1. Introduction
Diatomic molecules are important in many areas of modern science, such as atmospheric chemistry [
1,
2], astrobiology [
3], and ultracold physics [
4]. Despite their simplicity, diatomic molecules are valuable models for understanding the relationship between structure and thermodynamic properties in more complex systems. For this purpose, the development of high-quality theoretical methods to calculate these properties is of paramount importance. Two main strategies have usually been adopted to improve the accuracy of these methods: (i) improving the description of the electronic structure [
5,
6,
7,
8], and (ii) implementing better models to describe the (rotational and vibrational) nuclear motions [
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28].
Obtaining more reliable information about the electronic structure has been achieved by (i) a high-level quantum method [
5,
6,
7], (ii) a robust basis set [
5,
6,
7], (iii) the extrapolation to the complete basis set (CBS) limit [
5,
6,
7], (iv) the inclusion of relativistic and spin–orbit effects [
5,
6,
7,
8], and (v) a very good description of the electronic correlation [
5,
6,
7].
The rotational structure is usually described by a classical approach since the temperatures of interest are normally much higher than the characteristic rotational temperatures. However, at low temperatures, it is important to adopt a quantum description of this structure to obtain accurate values [
9].
At low and moderate temperatures, the vibrational structure can be reasonably described by the harmonic oscillator model [
10]. However, this approach is known to overestimate the vibrational frequencies [
10], leading to significant errors at high temperatures. To overcome this limitation, several correction methods have been applied [
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28]. Most of them can be classified into two groups: (i) methods that use empirical scaling factors to correct the harmonic frequencies [
13,
14,
15,
16,
17,
18], and (ii) methods that use anharmonic potentials [
19,
20,
21,
22,
23,
24,
25,
26,
27,
28].
The thermodynamic properties of a molecular system can be calculated using general quantum chemistry packages [
29,
30,
31,
32,
33,
34]. However, the required computational resources and level of expertise are considerable. The main difficulty in performing these calculations is probably the installation of such types of packages, which constitutes a very hard task for nonspecialized researchers or students.
In this work, I have developed an open-source Excel application to calculate thermodynamic properties for diatomic ideal gases. This application, called Diatomic, is very easy to use and requires only some basic knowledge of theoretical chemistry. In fact, a user of Diatomic only has to input a limited number of molecular constants (namely, the electronic ground state energy and the corresponding spin multiplicity, the atomic masses, the rotational symmetry number, the equilibrium bond length, the harmonic vibrational wave number, and the well depth of the Morse potential) to perform a calculation of this type. This information is freely available from online resources [
35]. Additionally, the pressure and a set of fourteen temperatures must also be inputted.
Diatomic provides many features, often not available in general quantum chemistry packages, to perform and interpret this type of calculation. These include (i) two models (classical and quantum) describing the rotational structure, (ii) two models (harmonic and Morse) describing the vibrational structure, (iii) energies and Boltzmann populations for the quantum rotational model, (iv) energies and Boltzmann populations for both vibrational models implemented in the application, (v) molar thermodynamic properties as a function of temperature available in very easy to interpret and process tables, and (vi) availability of these tables organized by different criteria.
A future user of Diatomic should be interested in diatomic ideal gases for pedagogical and/or scientific purposes. I envision three main applications for this tool: (i) educational applications: professors and students can use this application to explore fundamental concepts in quantum chemistry and/or statistical mechanics without requiring special skills in computer science; (ii) research in experimental chemistry: researchers in this field can use Diatomic to easily predict the thermodynamic properties of diatomic ideal gases; and (iii) research in computational chemistry: researchers can explore specific features of Diatomic to rationalize the results of computational calculations.
Most of the aforementioned refinements to the electronic structure can be easily incorporated into Diatomic, as they solely impact the electronic energy. For this purpose, one only needs to search the literature for these corrected values. However, the goal of this work is not to obtain highly accurate values for the molar thermodynamic properties under study. Rather, I aim to demonstrate that users can achieve reliable results for these properties using Diatomic and freely available molecular data online. Additionally, since Diatomic is an open-source collaborative project, anyone can contribute by adding new vibrational potentials to the application.
