature) ensembles at 300 K and 1.0 bar for 300 ps to ensure a stable and physiologically relevant simulation environment. The molecular dynamics simulations were then run for 50 ns, employing the Leapfrog integrator for numerical integration of the equations of motion, with 5000 frames saved throughout the simulation to provide a detailed and comprehensive representation of the system’s conformational space and dynamics.
3. Results
3.1. Prediction of Unregulated/Unstructured and Disorder Regions for SLITRK1 Protein
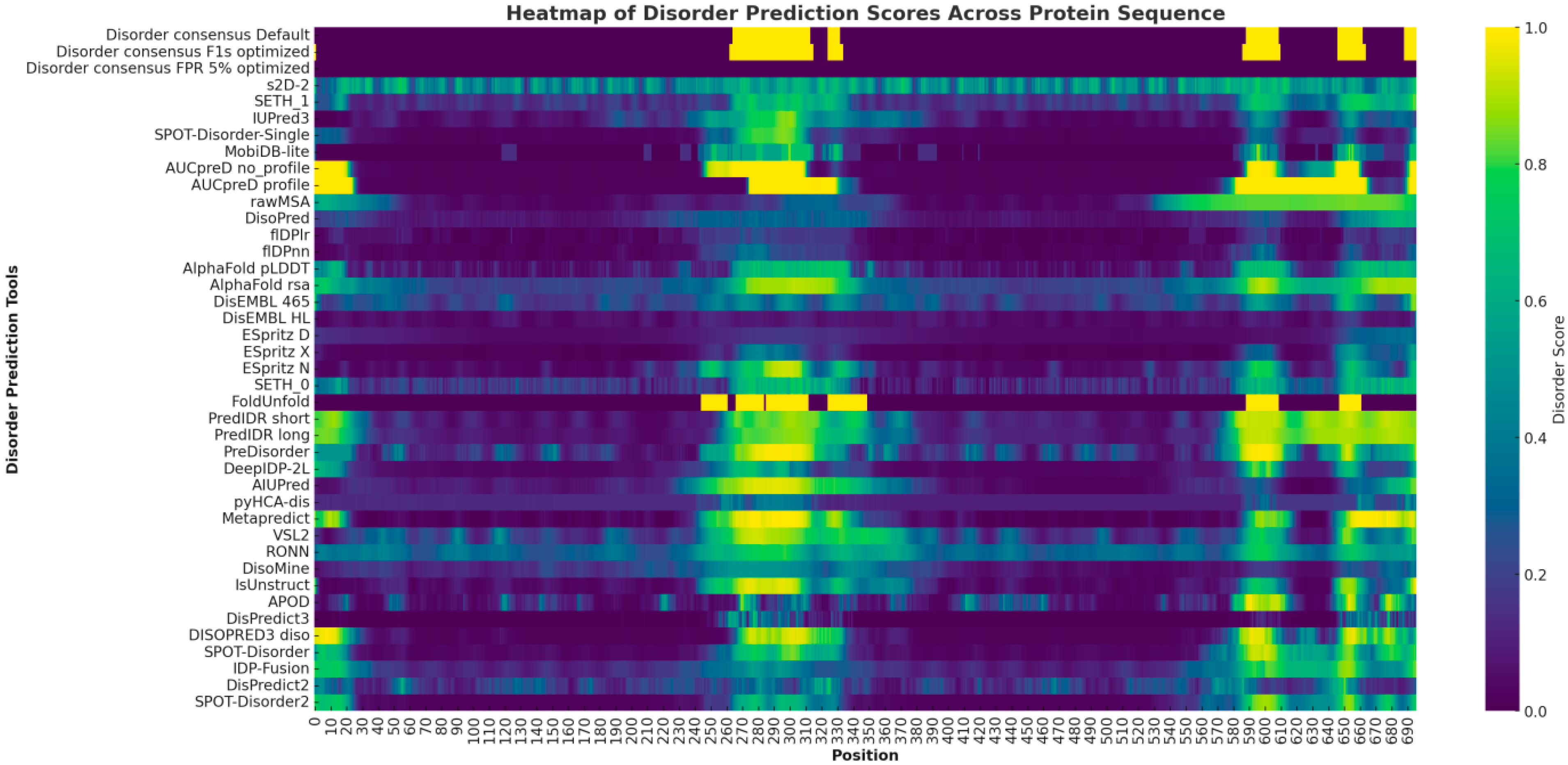
The heatmap prominently highlights several regions with disorder predictions for the SLITRK1 protein. At the N-terminus, approximately within positions 1–50, there is a display of low to moderate disorder predictions across various tools. The central region of the protein, particularly between positions 250 and 550, is characterized by intense yellow and green hues, indicating a high propensity for the disorder as consistently predicted by multiple algorithms. Lastly, around positions 850–900, the C-terminal region exhibits significant indications of disorder intensity (Figure 2).
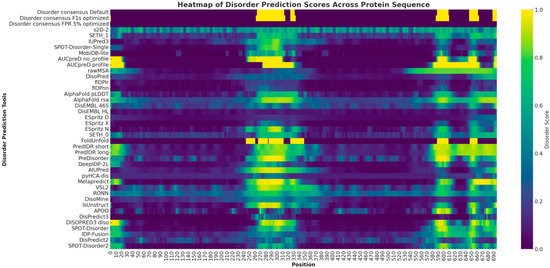
Figure 2.
The heatmap provides a comprehensive visualization of disorder prediction scores for the SLITRK1 protein, as determined by various bioinformatic tools. Each row represents a different predictive tool, while each column corresponds to a specific amino acid position within the protein sequence. The intensity of the yellow color in the heatmap reflects the degree of predicted disorder.
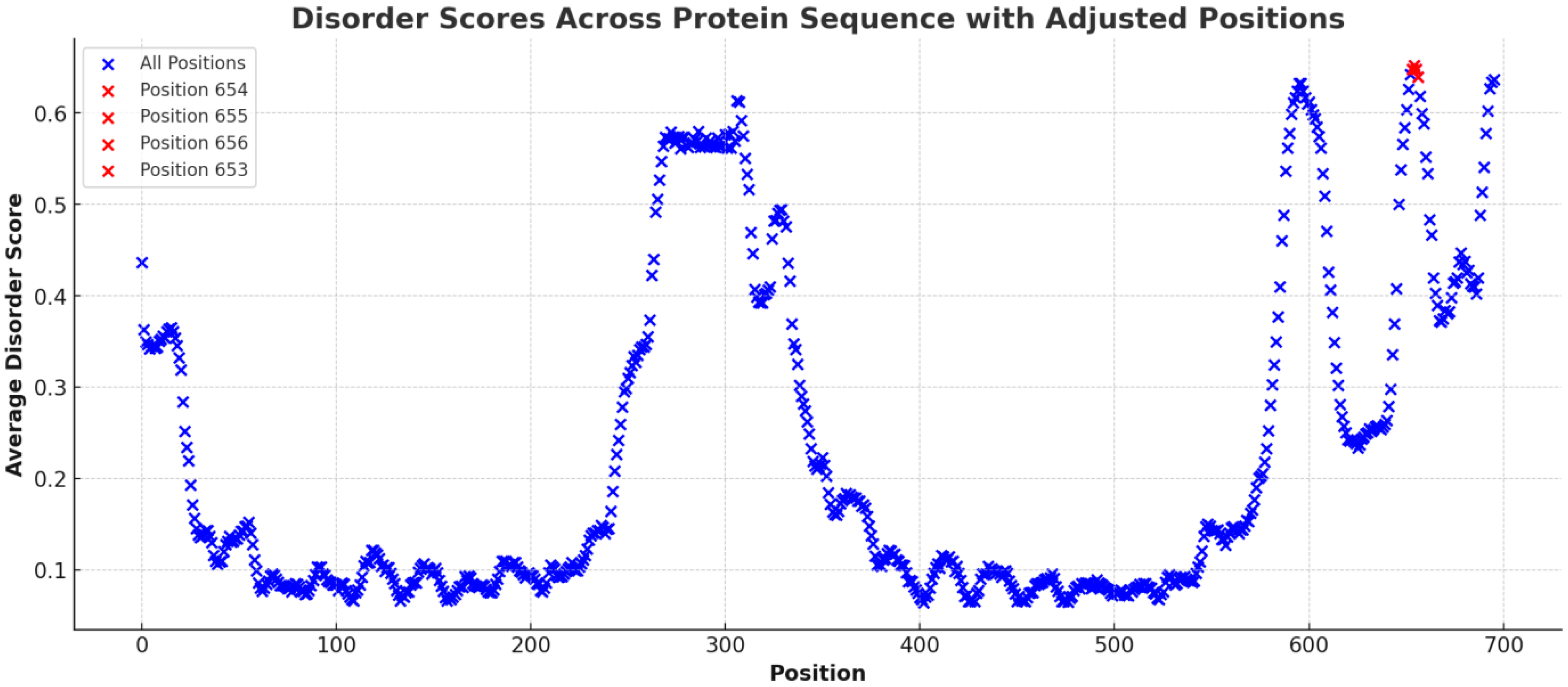
In Figure 2, the mean disorder scores for the amino acid sequence of the SLITRK1 protein were determined by averaging the data across various predictive tools. The results highlighted certain regions with elevated average scores, with position S656 notably standing out with a substantially higher mean disorder score than other residues (S656: Mean Disorder Score ≈ 0.67) (Figure 3). Regions with exceptionally high disorder scores, including positions 656 (Mean Disorder Score ≈ 0.67), 654 (Mean Disorder Score ≈ 0.656), 653 (Mean Disorder Score ≈ 0.656), and 652 (Mean Disorder Score ≈ 0.655), are emphasized in red in the graphic (Figure 3).
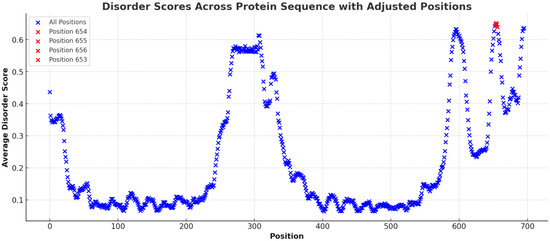
Figure 3.
The figure illustrates the distribution of average disorder scores across the amino acid sequence of the SLITRK1 protein. Each point on the plot corresponds to an amino acid position, with the associated average disorder score derived from various bioinformatic prediction tools. Notably, the graph highlights regions with significantly elevated disorder scores, indicated by red coloration. These regions include amino acid positions 656 (Average Disorder Score ≈ 0.67), 654 (Average Disorder Score ≈ 0.656), 653 (Average Disorder Score ≈ 0.656), and 652 (Average Disorder Score ≈ 0.655).