Diatomic was used here to calculate these properties for fifteen ideal gases as a function of temperature and at a constant pressure. The chemical species studied correspond to all diatomic molecules that can be obtained by combining these five elements: H, N, O, F, and Cl.
These molecules are classified according to their symmetry (homonuclear or heteronuclear) and spin multiplicity (singlet, doublet, or triplet) (see
Table 1).
2. Methods
2.1. General Formalism
According to the well-established formalism of statistical mechanics [
36,
37,
38], all the most relevant thermodynamic properties of an ideal gas can be calculated from its partition function. For a molar canonical ensemble, which is equivalent to an isothermal-isobaric ensemble for an ideal gas, and within the Born–Oppenheimer approach, this partition function (
Q(
NA,
V,
T)) can be calculated from its molecular components:
In Equation (1),
NA is the Avogadro number and
Z is the molecular partition function. In the same Equation,
Zel,
Ztransl,
Zrot, and
Zvib are the (electronic, translational, rotational and vibrational) components of the partition function. Within this formalism, the molar thermodynamic properties can be decomposed in a similar way:
In some of the preceding Equations (3), (5) and (8) an additional non-specific component (
RT or
R) must also be included. The components of the molar thermodynamic properties can be calculated as follows:
Equation (10) should be used for partition functions (
Zcomp) calculated analytically and Equation (11) should be used for partition functions calculated numerically. In the last equation,
is the energy of the quantum state
i and
Pi is the corresponding Boltzmann population given by the following formula:
in which
kb is the Boltzmann constant.
In Equation (15),
is the variance associated with the energy component (
Ecomp) calculated as
In Equation (16),
and
are, respectively, the average values of the energy component (
) and of its square (
) given by
2.2. Classification of the Molar Thermodynamic Properties
The molar thermodynamic properties are classified according to the criteria presented in
Table 2.
Some properties of this type depend on the zero-point energy (
ZPE), which corresponds to the molar energy at 0 K. This quantity can be calculated as the sum of two components: the zero-point electronic energy (
ZPEE) and the zero-point vibrational energy (
ZPVE):
In the last two equations, and are the energies of the electronic and vibrational ground states, respectively.
The type 1 molar thermodynamic properties correspond to the absolute values of the thermodynamic properties that are dependent on the zero-point energy. A list of these properties is given in
Table 3.
These properties are functions of temperature (
T) and a limited number of molecular constants, which will be presented in
Section 2.10. The molar entropy (
Sm) is also dependent on the pressure (
P) due to its translational component (
).
The type 2 molar thermodynamic properties correspond to the thermal values of the thermodynamic properties that depend on zero-point energy. A list of these properties is presented in
Table 4.
A generic type 2 molar thermodynamic property (
) can be calculated according to the following formula:
in which
is the molar value of the thermodynamic property (
X) at a generic temperature (
T) and
is the corresponding molar value at a reference temperature (
Tref). In this study, two distinct reference temperatures were used. The original molar thermal values, calculated from the Diatomic application, use the reference temperature of 0 K. Some thermal values, presented in
Section 3, use the reference temperature of 298.15 K for consistency with the corresponding experimental values.
The type 3 molar thermodynamic properties are those that are independent of zero-point energy. A list of these properties is presented in
Table 5.
2.3. Classical Limit
The general formalism, as presented in
Section 2.1, is based on a quantum formulation. In this case, the partition function associated with a component
comp is calculated as a summation over the respective quantum levels (
i),
in which
gi is the degeneracy of the quantum level
i and
is the corresponding energy.
However, when the temperature is high enough that the energy difference between two successive levels is much smaller than kBT, the summation (23) can be replaced by an appropriate integral (classical limit). In this case, it is valid the so-called equipartition principle, according to which each classical quadratic energy term contributes with R/2 for the molar heat capacity at constant volume (Cv,m) and RT/2 for the average value of the molar energy (<E>m).
Each energy component is associated with a characteristic temperature (Θcomp), which determines the applicability of the classical limit and the equipartition principle (T >> Θcomp). These characteristic temperatures vary considerably depending on the nature of the component. In general, the electronic temperatures (Θel) are very high, the vibrational temperatures (Θvib) are moderate or high, the rotational temperatures (Θrot) are low, and the translational temperatures (Θtransl) are very low.