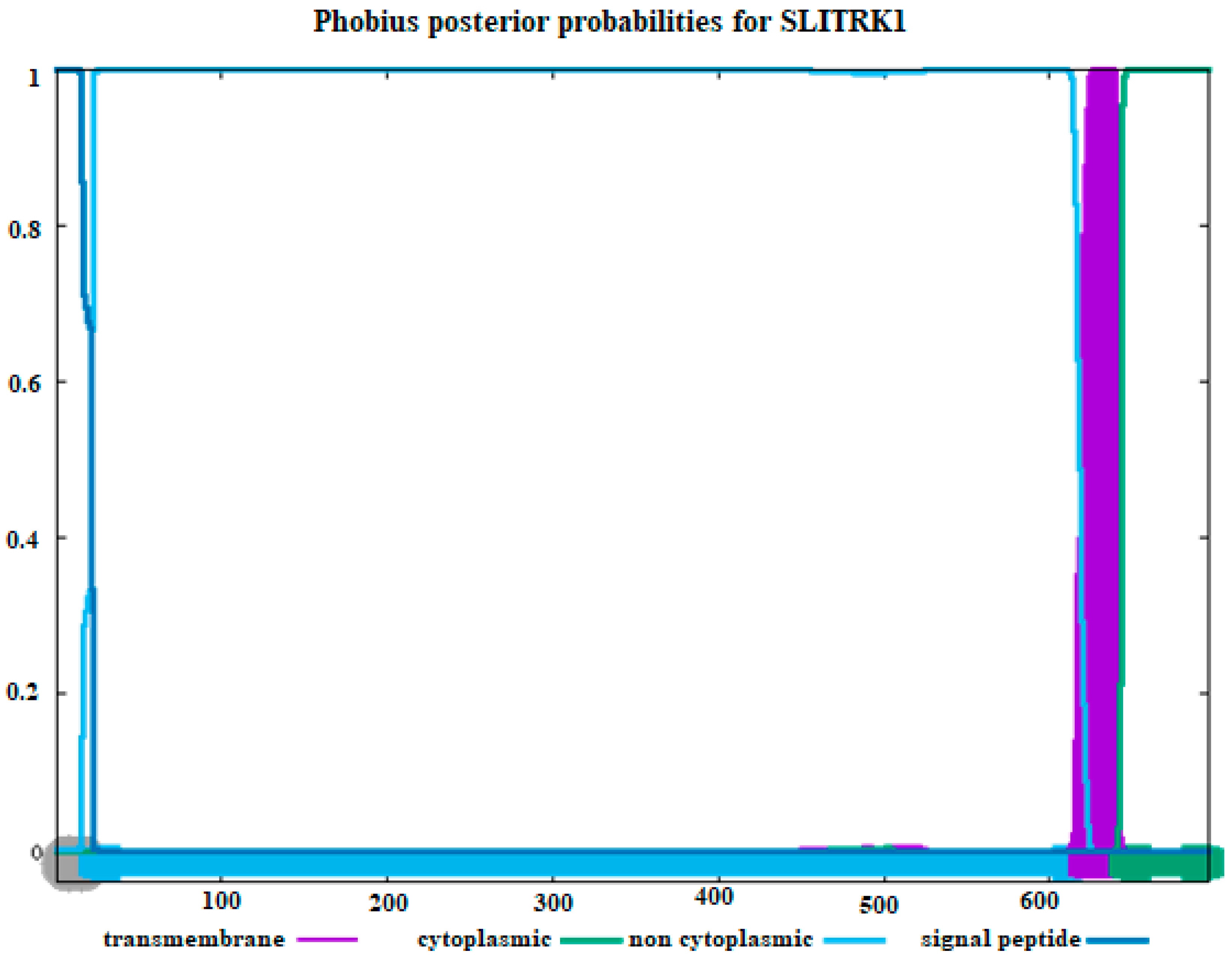
3.2. Prediction of the Transmembrane Topology of the SLITRK1 Protein
The amino acid sequence of our sample was used to determine the transmembrane topology and signal peptides of the SLITRK1 protein, as presented in Figure 4. It was found that the amino acids of SLITRK1 located beyond the 650th position are situated in the cytoplasm. Additionally, most of the amino acids studied in SLITRK1 are not situated in the cytoplasm, as depicted in Figure 4. The regions identified between positions 250 and 300 in Figure 2 and Figure 3 were not further emphasized because, according to Figure 4, they are located within the cytoplasmic area.
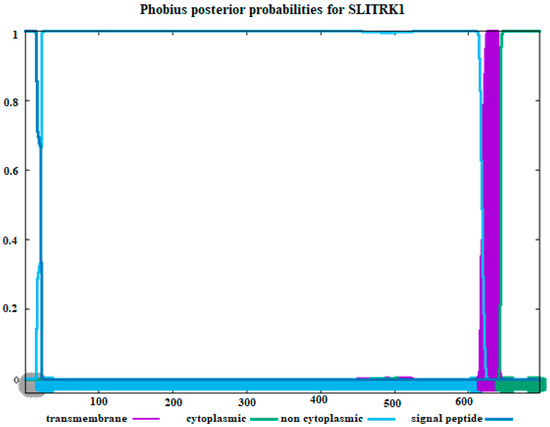
Figure 4.
Estimating transmembrane topology and signal peptides using amino acid sequences of SLITRK1. (Purple color: transmembrane, green color: cytoplasmic part, blue: non-cytoplasmic part, and dark blue color: signaling peptides). The X–axis represents amino acid positions along the SLITRK1 protein sequence, and the Y–axis indicates the posterior probability (0 to 1) for each functional region.
3.3. Prediction of Both Protein Stability Changes and Evolution of the SLITRK1 Protein, as Well as Obtaining SNP Prediction
To maintain scientific rigor, the consistent findings derived from both Figure 2 and Figure 3 were utilized in subsequent stages of the analysis. However, additional amino acid positions were also subjected to the full range of analyses. Most of the results corroborated the data presented in Figure 2 and Figure 3. Table 1 provides a detailed evaluation of various amino acid variants in the SLITRK1 protein using three computational tools: I-MUTANT, PANTHER, and PhD-SNP. I-MUTANT assesses the stability changes (‘Decrease’ or ‘Increase’) caused by each variant, assigning a specific score to quantify this effect. The highest decrease in stability is observed for the A654S variant (Score: 9), while several variants, including N655L and all S656 substitutions, are shown to increase stability, each receiving a uniform score of 6. Additionally, results were obtained for amino acid positions beyond those specified in this table. Further details can be found in Table S1.

Table 1.
Substitution or deletion of an amino acid is a demonstration of estimating whether it affects the function.
3.4. Prediction of Wild-Type and Mutant-Type SLITRK1 Amino Acid Properties
Table 2 evaluates the properties of wild-type and mutant amino acids in the SLITRK1 protein using the HOPE tool. The fundamental properties analyzed include size, charge, hydrophobicity, and the potential impact on protein conservation. The table reveals several notable differences between wild-type and mutant amino acids. For example, the size of the amino acid often changes in mutants (e.g., A654G, A654H), which can lead to interaction loss or the formation of bumps in the protein structure. The charge and hydrophobicity also vary in many variants, such as S656E and S656R, potentially affecting the protein’s stability and folding. Some variants, including N655L and S656M, are indicated to disrupt hydrogen bonding and proper folding, suggesting significant alterations in protein functionality. The HOPE tool’s assessment of the impact on protein conservation varies, with several mutations, such as A654S, N655H, and S656F, being classified as potentially more damaging, as indicated by ‘Probably Yes’ in the conservation column. Additionally, results were obtained for amino acid positions beyond those specified in this table. Further details can be found in Table S2.
Table 2.
The wild-type and mutant amino acid results from the Hope tool (SLITRK1: NP_001268432.1) properties. a = no results were obtained using the HOPE tool.
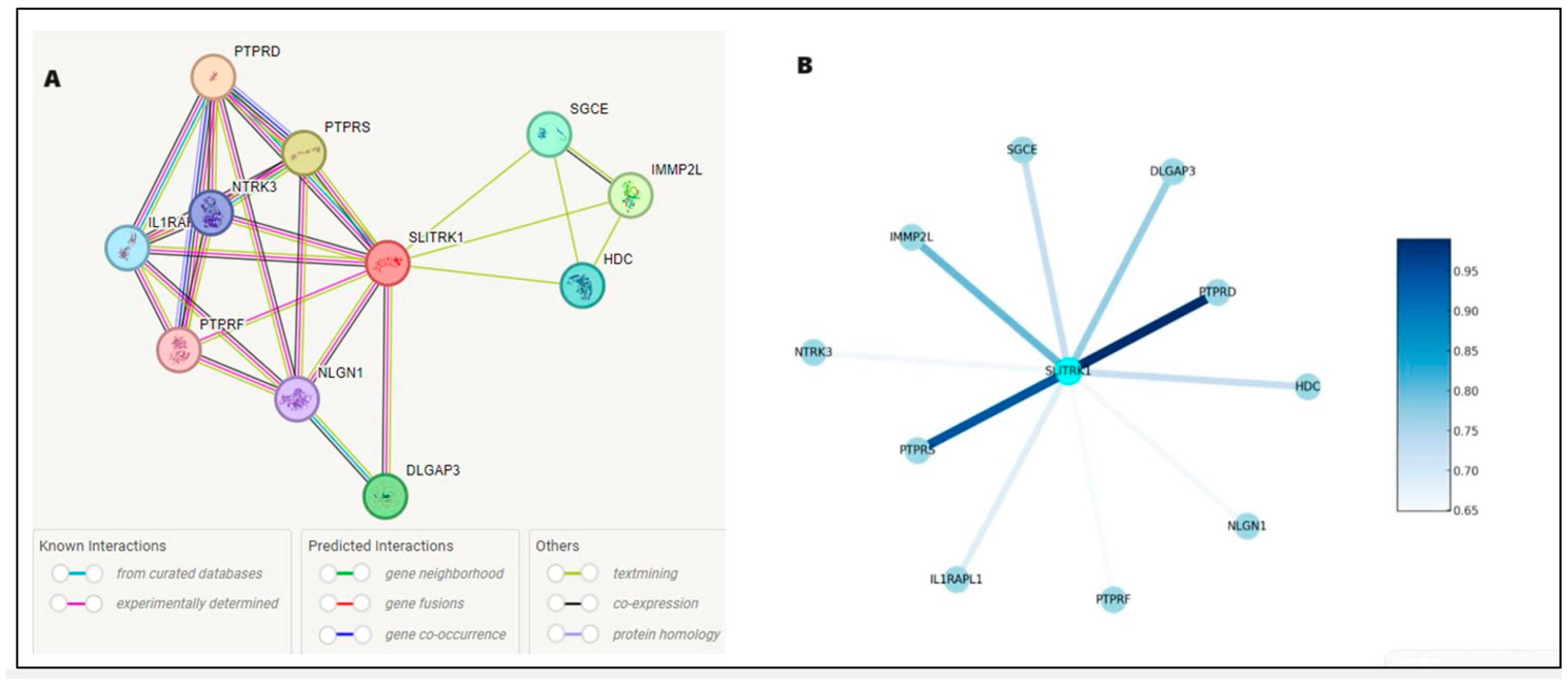
3.5. Interaction of SLITRK1’s Protein with Possible Proteins
Figure 5 illustrates the protein–protein interaction network for SLITRK1, with SLITRK1 positioned at the core as the principal node, interconnected with various proteins, including SGCE, NPTX1, IGHMBP2, PTPRS, PTPRD, GRIN1, DLGAP3, NRXN1, OPCML, and IL1RAP. The interaction between SLITRK1 and PTPRD is well-documented in databases. Additionally, experimental evidence shows that SLITRK1 interacts directly with PTPRS and PTPRD and indirectly with IL1RAP, NRXN1, DLGAP3, and GRIN1. The separate analysis of normal and mutated FASTA sequences did not alter these interactions (Figure 5A) but only affected the binding scores. Results predicted using bioinformatics tools were downloaded as a TSV (Tab-Separated Values) file, and Python was used to analyze these interactions based on their scores.