This principle is rarely applicable to the electronic component, because the energy differences between two successive electronic levels are usually too large [
36,
37]. For diatomic molecules, when this principle is fully applied to the nuclear motion components (translational, rotational, and vibrational), these molar thermodynamic properties can be estimated as follows:
The translational component, associated with three normal modes, contributes with 3R/2 to Cv,m and with 3RT/2 to <E>m.
The rotational component, associated with two normal modes, contributes with R to Cv,m and with RT to <E>m.
The vibrational component, associated with one normal mode, contributes with
R to
Cv,m and with
RT to <
E>
m. It is important to state that the equipartition principle can only be applied to the vibrational component for the harmonic oscillator model (see
Section 2.8). This principle does not apply to the Morse oscillator model.
A translational or rotational normal mode contributes with a single value to the referred molar thermodynamic properties (R/2 to Cv,m and RT/2 to <E>m), because it is associated with a single quadratic kinetic energy term in classical mechanics. The vibrational normal mode contributes twice as much to the aforementioned molar thermodynamic properties (R to Cv,m and with RT to <E>m) due to its association with two quadratic energy terms (one kinetic and one potential) in classical mechanics.
For a temperature interval [
Tref,
T], where the classical limit and the equipartition principle are applicable to the nuclear motion components but not to the electronic component, the following approximate equations are valid:
In Equation (28), is the molar entropy at a reference temperature Tref.
2.4. Theoretical Models
In the Diatomic application, the translational structure is always treated using the classical approach (see
Section 2.6), while the electronic structure is handled in a distinctive manner (see
Section 2.5). Two models (classical and quantum) have been implemented for the rotational structure (see
Section 2.7). For the vibrational structure, two models (harmonic oscillator and Morse oscillator) are also available (see
Section 2.8). Additionally, an almost fully classical model (classical rotation and classical vibration; see
Section 2.3) is used in this work. These theoretical models are summarized in
Table 6.
The quantum rotational (
quant) and classical rotational (
class) models yield significantly different results only at temperatures around or below the rotational temperature (Θ
rot) [
9,
36,
37]. In this study, however, the temperatures in question (
T ≥ 298.15 K) are considerably higher than the highest rotational temperature (Θ
rot = 87.44 K for H
2). Consequently, it is reasonable to expect that the theoretical model (
class or
quant) used to describe the rotational structure has a minimal impact on the results.
The harmonic and Morse potentials are very similar for low vibrational energies and bond lengths close to the equilibrium. However, they diverge significantly in opposite situations (high vibrational energies and/or bond lengths far from the equilibrium) [
36,
37]. From the perspective of statistical mechanics, this means that the values of molar thermodynamic properties calculated using the two associated vibrational models (
harm and
Morse) should be similar at low temperatures [
36,
37]. As the temperature increases, these differences are expected to become more pronounced due to the rise in Boltzmann populations associated with the vibrational excited states [
36,
37]. However, this effect can be mitigated by the increase in vibrational frequency [
36,
37].
The performance of the (
class,
class) model should be associated with the applicability of the equipartition principle [
36,
37]. For molar thermodynamic properties directly associated with this principle (such as
Cp,m or
<Ho>m, therm), this performance should result from a competition between harmonic and anharmonic factors. From a harmonic perspective, this performance should increase with temperature by the convergence to the classical limit (where the equipartition principle is fully applied). Conversely, from an anharmonic perspective, this performance is expected to decrease with temperature.
2.5. Electronic Structure
For the molecules studied, it was assumed that the energy differences between the electronic ground state and the electronic excited states are very large. Consequently, only the first state is occupied at the temperatures under consideration. Furthermore, in the calculation of the electronic partition function (
Zel), it is usually assumed that the energy of the electronic ground state (
E0) is the zero of the energy scale,
in which
g0 is the spin multiplicity of the electronic ground state.
This implies that some corrections have to be introduced to the general formalism presented in
Section 2.1 and
Section 2.3:
2.6. Translational Structure
The translational structure is fully treated by the classical approach. Thus, the translational partition function and the translational components of some relevant molar thermodynamic properties can be calculated, respectively, as follows:
In Equations (34), (35) and (38), Λ is the thermal de Broglie wavelength given by
in which
m is the molecular mass and
h is the Planck constant.
2.7. Rotational Structure
The rotational structure is treated using two alternative approaches (quantum and classical). Within the quantum formalism, the rotational partition function can be calculated as
In the Diatomic application, a finite number of terms (Jmax = 2000) are used in the summations included in Equation (40). However, this was enough to obtain convergence in the rotational partition for all the calculations performed here.
In this equation,
σrot is the rotational symmetry number (
σrot = 2 for diatomic homonuclear molecules and
σrot = 1 for diatomic heteronuclear molecules),
is the energy of the rotational quantum level
J,
is the respective degeneracy,
B is the rotational constant, and
is the rotational temperature. The last two quantities can be calculated using the following equations:
In Equation (41),
I is the moment of inertia given by
where
μ is the reduced mass of the diatomic molecule,
m1 is the mass of its first atom,
m2 is the mass of its second atom, and
req is its equilibrium bond length.
Within this formalism, the rotational components of some relevant molar thermodynamic properties can be calculated as
In Equation (46),
and
are the average values of the rotational energy (
) and of its square (
), respectively, given by
Within the classical formalism, Equations (40)–(49) must be reformulated as
2.8. Vibrational Structure
The vibrational structure is treated using two alternative approaches (harmonic oscillator and Morse oscillator). Within the harmonic oscillator formalism, the vibrational partition function can be calculated as
where
νvib is the vibrational frequency and Θ
vib the vibrational temperature, given, respectively, by
in which
K is the force constant of the diatomic molecule,
Within this formalism, the vibrational components of some relevant molar thermodynamic properties can be calculated as follows:
Within the Morse oscillator formalism, Equations (55)–(61) must be reformulated as
where
is the vibrational energy of a diatomic molecule according to this formalism given by Equation (63):
A harmonic vibrational frequency is used here.
In Equation (63),
v is the vibrational quantum number,
vmax + 1 is the number of bound states associated with the Morse potential, and χ
e is the anharmonicity constant. The last two quantities can be calculated by Equations (64) and (65), respectively:
In Equation (65),
De is the well depth of the Morse potential.
In Equation (68),
and
are the average values of the vibrational energy (
) and of its square (
), respectively, given by
2.9. Standard Molar Enthalpy of Formation
The standard molar enthalpy of formation of a diatomic heteronuclear gas AB (ΔfHo(AB, g)) is defined as the enthalpy variation associated with the following chemical equation under standard conditions:
In Equation (73), the standard molar enthalpy of a diatomic gas X (<Ho (X, g)>m; X = AB, A2, or B2) is the average value of the standard molar enthalpy of this chemical species at a pressure 1 bar and a specific temperature (usually 298.15 K).
2.10. Molecular Constants
In the theoretical models considered, the molar thermodynamic properties depend only on a limited number of molecular constants (primary molecular constants). However, in many cases, the formalism previously presented in this section was expressed using other molecular constants (secondary molecular constants) that can be obtained from the primary ones. A list of the molecular constants used in this work is presented in
Table 7. The values of these constants used in the calculations are provided in the
Tables S3 and S4 in Supplementary Materials.
2.11. Diatomic Application
A general flowchart of the Diatomic application is shown in
Figure 1 (a detailed flowchart is provided in
Figure S1).
This application is organized into three different blocks of spreadsheets: the block of data, the block of preliminary calculations, and the block of results.
The block of data provides the universal constants, whose values were also adopted by Gaussian 09 and recommended by Mohr et al. [
39]. The users are then required to input the primary molecular constants (see
Table 7 and
Table S3), and the secondary molecular constants (see
Table 7 and
Table S4) are further calculated in this block. The block of preliminary calculations characterizes the rotational and vibrational structures within the different theoretical models available in Diatomic and calculates the partition functions. In the block of results, the final results are presented/calculated using different criteria (see details in
Supplementary Materials).