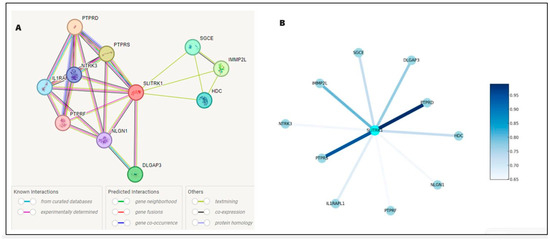
Figure 5.
Delineates the protein–protein interaction network for SLITRK1, mapping its extensive connections within the cellular framework. The interactions are distinguished by a color scheme: gene neighborhood is marked in yellow, known database interactions in blue, experimentally determined interactions in purple, and gene coexistence in dark blue Panel (A). Panel (B) elucidates the interaction strengths, employing a color gradient where lighter shades of blue represent interactions with lower confidence scores, transitioning to darker shades for interactions with higher confidence. A notable example is the interaction between SLITRK1 and PTPRD, depicted in dark blue, indicating a high-confidence association and underscoring its potential significance in the protein–protein interaction landscape.
The intensification of color in the network indicates a greater number of interactions. The connecting lines (edges) represent the interactions between SLITRK1 and these proteins, with the intensity of the line color reflecting the strength of the evidence supporting each interaction—the darker the line, the higher the confidence in the data. The accompanying legend quantifies the color gradient with numerical values, translating lighter blues to lower confidence scores around 0.65, progressing to darker blues for higher confidence levels, reaching up to 0.95 (Figure 5B). For instance, the interaction of SLITRK1 with PTPRD is indicated by a darker shade, signifying a more robust confidence score, in contrast to its interaction with IL1RAPL1, represented by a lighter shade.
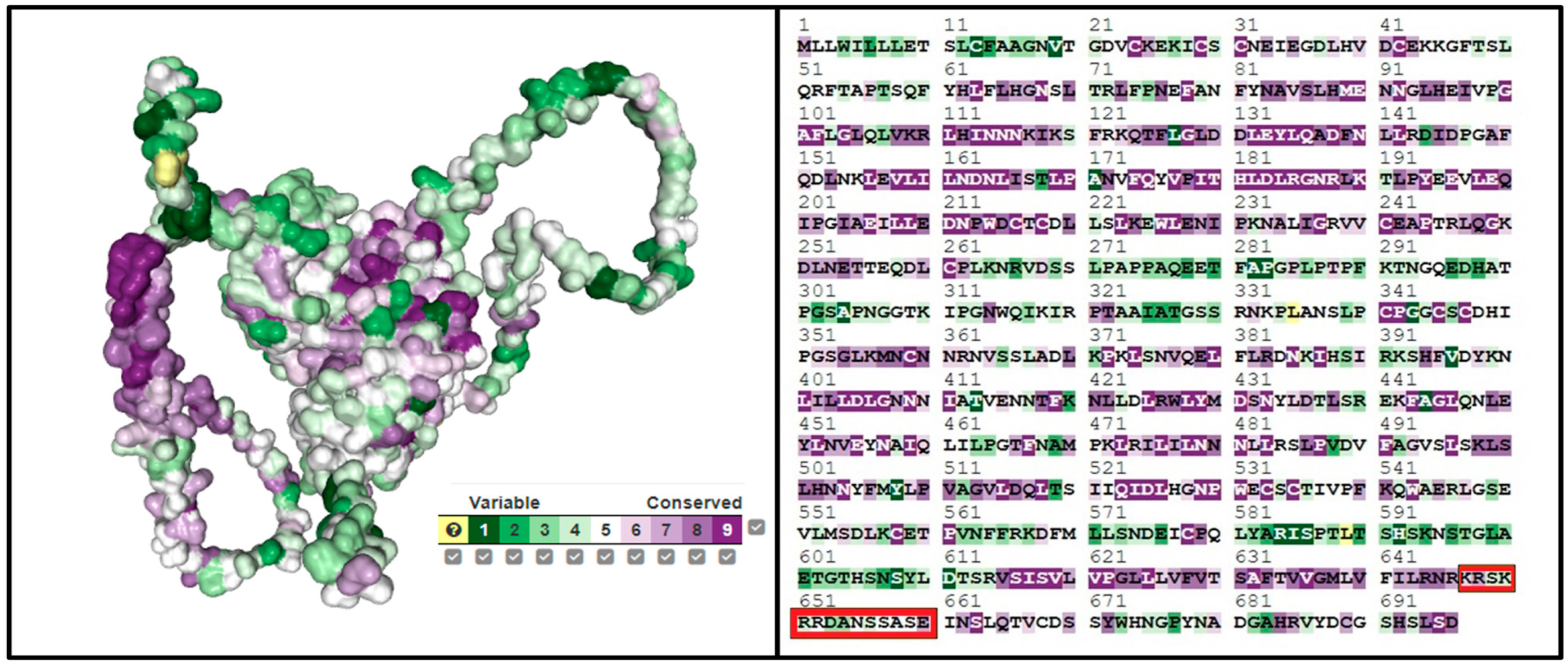
3.6. Prediction of the Evolutionary Pattern of Amino Acids on SLITRK1 Protein
We employed the ConSurf tool (consurf.tau.ac.il, accessed on 5 February 2024) to investigate conserved domains within the SLITRK1 protein, with the results presented in Figure 6. An analysis via the ConSurf server revealed a general absence of conserved amino acids in SLITRK1, except for specific regions encompassing residues K647, R648, and E660. The scores assigned to the amino acids from K647 to E660 are as follows: K647 scored 6, indicating moderate conservation; R648 scored 7, suggesting higher conservation; while S649, K650, and R651 each scored 4, reflecting lower conservation. R652 and N655 were moderately conserved with scores of 5. D653 and A654, with the lowest scores of 3, are the most variable within this segment. The highest score, indicating significant conservation, was given to E660 with a score of 8, approaching the maximum score of 9 on the ConSurf scale. This scoring reflects a gradient of conservation within this stretch of the SLITRK1 protein, identifying highly conserved and highly variable regions that may be crucial to the protein’s function and interactions.
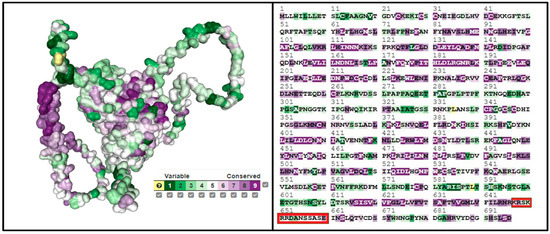
Figure 6.
Estimation of the detection of conserved regions of the SLITRK1 protein using ConSurf. It is the sequences we have determined, indicated by the red color.
3.7. Determination of Estimated Physico-Chemical Parameter of SLITRK1
Our findings indicate that the regular SLITRK1 protein has a molecular weight of 77,734.88 kDa. Among the potential outcomes, the A654W and A658W mutations displayed the highest molecular weights, approximately 77,850.01 kDa. In contrast, the R648G and R651G mutations had the lowest molecular weights, roughly 77,635.74 kDa, among all the mutant and regular forms. Table S3 provides detailed results, including these findings. The theoretical isoelectric point (pI) of normal SLITRK1 was estimated to be 6.00, while the theoretical pI of all other SLITRK1 variants ranged from 5.86 to 6.16 (Table 3). All normal and variant forms of SLITRK1 were predicted to be negatively charged. The estimated half-lives were 30 h for all proteins in mammalian reticulocytes (in vitro), over 20 h in yeast (in vivo), and over 10 h in E. coli (in vivo). All results were deemed unstable according to the instability index (Table 3). Table 4 presents potential changes (mutations or deletions) that may affect the stability of SLITRK1. The aliphatic index exhibited significant variation among all variant proteins, ranging from 97.36 to 98.06, while the aliphatic index of the normal SLITRK1 was 97.50. The overall weighted aliphatic index ranged from 97.50 to 98.06 (Table 3). The overall mean hydropathy value of SLITRK1 was negative across all variations, with the normal value being −0.173. The remaining GRAVY values ranged between −0.160 and −0.174 (Table 3). These predicted physicochemical parameters are significant in terms of their potential impact on the stability of SLITRK1, alterations in the aliphatic index, potential in vitro and in vivo half-life studies, and the role of hydrophobicity in SLITRK1.
Table 3.
Presents the estimated physicochemical parameters of the SLITRK1 protein, as analyzed by the ExPASy ProtParam tool, with the reference sequence NP_001268432.1 designated as ‘Normal’, indicating the absence of substitutions. The predicted half-life for all estimates vary across different biological systems: for mammalian reticulocytes, the half-life is approximately 30 h; in yeast, the in vivo half-life is greater than 20 h; and in E. coli, the in vivo half-life exceeds 10 h.
Table 4.
Encapsulates the molecular docking scores quantifying the binding affinities between the wild-type SLITRK1 protein and its variants against four drugs—Pimozide, Aripiprazole, Risperidone, and Haloperidol—employed in the management of Tourette Syndrome. The affinities are calculated using the HDOCK server. They are expressed in kilocalories per mole (kcal/mol), with ‘Normal’ indicating the scores for the wild-type protein without any amino acid substitutions.
3.8. Molecular Docking Scores Between SLITRK1 Protein and Pimozide, Aripiprazole, Risperidone, and Haloperidol Drugs
Table 4 provides an overview of molecular docking scores between the SLITRK1 protein and a set of drugs commonly used in the treatment of Tourette Syndrome, including Pimozide, Aripiprazole, Risperidone, and Haloperidol. These scores, measured in kcal/mol, were obtained using the HDOCK server and are used to predict the binding affinity between the protein and the drugs; lower scores generally indicate stronger binding affinity. The ‘Normal’ condition refers to the wild-type SLITRK1 protein without any amino acid substitutions. Across the different drug interactions, the Normal variant consistently shows specific docking scores for each drug, serving as a baseline for comparisons.
Upon examining the variants, most substitutions—such as D653N/Q, A654G/H/S/T, N655H/L, and S656E/I/R/W—maintain similar docking scores to the Normal, indicating that these mutations do not significantly alter the binding affinity between SLITRK1 and the drugs. However, there are notable exceptions. For example, the S656M variant demonstrates significantly stronger docking scores for all drugs, particularly for Pimozide (from −140.77 to −157.90 kcal/mol) and Risperidone (from −167.96 to −172.66 kcal/mol), suggesting an increased binding affinity due to this mutation.
These findings imply that, while most SLITRK1 variants do not affect the molecular interaction with these drugs, certain mutations, specifically S656M, could potentially influence the efficacy of Aripiprazole and the overall binding affinity of SLITRK1 to the studied drugs. This could have implications for the pharmacodynamics of these medications in individuals with these specific SLITRK1 mutations. Table S4 includes all other predicted results. The S656M mutation altered the docking scores for all drugs.