3. Results
In this study, the Diatomic application was used to calculate the molar thermodynamic properties presented in
Table 2,
Table 3,
Table 4 and
Table 5 for the molecules listed in
Table 1. The first four models presented in
Table 6 (
harm,
class;
Morse,
class;
harm,
quant; and
Morse,
quant) were employed in these calculations. The molecular constants, calculated at a CCSD(T)/cc-pVQZ quantum level, were taken from the Computational Chemistry Comparison and Benchmark DataBase [
35]. These thermodynamic properties were calculated for 14 different temperatures (ranging from 298.15 K to 1573.15 K) at a constant pressure of 1 bar. Thus, these are standard molar thermodynamic properties at different temperatures. These results are available in
Supplementary Materials (see Table S1 and the files referred to therein).
For comparison, these properties were also calculated using Gaussian 09 [
29] at a CCSD(T)/cc-pVQZ quantum level. For each molecule studied, a geometry optimization with harmonic frequency calculation was performed. The maximum deviations between the molar thermodynamic properties, calculated by Diatomic and Gaussian 09, were 0.002 calorie-based units (see folder “04_G09_vs_Diatomic_Comparisions” in
Supplementary Materials).
A special focus was placed on the analysis of four molar thermodynamic properties, which can be directly compared with experimental values. The selected properties were (i) the standard molar enthalpy of formation at 298.15 K (Δ
fHo), (ii) the molar heat capacity at constant pressure (
Cp,m), (iii) the average value of the standard molar thermal enthalpy (<
Ho>
m, therm), and (iv) the standard molar entropy (
Som). The five theoretical models presented in
Table 6 were tested in these calculations. The methodology and the thermodynamic constraints (in terms of temperature and pressure) were consistent with those used in the global calculations.
For most of the molecules under study, the experimental values for Δ
fHo were provided by Chase Jr. [
40] and are freely available in the NIST Chemistry WebBook database [
41]. For the nitrogen monochloride (NCl) molecule, the corresponding experimental value was provided by Shamasundar and Arunan [
42].
The experimental values for the other molar thermodynamic properties (
Cp,m, <
Ho>
m, therm and
Som) were estimated using fittings to the Shomate equations, performed within the NIST Chemistry WebBook database [
41], from original values provided by Chase Jr. [
40]. A reference temperature of 298.15 K was chosen for both the experimental and calculated values of <
Ho>
m, therm. The relative errors for the calculated values were determined using the experimental values as a reference. No experimental values are available for the nitrogen monochloride (NCl) molecule.
Finally, Diatomic was used to address an interesting pedagogical issue: the molecular interpretation of the molar heat capacity at constant pressure
(Cp,m). For this purpose, a representative group of three molecules (H
2, O
2, and Cl
2) was selected. The methodology and the thermodynamic constraints (in terms of temperature and pressure) were consistent with those used in the preceding calculations. Some relevant molecular constants of these molecules are presented in
Table 8.
Table 9 presents the values for the vibrational quantity
(where
T is the working temperature and
is the variance of vibrational energy) and
Cp,m as functions of temperature for the three representative molecules (H
2, O
2, and Cl
2), calculated using the (
harm,
class) theoretical model.