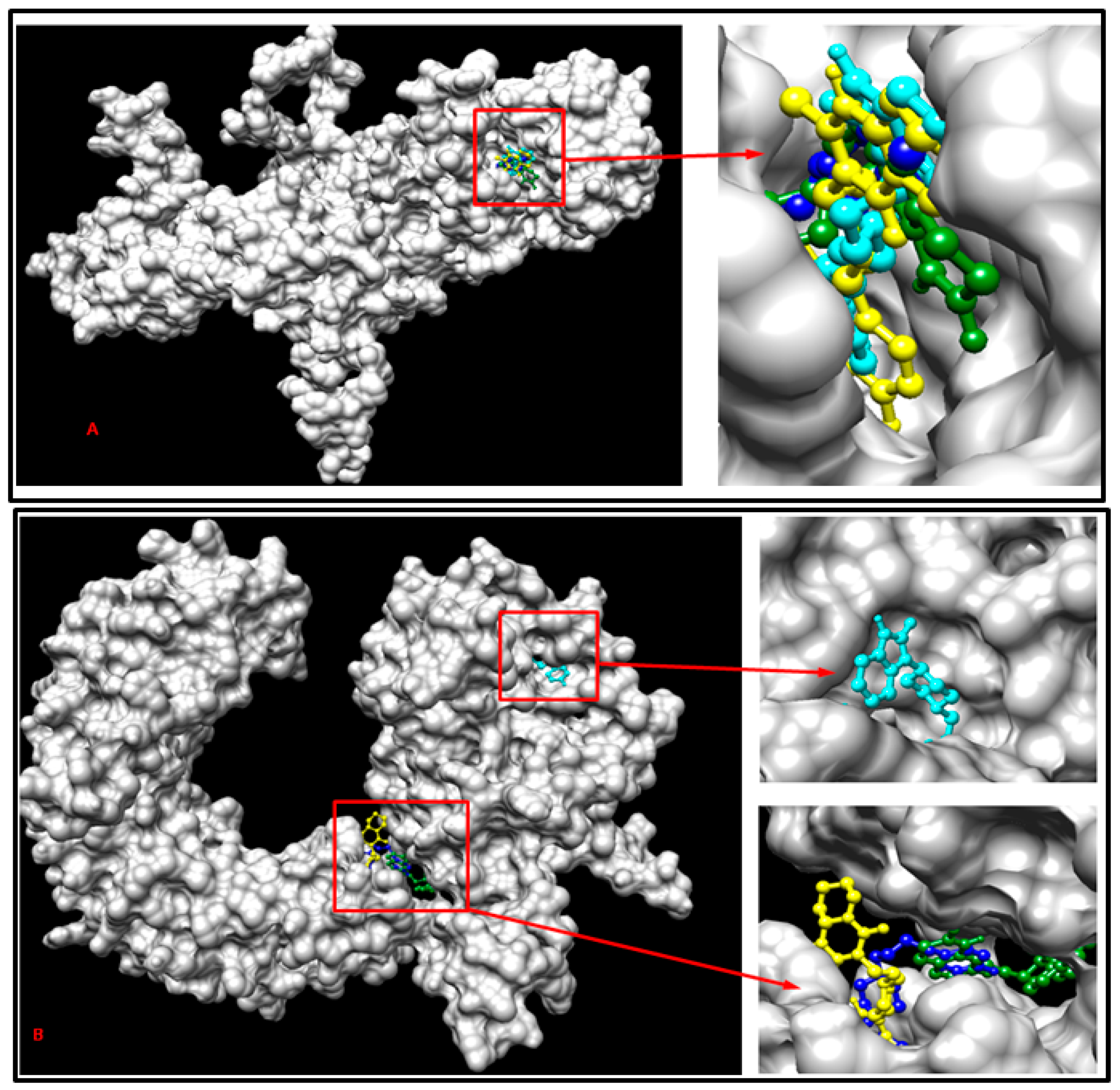
The normal SLITRK1 protein and the SLITRK1 protein with the S656M mutation underwent batch docking with the drugs Pimozide, Aripiprazole, Risperidone, and Haloperidol. The findings suggest that the S656M mutation induced structural modifications in the protein’s 3D conformation and influenced the drug-binding sites (Figure 7). Notably, the binding site of Pimozide on both the normal and mutant proteins closely resembled each other. At the same time, considerable alterations were observed in the binding sites for all the other ligands (Figure 7).
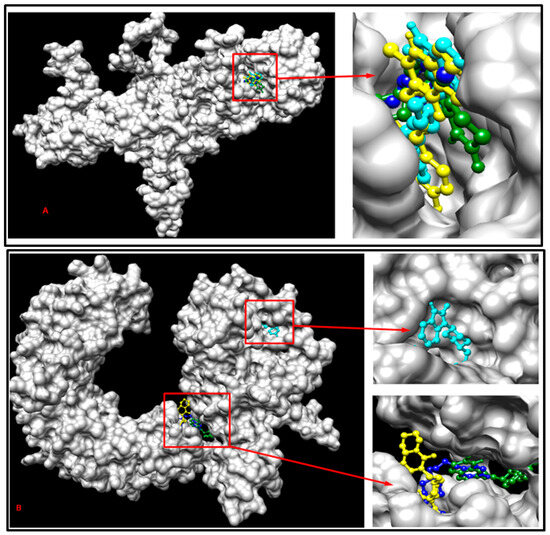
Figure 7.
Demonstration of multiple docking results with SLITRK1 protein-related drugs containing the S656M mutation (A) and with normal SLITRK1 protein and related drugs (B). The cyan color represents Pimozide, the yellow color represents Risperidone, the dark green represents Aripiprazole, and the blue color represents Haloperidol for both (A) and (B).
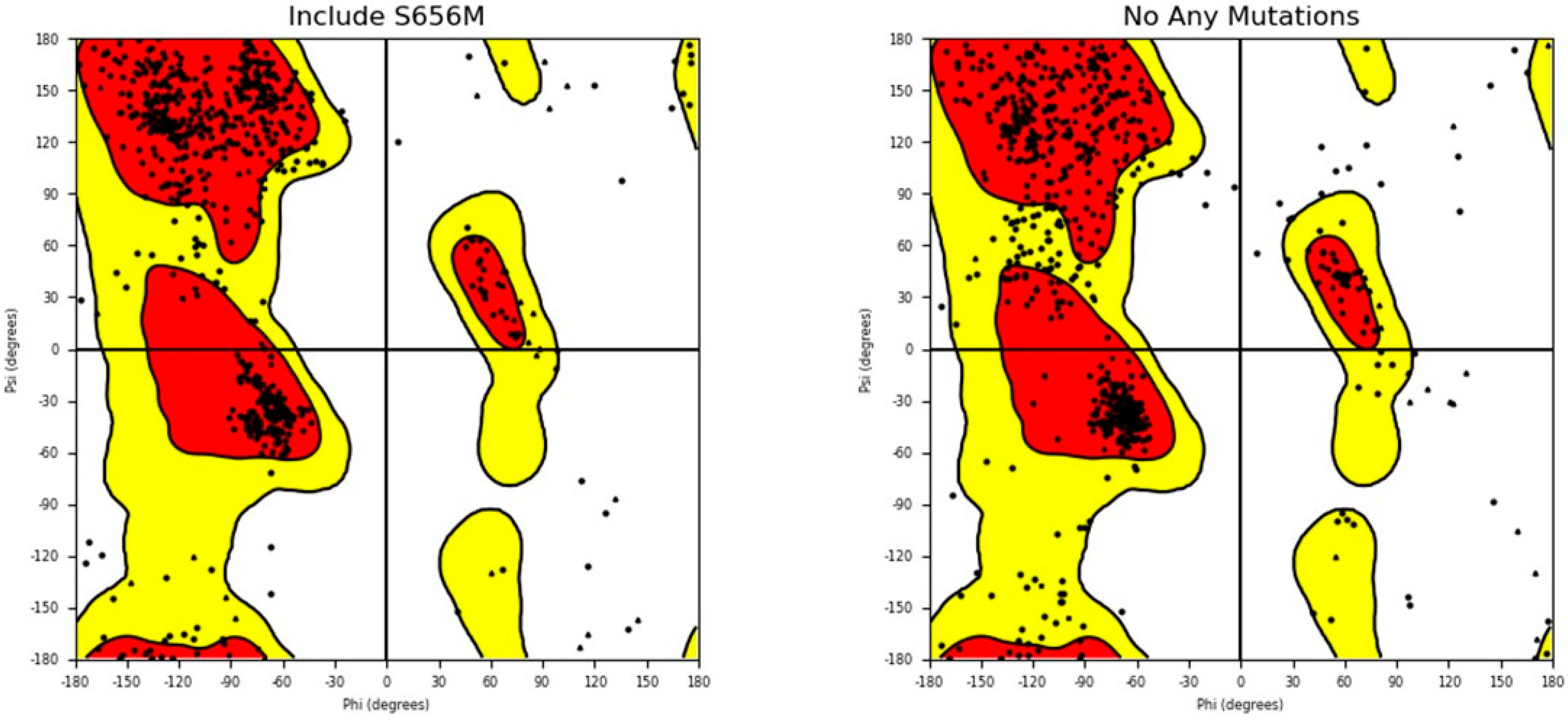
Additionally, the structures of both the wild-type and mutant forms of the SLITRK1 protein were analyzed using the Ramachandran plot to determine whether the S656M mutation altered the amino acid configuration within the structure (Figure 8). The results indicate that the S656M mutation significantly affected the amino acid arrangement (Figure 8).
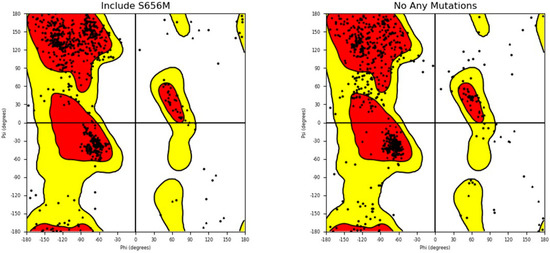
Figure 8.
Ramachandran plot analysis of both wild - type SLITRK1 and S656M mutant proteins. The plot illustrates the distribution of phi and psi angles for the amino acid residues in the protein structures. The comparison allows for assessing any structural changes resulting from the S656M mutation.