Table 10 presents the corresponding values, calculated using the (
Morse,
class) theoretical model. Additionally, it includes the experimental values for
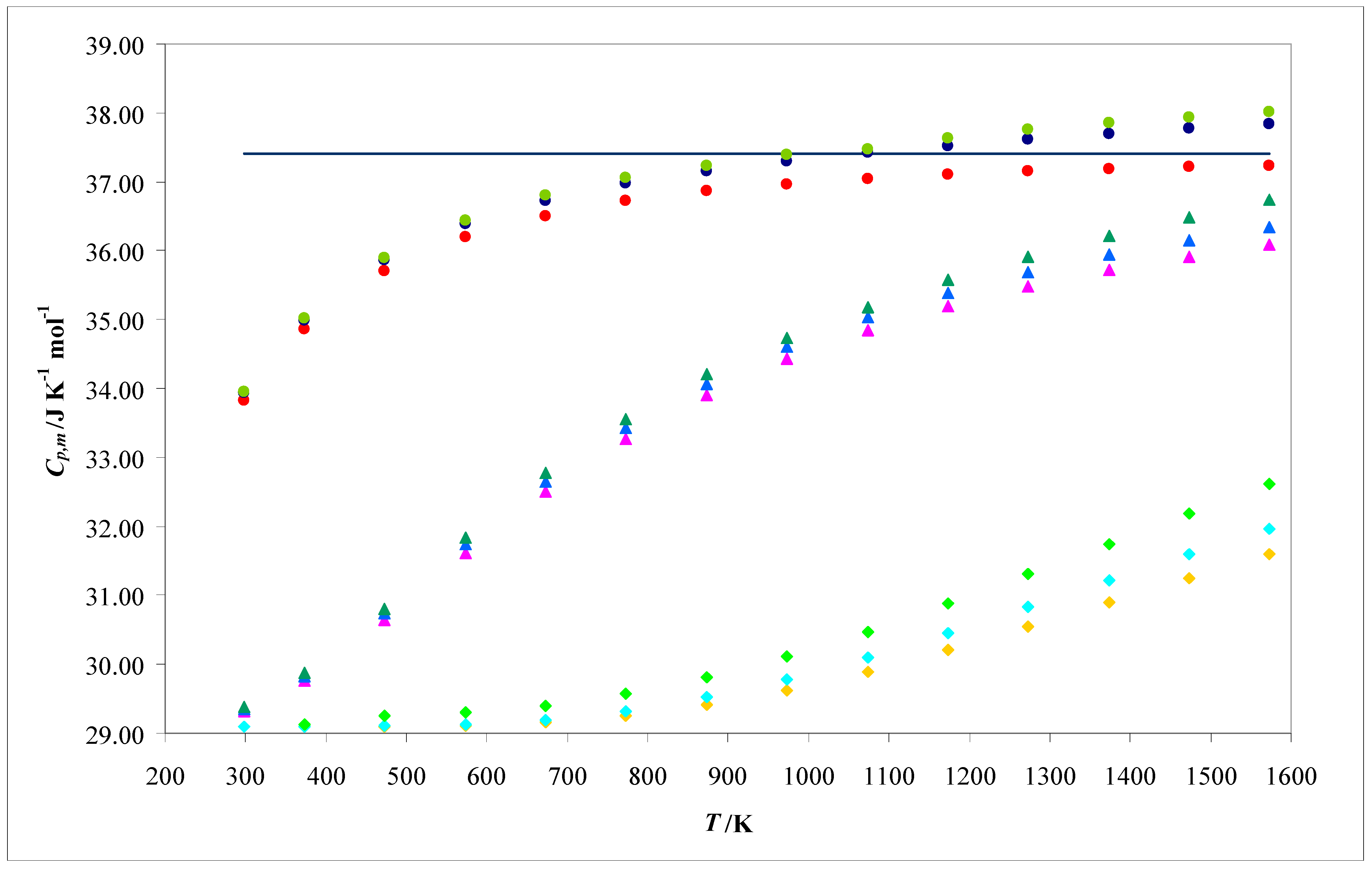
Cp,m. In
Figure 2, the calculated (using the (
harm,
class), (
Morse,
class), and (
class,
class) theoretical models) and experimental values for
Cp,m are represented as functions of temperature.
These quantities (
and
Cp,m) are directly proportional, as can be demonstrated by combining Equations (8) and (60) for the (
harm,
class) theoretical model or Equations (8) and (68) for the (
Morse,
class) theoretical model. This way a general equation representing the referred proportionally can be obtained:
Equation (74) is valid for both theoretical models and all the molecules since the working temperatures are significantly larger than the characteristic rotational temperatures (Θ
rot) for the molecules under study (the equipartition principle is valid for both translational and rotational degrees of freedom). This condition is fully applied in the present case, as the minimum working temperature (298.15 K) is much larger than the maximum rotational temperature (Θ
rot(H
2) = 87.46 K). In
Figure 3,
Cp,m is represented as a function of
. The dependence of this vibrational quantity on temperature is represented in
Figure 4.