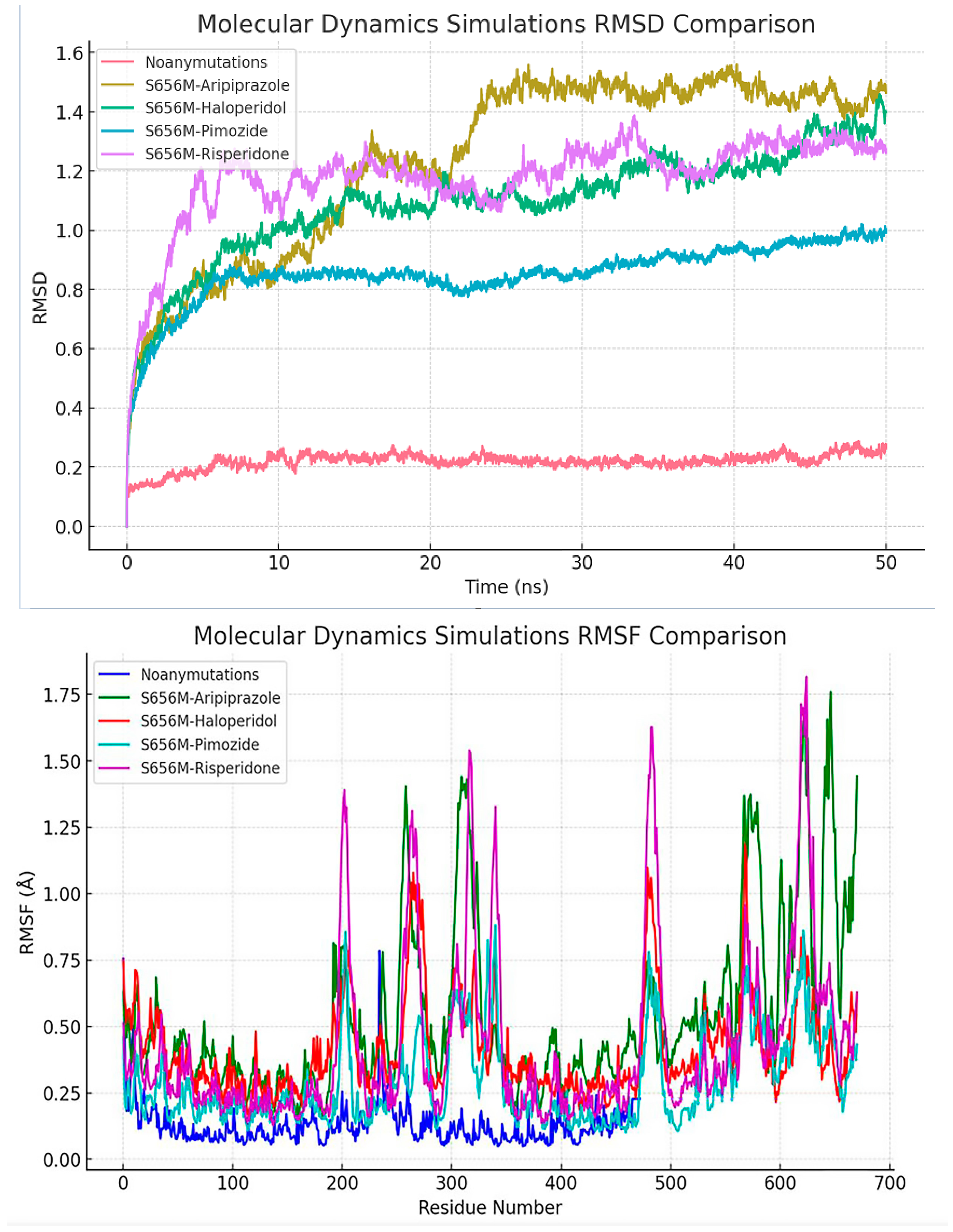
In comparison to the other simulations, the ‘Noanymutations’ scenario manifests a more stable conformation, as evidenced by its lower RMSD (Root Mean Square Deviation) values, indicating a substantial degree of structural constancy in the protein. On the other hand, the ‘S656M-Aripiprazole’ simulation exhibits a rapid increase in RMSD at the outset, subsequently plateauing to a stable state, denoting an initial phase of conformational adjustments followed by stabilization. In contrast, the ‘S656M-Haloperidol’, ‘S656M-Pimozide’, and ‘S656M-Risperidone’ simulations present higher RMSD values accompanied by fluctuations over time, reflecting a more dynamic behavior of the protein and suggesting susceptibility to conformational changes under these conditions (Figure 9).
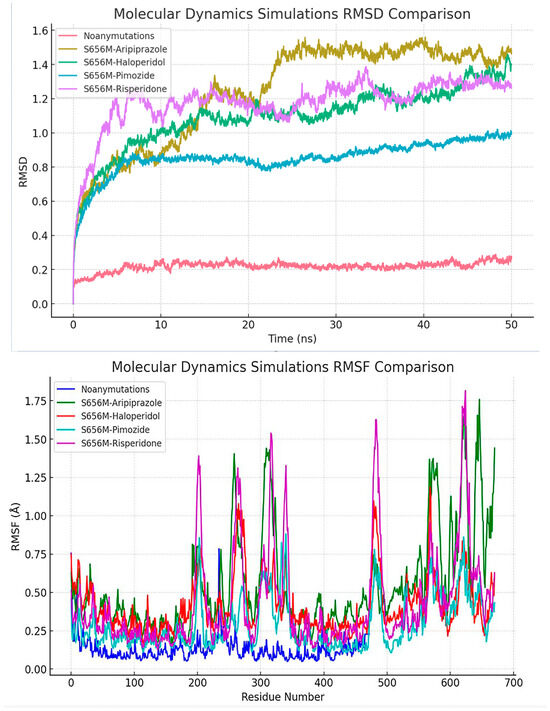
Figure 9.
Provides a comprehensive comparison of the RMSD (Root Mean Square Deviation) values obtained from various molecular dynamics simulations. The simulations include ‘Noan - ymutations’, ‘S656M - Aripiprazole’, ‘S656M - Haloperidol’, ‘S656M - Pimozide’, and ‘S656M - Risperidone’. The ‘Noanymutations’ simulation exhibits a notably stable behavior with lower RMSD values, indicating higher structural stability throughout the simulation. In contrast, ‘S656M - Haloperidol’, ‘S656M - Pimozide’, and ‘S656M - Risperidone’ simulations demonstrate more dynamic behaviors with higher RMSD values and larger fluctuations, suggesting potential conformational changes during the simulation period. ‘S656M - Aripiprazole’ shows a balanced behavior with a moderate range of RMSD values. Also, this figure illustrates the comparative analysis of root mean square fluctuation (RMSF) values across different molecular dynamics simulations, including ‘Noanymutations’, ‘S656M - Aripiprazole’, ‘S656M - Haloperidol’, ‘S656M - Pimozide’, and ‘S656M - Risperidone’. RMSF values provide insights into the flexibility and dynamic behavior of the protein residues throughout the simulation time.
While ‘S656M-Pimozide’ and ‘S656M-Risperidone’ share similarities in their oscillatory patterns and elevated RMSD levels, ‘S656M-Haloperidol’ stands out with its more pronounced fluctuations and generally higher RMSD values, highlighting distinct conformational dynamics within this particular simulation. Figure 9 presents all results, where the ‘Noanymutations’ scenario generally shows lower RMSF (Root Mean Square Fluctuation) values, indicating higher structural stability and less flexibility across the protein residues. The ‘S656M-Aripiprazole’ simulation exhibits moderate fluctuations, with certain regions showing higher RMSF values, pointing to localized flexibility. Meanwhile, ‘S656M-Haloperidol’, ‘S656M-Pimozide’, and ‘S656M-Risperidone’ demonstrate higher RMSF values across various regions, suggesting enhanced flexibility and dynamic behavior, potentially leading to conformational changes (Figure 9).
3.9. Assessing SLITRK1 Protein Variants and Drug Interactions Through Reinforcement Learning
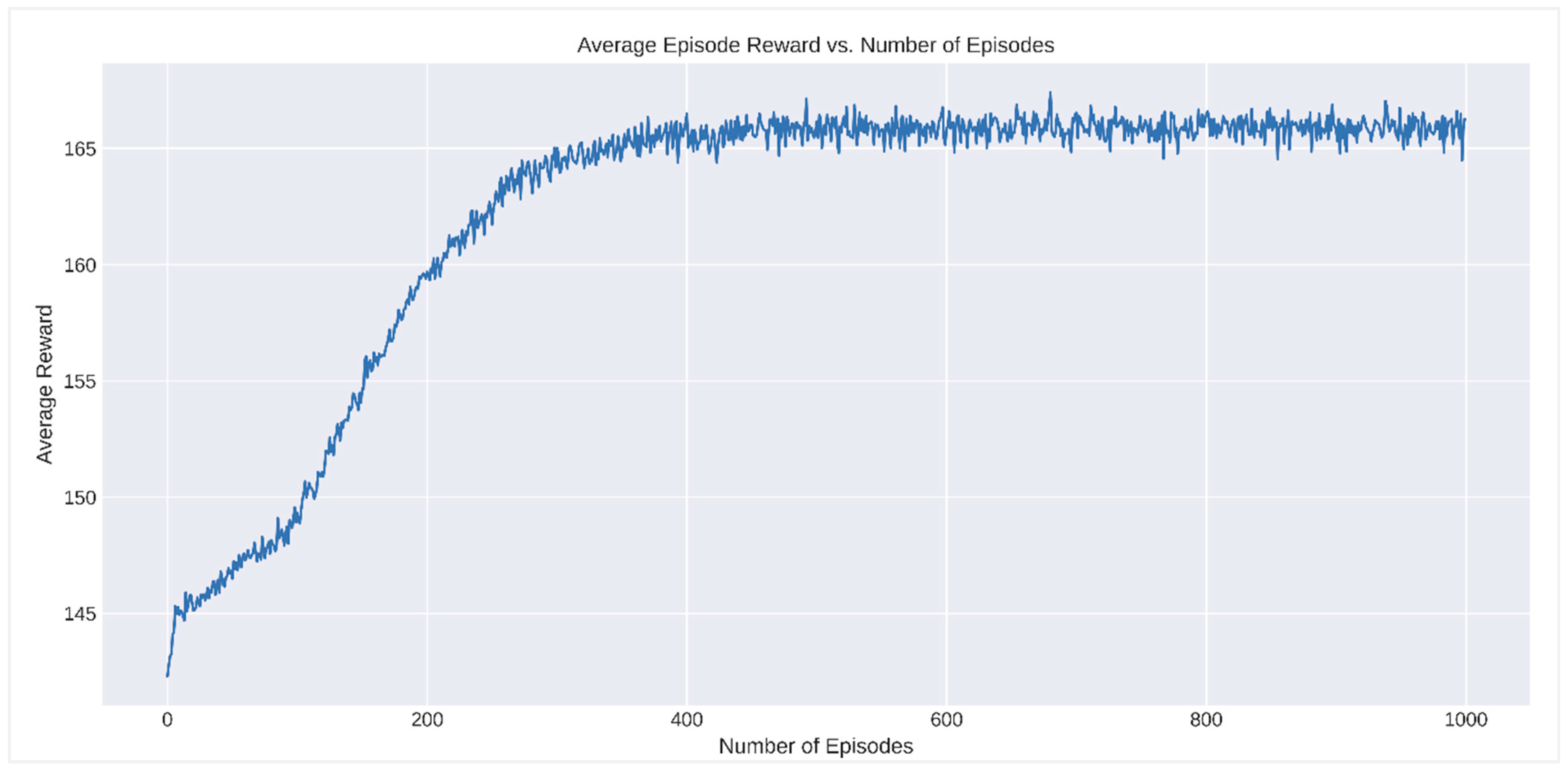
RL algorithms assessed the interactions between SLITRK1 protein variants and various drugs. The analysis identified the optimal drug for each variant based on cumulative reward metrics. Figure 10 showcases the progression of the average episode reward across the number of episodes. A marked reward increase is observed in the initial stages, indicating that the algorithm is in the learning and exploration phase. Around the 200th episode, reward fluctuations diminish, and a more consistent upward trend is noted, reflecting the algorithm’s transition to making more optimized decisions based on accumulated knowledge. From the 600th episode onwards, the average episode reward plateaus, suggesting that the algorithm has stabilized and is following an optimal or near-optimal policy.
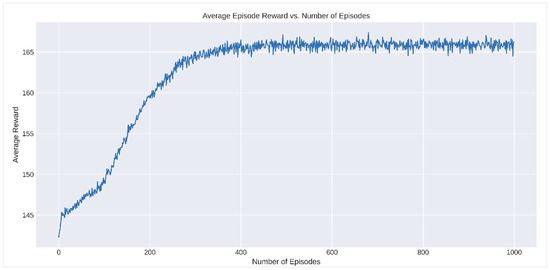
Figure 10.
Analysis of average episode reward vs. number of episodes in reinforcement learning.
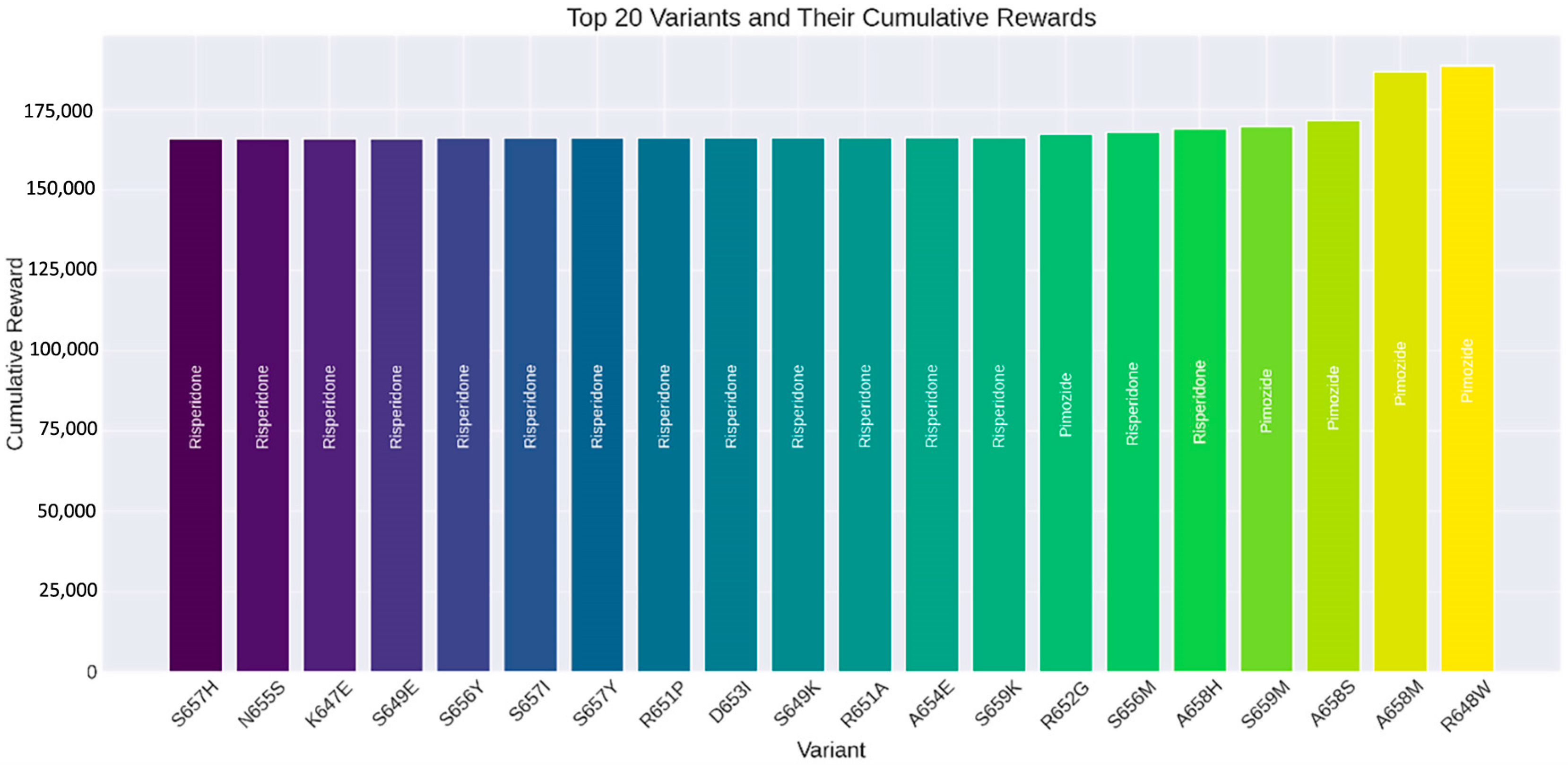
These trends underscore the efficacy of the RL algorithm in analyzing drug interactions for SLITRK1 protein variants and pinpointing the most effective therapeutic options. As depicted in Figure 11, the S657H variant’s most effective drug is Risperidone, yielding a cumulative reward of 166,028.48. Similarly, Risperidone is the best-performing drug for the N655S variant, with a cumulative reward of 166,034.86. For the K647E variant, Risperidone again proved to be the superior choice, achieving a cumulative reward of 166,041.21. The S649E variant also favored Risperidone, with a cumulative reward of 166,042.94. Lastly, for the S656Y variant, Risperidone was determined to be the most beneficial, with a cumulative reward of 166,090.22.
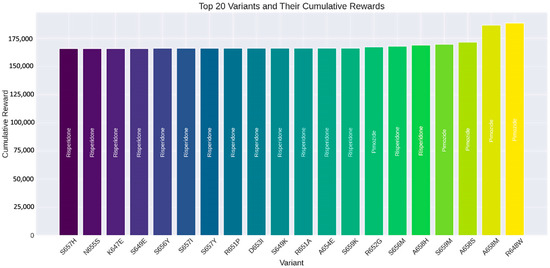
Figure 11.
Top 20 SLITRK1 protein variants and their cumulative rewards determined by reinforcement learning algorithms.
4. Discussion
The comprehensive analysis of the SLITRK1 protein presented in this study provides an in-depth understanding of its functional dynamics, particularly in relation to neurological disorders. This includes an exploration of its structure at the amino acid level, unstructured regions, transmembrane topology, stability changes, protein–protein interactions, and its role in complex protein networks. Such an encompassing examination sheds light on the multifaceted aspects of SLITRK1, highlighting its significance in the context of its structural and functional properties. The intrinsic disorder identified in the N-terminus, central, and C-terminal regions (Figure 1) underscores the protein’s flexible and dynamic nature. This characteristic aligns with the functional versatility of IDPs in cellular signaling and regulation [4,5,69]. These disordered regions are crucial for the adaptive functioning of proteins in complex cellular environments, playing significant roles in synaptic plasticity and neurotransmission [6,7,70]. The IDRs in SLITRK1 likely contribute to its multifaceted roles in synapse formation and neural development.
Based on our results, position S656 emerges as a focal point with a high mean disorder score (Figure 3), indicating that single amino acid changes can significantly impact a protein’s disorder profile and function [10,71]. The study also reveals the transmembrane topology of SLITRK1, with key segments located in the cytoplasmic area (Figure 4). This finding is significant, as transmembrane proteins are vital in cell signaling and interactions with the extracellular environment, impacting neuronal communication and synaptic formation [2,72,73,74]. The localization of SLITRK1’s segments suggests its involvement in membrane-associated signaling pathways essential for neural development and function.
Additionally, the stability changes induced by amino acid substitutions in SLITRK1 (Table 1) highlight the importance of protein stability in maintaining proper function. Variants causing stability decreases might lead to misfolding or aggregation, which are associated with neurodegenerative diseases while stability-increasing variants could protect against such effects [8,9,75,76]. The evolutionary analysis using ConSurf (Figure 5) further adds to this understanding by identifying conserved regions within SLITRK1. These regions, like S656, are likely crucial for maintaining structural integrity and functional specificity, possibly mediating interactions and signaling processes in the nervous system [27].
Moreover, the protein–protein interaction network of SLITRK1 (Figure 5) reveals its potential role in complex signaling cascades. Interactions with proteins like PTPRS and PTPRD underscore SLITRK1’s involvement in neural development and synaptic function [11,12]. The effects of mutations, especially the S656M variant, on these interactions and drug-binding affinities (Table 4) suggest potential alterations in SLITRK1’s functionality and its role in neurological conditions such as Tourette Syndrome.
Studies supporting the association between the SLITRK1 and IMMP2L genes and neurological disorders play a critical role in elucidating the molecular mechanisms underlying Tourette syndrome and other neurodevelopmental disorders. For instance, Zhang et al. reported that mutations observed in the IMMP2L gene are strongly associated with neurodevelopmental disorders such as autism spectrum disorder, attention-deficit/hyperactivity disorder, and Gilles de la Tourette syndrome [77]. These mutations are suggested to contribute to the pathophysiology of these conditions, particularly by disrupting mitochondrial function and neuronal development processes. Similarly, studies conducted using mouse models by Kreilaus et al. demonstrated that the knockdown of the IMMP2L gene leads to behavioral changes resembling autism spectrum disorder and Tourette syndrome, further supporting the potential role of this gene in neurological functions [78]. Additionally, Clarke et al. provided a detailed account of how IMMP2L mutations influence mitochondrial dynamics, synaptic connections, and neurotransmission, emphasizing the significance of these processes in the pathogenesis of conditions such as Tourette syndrome [79]. Collectively, these comprehensive studies highlight the multifaceted roles of SLITRK1 and IMMP2L in synaptic functions and mitochondrial activities, providing critical insights into the molecular basis of neurodevelopmental disorders like Tourette syndrome. These findings also underscore the potential for developing novel therapeutic strategies targeting these genes.
The molecular docking analysis, particularly with respect to the S656M mutation in SLITRK1, provides significant insights into the interaction dynamics between the protein and various drugs used in treating neurological disorders, such as Tourette Syndrome [15,32]. The notable alteration in the binding affinity of SLITRK1 to drugs like Pimozide and Risperidone (Table 4, Figure 7) underscores the potential impact of amino acid substitutions on drug efficacy. This finding is consistent with recent studies emphasizing the importance of molecular docking in predicting drug–protein interactions and assessing the implications of genetic variations on pharmacodynamics [3,14]. The S656M mutation, by altering the docking scores, suggests a structural change in the binding site, which could either enhance or inhibit the interaction with the drugs. The Ramachandran plot analysis (Figure 8) further assesses the structural integrity of SLITRK1, highlighting the impact of the S656M mutation on the protein’s structure and functionality [19]. Such alterations are critical, as they can directly influence medications’ therapeutic effectiveness and potential side effects [14,25]. This aligns with growing evidence on the role of structural protein variations in modulating drug responses, highlighting the need for personalized medicine approaches in neurological disorders [26].
The molecular dynamics simulations, as illustrated in Figure 9, provide a deeper understanding of the dynamic behavior and structural stability of the SLITRK1 protein. The increased RMSD and RMSF values observed in the S656 M variants indicate enhanced flexibility and dynamic behavior, which can have profound implications for the protein’s functional repertoire [16,17]. This observation aligns with recent advancements in molecular dynamics simulations, which have become pivotal in elucidating the impact of mutations on protein structure and function [18,20,80]. The structural changes induced by mutations like S656M could lead to altered protein–protein interactions, affecting signaling pathways and synaptic functions. This is particularly relevant in the context of neurological disorders, where protein misfolding and aggregation play a critical role [10,25]. Recent studies have demonstrated the utility of molecular dynamics in understanding the conformational landscape of proteins in diseases, providing insights into potential therapeutic targets and intervention strategies [28,81].
In summary, while the focus of this study has been on neurological disorders, the implications of our findings are broad, highlighting the importance of SLITRK1 in a range of biological mechanisms and diseases. The versatility and complexity of SLITRK1 underscore the need for continued research into its various roles and the potential for targeted therapies in diverse pathological conditions.
This study also demonstrates that using RL to evaluate drug interactions with SLITRK1 protein variants offers significant advantages over traditional methods. RL provides a dynamic approach that learns optimal drug selections through iterative interactions, efficiently determining the most effective treatment options based on cumulative rewards. This capability allows RL to not only improve the accuracy of predictions but also to accelerate the identification of beneficial drug–variant interactions, making it a powerful tool in the field of precision medicine [82]. This research highlights the potential of RL in bioinformatics and drug discovery, presenting a framework that can be extended to other proteins and diseases. The results underscore the effectiveness of RL in identifying optimal drugs for specific protein variants. Figure 10 demonstrates the learning process of the RL algorithm by showing the average episode reward against the number of episodes. Initially, there is a significant increase in rewards, indicating the exploration phase of the algorithm. As the number of episodes progresses, the rewards stabilize, reflecting the algorithm’s ability to make more consistent and optimized decisions using the learned policy. This pattern confirms the effectiveness of the RL approach in achieving a stable and optimized decision-making process. Figure 11 displays the top 20 SLITRK1 protein variants with the highest cumulative rewards. Identifying high-reward drug–variant pairs emphasizes the model’s precision in drug selection, which is crucial for personalized medicine. By using RL, researchers can simulate numerous drug–variant interactions, reducing reliance on extensive in vitro or in vivo experiments, which are often time-consuming and costly. Integrating RL into bioinformatics aligns with the increasing use of ML and artificial intelligence (AI) in drug discovery and development [21]. Research has shown that RL can significantly enhance the efficiency and accuracy of predicting drug efficacy, supporting its application in precision medicine [80]. As a result, this study highlights the potential of RL in evaluating drug interactions of SLITRK1 protein variants. The findings provide a strong foundation for future research and encourage the adoption of RL methodologies in bioinformatics applications. As the field progresses, the integration of RL and other AI-assisted approaches will lead to more personalized and effective treatment strategies.
5. Conclusions
This study offers a comprehensive analysis of the SLITRK1 protein, emphasizing its structural dynamics, disorder propensity, and pharmacological interactions, particularly in relation to neurological disorders. We identified key features, such as intrinsic disorder regions, transmembrane topology, and the significant impact of amino acid substitutions like the S656M mutation on SLITRK1’s 3D structure and biological functions. These findings demonstrate how such mutations alter drug binding affinities, with Aripiprazole consistently showing high docking scores across various SLITRK1 variants, as revealed through RL analysis. Additionally, our study highlights SLITRK1’s involvement in critical neuronal processes and its potential role in synaptic plasticity and neural development. The integration of molecular dynamics simulations provided further insights into the structural changes induced by mutations, underscoring the need for personalized medicine approaches to treat neurological disorders. The novel use of RL in evaluating drug interactions with SLITRK1 variants showcases its potential to enhance drug discovery and development. In summary, this research deepens our understanding of SLITRK1’s multifaceted roles in neural function and neurological conditions and highlights the effectiveness of advanced computational methods in developing targeted therapeutic strategies. Our findings pave the way for future studies to exploit the therapeutic potential of SLITRK1 and refine precision medicine approaches for neurological disorders.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/computation13020029/s1, Table S1: continuation of the demonstration to predict whether substitution or deletion of an amino acid affects function; Table S2: a continuation of the results of the characteristics of wild-type and mutant-type amino acids obtained from the Hope tool (SLITRK1: NP_001268432.1); Table S3: a continuation of the physicochemical parameter estimation of SLITRK1 using ExPASyProtParam; Table S4: the continuation of the molecular insertion scores between the SLITRK1 protein obtained using the HDOCK server and the drugs Pimozide, Aripiprazole, Risperidone, and Haloperidol.
Author Contributions
Conceptualization, E.A.; formal analysis, E.A. and K.K.K.; investigation, E.A.; methodology, E.A. and K.K.K.; software, E.A. and K.K.K.; validation, K.K.K.; writing—original draft, E.A., A.İ., D.Y., N.Ö.Ö., M.R., J.K., R.R.A., E.S., N.M. and V.S.; writing—review and editing, K.K.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article; further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| TS | Tourette syndrome |
| SLITRK1 | SLIT and NTRK like protein 1 |
| CDC | Centers for Disease Control and Prevention |
| NP | National Provider Identifier |
| PhD SNP | Prediction of Human Deleterious Single Nucleotide Polymorphisms |
| PANTHER | Protein ANalysis THrough Evolutionary Relationships |
| FASTA | Fast All DNA Sequence Comparison |
| STRING | Search Tool for the Retrieval of Interacting Genes/Proteins |
| ConSurf | Consurf Server for the Identification of Functional Regions in Proteins |
| GRAVY | Grand Average of Hydropathicity |
| I Mutant 2.0 | Protein Stability Change Prediction Server |
| MD | Molecular Dynamics |
| RMSD | Root Mean Square Deviation |
| RMSF | Root Mean Square Fluctuation |
| RL | Reinforcement Learning |
| ML | Machine Learning |
| AI | Artificial Intelligence |
References
- Hatayama, M.; Aruga, J. Developmental control of noradrenergic system by SLITRK1 and its implications in the pathophysiology of neuropsychiatric disorders. Front. Mol. Neurosci. 2023, 15, 1080739. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Colangelo, A.M.; Virtuoso, A.; Alberghina, L.; Papa, M. Neurons, Glia, Extracellular Matrix and Neurovascular Unit: A Systems Biology Approach to the Complexity of Synaptic Plasticity in Health and Disease. Int. J. Mol. Sci. 2020, 21, 1539. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically Disordered Proteins and Their “Mysterious” (Meta)Physics. Front. Phys. 2019, 7, 10. [Google Scholar] [CrossRef]
- Babu, M.M.; Kriwacki, R.W.; Pappu, R.V. Structural biology. Versatility from protein disorder. Science 2012, 337, 1460–1461. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef]
- Parthiban, V.; Gromiha, M.M.; Schomburg, D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006, 34, W239–W242. [Google Scholar] [CrossRef]
- Tokuriki, N.; Tawfik, D.S. Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 2009, 19, 596–604. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Kim, E.; Sheng, M. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 2004, 5, 771–781. [Google Scholar] [CrossRef]
- Um, J.W.; Ko, J. LAR-RPTPs: Synaptic adhesion molecules that shape synapse development. Trends Cell Biol. 2013, 23, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Dyke, K.; Jackson, G.; Jackson, S. Non-invasive brain stimulation as therapy: Systematic review and recommendations with a focus on the treatment of Tourette syndrome. Exp. Brain Res. 2022, 240, 341–363. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Eastwood, M.P.; Bank, J.A.; Jumper, J.M.; Salmon, J.K.; Shan, Y.; et al. Atomic-Level Characterization of the Structural Dynamics of Proteins. Science 2010, 330, 341–346. [Google Scholar] [CrossRef]
- Piana, S.; Donchev, A.G.; Robustelli, P.; Shaw, D.E. Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119, 5113–5123. [Google Scholar] [CrossRef]
- Prieto-Martínez, F.; Arciniega, M.; Medina-Franco, J. Molecular docking: Current advances and challenges. TIP Rev. Espec. En Cienc. Químico-Biológicas 2018, 21, 65–87. [Google Scholar] [CrossRef]
- Sobolev, O.V.; Afonine, P.V.; Moriarty, N.W.; Hekkelman, M.L.; Joosten, R.P.; Perrakis, A.; Adams, P.D. A Global Ramachandran Score Identifies Protein Structures with Unlikely Stereochemistry. Structure 2020, 28, 1249–1258.e1242. [Google Scholar] [CrossRef]
- Vakser, I.A. Challenges in protein docking. Curr. Opin. Struct. Biol. 2020, 64, 160–165. [Google Scholar] [CrossRef]
- Dhudum, R.; Ganeshpurkar, A.; Pawar, A. Revolutionizing Drug Discovery: A Comprehensive Review of AI Applications. Drugs Drug Candidates 2024, 3, 148–171. [Google Scholar] [CrossRef]
- Sutton, R.S.; Barto, A.G. Reinforcement Learning: An Introduction. IEEE Trans. Neural Netw. 2018, 9, 1054. [Google Scholar] [CrossRef]
- Gjærum, V.B.; Rørvik, E.L.H.; Lekkas, A.M. Approximating a deep reinforcement learning docking agent using linear model trees. In Proceedings of the 2021 European Control Conference (ECC), Naples, Italy, 29 June–2 July 2021; pp. 1465–1471. [Google Scholar]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. TensorFlow: A System for Large-Scale Machine Learning. arXiv 2016, arXiv:1605.08695. Available online: https://arxiv.org/abs/1605.08695 (accessed on 25 November 2024).
- Patel, M.N.; Halling-Brown, M.D.; Tym, J.E.; Workman, P.; Al-Lazikani, B. Objective assessment of cancer genes for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.H.; Alanko, I.; Bhadane, R.; Bonvin, A.M.J.J.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S.; et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2021, 9, 71. [Google Scholar] [CrossRef]
- Jeyasri, R.; Muthuramalingam, P.; Suba, V.; Ramesh, M.; Chen, J.T. Bacopa monnieri and Their Bioactive Compounds Inferred Multi-Target Treatment Strategy for Neurological Diseases: A Cheminformatics and System Pharmacology Approach. Biomolecules 2020, 10, 536. [Google Scholar] [CrossRef]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Rizzo, R.; Eddy, C.M.; Calí, P.; Gulisano, M.; Cavanna, A.E. Metabolic Effects of Aripiprazole and Pimozide in Children With Tourette Syndrome. Pediatr. Neurol. 2012, 47, 419–422. [Google Scholar] [CrossRef]
- Roessner, V.; Schoenefeld, K.; Buse, J.; Bender, S.; Ehrlich, S.; Münchau, A. Pharmacological treatment of tic disorders and Tourette Syndrome. Neuropharmacology 2013, 68, 143–149. [Google Scholar] [CrossRef]
- Pringsheim, T.; Holler-Managan, Y.; Okun, M.S.; Jankovic, J.; Piacentini, J.; Cavanna, A.E.; Martino, D.; Müller-Vahl, K.; Woods, D.W.; Robinson, M.; et al. Comprehensive systematic review summary: Treatment of tics in people with Tourette syndrome and chronic tic disorders. Neurology 2019, 92, 907–915. [Google Scholar] [CrossRef]
- Besag, F.M.; Vasey, M.J.; Lao, K.S.; Chowdhury, U.; Stern, J.S. Pharmacological treatment for Tourette syndrome in children and adults: What is the quality of the evidence? A systematic review. J. Psychopharmacol. 2021, 35, 1037–1061. [Google Scholar] [CrossRef] [PubMed]
- Lieutaud, P.; Ferron, F.; Uversky, A.V.; Kurgan, L.; Uversky, V.N.; Longhi, S. How disordered is my protein and what is its disorder for? A guide through the “dark side” of the protein universe. Intrinsically Disord. Proteins 2016, 4, e1259708. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Gsponer, J.; Kurgan, L. DEPICTER2: A comprehensive webserver for intrinsic disorder and disorder function prediction. Nucleic Acids Res. 2023, 51, W141–W147. [Google Scholar] [CrossRef] [PubMed]
- Del Conte, A.; Bouhraoua, A.; Mehdiabadi, M.; Clementel, D.; Monzon, A.M.; Predictors, C.; Tosatto, S.C.E.; Piovesan, D. CAID prediction portal: A comprehensive service for predicting intrinsic disorder and binding regions in proteins. Nucleic Acids Res. 2023, 51, W62–W69. [Google Scholar] [CrossRef]
- Meng, F.; Uversky, V.N.; Kurgan, L. Comprehensive review of methods for prediction of intrinsic disorder and its molecular functions. Cell. Mol. Life Sci. 2017, 74, 3069–3090. [Google Scholar] [CrossRef]
- Davey, N.; Babu, M.; Blackledge, M.; Bridge, A.; Capella-Gutierrez, S.; Dosztanyi, Z.; Drysdale, R.; Edwards, R.; Elofsson, A.; Felli, I.; et al. An intrinsically disordered proteins community for ELIXIR [version 1; peer review: 2 approved]. F1000Research 2019, 8, ELIXIR-1753. [Google Scholar] [CrossRef]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef]
- Vincent, M.; Uversky, V.N.; Schnell, S. On the Need to Develop Guidelines for Characterizing and Reporting Intrinsic Disorder in Proteins. Proteomics 2019, 19, 1800415. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.-P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P. PhD-SNPg: A webserver and lightweight tool for scoring single nucleotide variants. Nucleic Acids Res. 2017, 45, W247–W252. [Google Scholar] [CrossRef]
- Mohanty, G.; Jena, S.R.; Kar, S.; Samanta, L. Paternal factors in recurrent pregnancy loss: An insight through analysis of non-synonymous single-nucleotide polymorphism in human testis-specific chaperone HSPA2 gene. Environ. Sci. Pollut. Res. 2022, 29, 62219–62234. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.-J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.M.; Myhre, P.L.; Arthur, V.; Dorbala, P.; Rasheed, H.; Buckley, L.F.; Claggett, B.; Liu, G.; Ma, J.; Nguyen, N.Q.; et al. Large scale plasma proteomics identifies novel proteins and protein networks associated with heart failure development. Nat. Commun. 2024, 15, 528. [Google Scholar] [CrossRef]
- Henricson, A.; Käll, L.; Sonnhammer, E.L.L. A novel transmembrane topology of presenilin based on reconciling experimental and computational evidence. FEBS J. 2005, 272, 2727–2733. [Google Scholar] [CrossRef]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. Advantages of combined transmembrane topology and signal peptide prediction—The Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef]
- Yariv, B.; Yariv, E.; Kessel, A.; Masrati, G.; Chorin, A.B.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. Using evolutionary data to make sense of macromolecules with a “face-lifted” ConSurf. Protein Sci. 2023, 32, e4582. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.e.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Pramanik, K.; Ghosh, P.K.; Ray, S.; Sarkar, A.; Mitra, S.; Maiti, T.K. An in silico structural, functional and phylogenetic analysis with three dimensional protein modeling of alkaline phosphatase enzyme of Pseudomonas aeruginosa. J. Genet. Eng. Biotechnol. 2017, 15, 527–537. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Rapp, C.; Bai, X.; Reithmeier, R.A.F. Molecular analysis of human solute carrier SLC26 anion transporter disease-causing mutations using 3-dimensional homology modeling. Biochim. Biophys. Acta (BBA)—Biomembr. 2017, 1859, 2420–2434. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.; Goddard, T.; Huang, C.; Couch, G.; Greenblatt, D.; Meng, E.; Ferrin, T. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Agnihotry, S.; Pathak, R.K.; Singh, D.B.; Tiwari, A.; Hussain, I. Chapter 11—Protein structure prediction. In Bioinformatics; Singh, D.B., Pathak, R.K., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 177–188. [Google Scholar]
- Vasudevan, K.; Udhaya Kumar, S.; Mithun, A.; Raghavendra, B.; George Priya Doss, C. Chapter Ten—Structure-based virtual screening to identify potential lipase inhibitors to reduce lipid storage in Wolman disorder. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2023; Volume 133, pp. 351–363. [Google Scholar]
- Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK server for integrated protein–protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- K, K.; Nagarajan, S.; R, S.; Mohan, S. In Silico Identification of Potential Inhibitors of Substituted Quinazolin-4-One against Main Protease and Spike Glycoprotein of Sars Cov-2. Res. J. Pharm. Technol. 2023, 16, 2470–2476. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- Mnih, V.; Kavukcuoglu, K.; Silver, D.; Rusu, A.A.; Veness, J.; Bellemare, M.G.; Graves, A.; Riedmiller, M.; Fidjeland, A.K.; Ostrovski, G.; et al. Human-level control through deep reinforcement learning. Nature 2015, 518, 529–533. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, S.; Li, Y.; Guo, J.; Wei, Y.; Mu, Y.; Zheng, L.; Li, W. A new paradigm for applying deep learning to protein-ligand interaction prediction. Brief. Bioinform. 2024, 25, bbae145. [Google Scholar] [CrossRef]
- Chong, B.; Yang, Y.; Wang, Z.-L.; Xing, H.; Liu, Z. Reinforcement learning to boost molecular docking upon protein conformational ensemble. Phys. Chem. Chem. Phys. 2021, 23, 6800–6806. [Google Scholar] [CrossRef]
- Loschwitz, J.; Jäckering, A.; Keutmann, M.; Olagunju, M.; Olubiyi, O.O.; Strodel, B. Dataset of AMBER force field parameters of drugs, natural products and steroids for simulations using GROMACS. Data Brief. 2021, 35, 106948. [Google Scholar] [CrossRef]
- Kutzner, C.; Kniep, C.; Cherian, A.; Nordstrom, L.; Grubmüller, H.; de Groot, B.L.; Gapsys, V. GROMACS in the Cloud: A Global Supercomputer to Speed Up Alchemical Drug Design. J. Chem. Inf. Model. 2022, 62, 1691–1711. [Google Scholar] [CrossRef] [PubMed]
- Harmon, T.S.; Holehouse, A.S.; Rosen, M.K.; Pappu, R.V. Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. eLife 2017, 6, e30294. [Google Scholar] [CrossRef] [PubMed]
- Metskas, L.A.; Rhoades, E. Single-Molecule FRET of Intrinsically Disordered Proteins. Annu. Rev. Phys. Chem. 2020, 71, 391–414. [Google Scholar] [CrossRef] [PubMed]
- Aktaş, E.; Özdemir Özgentürk, N. A comprehensive examination of ACE2 receptor and prediction of spike glycoprotein and ACE2 interaction based on in silico analysis of ACE2 receptor. J. Biomol. Struct. Dyn. 2024, 42, 4412–4428. [Google Scholar] [CrossRef]
- Aruga, J.; Mikoshiba, K. Identification and characterization of Slitrk, a novel neuronal transmembrane protein family controlling neurite outgrowth. Mol. Cell. Neurosci. 2003, 24, 117–129. [Google Scholar] [CrossRef]
- Sanders, C.R.; Myers, J.K. Disease-Related Misassembly of Membrane Proteins. Annu. Rev. Biophys. 2004, 33, 25–51. [Google Scholar] [CrossRef]
- Nugent, T.; Jones, D.T. Transmembrane protein topology prediction using support vector machines. BMC Bioinform. 2009, 10, 159. [Google Scholar] [CrossRef]
- Robinson, P.A. Protein stability and aggregation in Parkinson’s disease. Biochem. J. 2008, 413, 1–13. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Zarrei, M.; Tong, W.; Dong, R.; Wang, Y.; Zhang, H.; Yang, X.; MacDonald, J.R.; Uddin, M.; et al. Association of IMMP2L deletions with autism spectrum disorder: A trio family study and meta-analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 93–100. [Google Scholar] [CrossRef]
- Kreilaus, F.; Chesworth, R.; Eapen, V.; Clarke, R.; Karl, T. First behavioural assessment of a novel Immp2l knockdown mouse model with relevance for Gilles de la Tourette syndrome and Autism spectrum disorder. Behav. Brain Res. 2019, 374, 112057. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.A.; Furlong, T.M.; Eapen, V. Tourette Syndrome Risk Genes Regulate Mitochondrial Dynamics, Structure, and Function. Front. Psychiatry 2021, 11, 556803. (In English) [Google Scholar] [CrossRef] [PubMed]
- Aktaş, E.; Saygılı, İ.; Kahveci, E.; Tekbıyık, Z.; Özgentürk, N.Ö. Bioinformatic investigation of Nipah virus surface protein mutations: Molecular docking with Ephrin B2 receptor, molecular dynamics simulation, and structural impact analysis. Microbiol. Immunol. 2023, 67, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Firoz, A.; Talwar, P. Role of death-associated protein kinase 1 (DAPK1) in retinal degenerative diseases: An in-silico approach towards therapeutic intervention. J. Biomol. Struct. Dyn. 2024, 42, 5686–5698. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Y.; Zhang, Y.; Li, K.; Lin, M.; Pan, F.; Wu, W.; Zhang, J. A reinforcement learning approach for protein–ligand binding pose prediction. BMC Bioinform. 2022, 23, 368. [Google Scholar] [CrossRef]
- Sethi, P.S.; Kaur, G.; Vasanth, D.S.; Ramakrishnan, M.; Kote, N. Applications of deep reinforcement learning for drug discovery. In Machine Intelligence and Data Science Applications, Singapore; Ramdane-Cherif, A., Singh, T.P., Tomar, R., Choudhury, T., Um, J.-S., Eds.; Springer: Singapore, 2023; pp. 133–141. [Google Scholar]
- Nada, H.; Choi, Y.; Kim, S.; Jeong, K.S.; Meanwell, N.A.; Lee, K. New insights into protein–protein interaction modulators in drug discovery and therapeutic advance. Sig. Transduct. Target. Ther. 2024, 9, 341. [Google Scholar] [CrossRef]
- Weaver, D.T.; King, E.S.; Maltas, J.; Scott, J.G. Reinforcement learning informs optimal treatment strategies to limit antibiotic resistance. Proc. Natl. Acad. Sci. USA 2024, 121, e2303165121. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).