Equation (74) establishes a linear relationship between
Cp,m and
. A least squares fitting, performed on these data (see
Figure 3), confirmed that the respective slope and intercept are
NA/
kB = 4.36 × 10
46 J
−1 K mol
−1 and 7
R/2 = 29.10 J K
−1 mol
−1, respectively. The individual point associated with the (
class,
class) model, identified by the symbol
![Computation 12 00229 i010]()
in
Figure 3, corresponds to the following coordinates:
The variance of vibrational energy (
) should reflect the Boltzmann populations for both the ground and excited vibrational states. These populations are presented in
Table 11 (for the (
harm,
class) theoretical model) and in
Table 12 (for the (
Morse,
class) theoretical model).
4. Discussion
The molecular interpretation for
Cp,m, as presented in this paper, is based on its linear relationship with
(see Equations (74)–(76) and
Figure 2). This indicates that both quantities exhibit analogous tendencies with respect to their variations with relevant factors such as the temperature (
T) or the vibrational frequency (ν
vib). This is evident by comparing
Figure 2 and
Figure 4.
The temperature dependence of the aforementioned quantities (
Cp,m and
) can be understood as a competition between two opposing tendencies: the increase in the variance of the vibrational energy (
) with
T and the decrease in the
T−2 term with the same variable. The overall result of this competition is that both quantities increase with
T, tending to constant values at high temperatures (see
Figure 2 and
Figure 4). For the (
harm,
class) theoretical model, these limits are derived from the equipartition principle (9
R/2 for
Cp,m and 1.91 × 10
−46 J
2 K
−2 for
). For the (
Morse,
class) theoretical model, these limits exceed those predicted by this principle. These convergences at high temperatures are evident for Cl
2 (lowest vibrational temperature: Θ
vib = 554.6 K), can be anticipated for O
2 (intermediate vibrational temperature: Θ
vib = 2999.6 K), and are not obvious for H
2 (highest vibrational temperature: Θ
vib = 6336.8 K).
The quantities under study (
Cp,m and
) clearly decrease with the ν
vib (see
Figure 2 and
Figure 4 and
Table 8,
Table 9 and
Table 10). This tendency originates from the decrease in the variance of the vibrational energy (
) with ν
vib (see
Figure 4).
The variance of the vibrational energy (
) is strongly correlated with the distribution of the Boltzmann populations across different vibrational states (see
Table 9,
Table 10,
Table 11 and
Table 12).
is zero when only the vibrational ground state is occupied (
P0 = 1.0000,
Pv>0 = 0.0000). Promoting a more balanced distribution of the vibrational populations, by decreasing the ground state population and increasing the populations of the vibrational excited states, will increase
. Given the exponential nature of the Boltzmann populations (see Equations (12) and (67)), this can be achieved by increasing the temperature or decreasing the vibrational frequency (reducing the energy difference between two consecutive vibrational states). These tendencies are confirmed in
Table 9,
Table 10,
Table 11 and
Table 12.
5. Conclusions
In this paper, I presented Diatomic as an alternative to quantum chemistry packages for calculating the thermodynamic properties of diatomic molecules. Diatomic is an open-source Excel application that requires only a limited number of molecular constants to perform these calculations. In addition, these constants can be easily obtained from freely available online databases.
This new application offers two models (Morse and harmonic) to describe the vibrational structure and two models (classical and quantum) to describe the rotational structure. By combining the rotational and vibrational structures, four theoretical models can be used within Diatomic (harm, class; Morse, class; harm, quant; and Morse, quant). Diatomic presents a number of distinctive features that enable its application for different purposes.
Diatomic was used to calculate four standard molar thermodynamic properties (standard molar enthalpy of formation, molar heat capacity at constant pressure, average value of the standard molar thermal enthalpy, and standard molar entropy) for a set of fifteen diatomic molecules. The results obtained with this application were compared with experimental data.
A molecular interpretation for the molar heat capacity at constant pressure (Cp,m), as an educational application of Diatomic, was also discussed here. This interpretation utilizes certain vibrational quantities provided by this application, such as the variance of the vibrational energy and the vibrational Boltzmann populations, which are typically unavailable in general quantum chemistry packages.


 in Figure 3, corresponds to the following coordinates:
in Figure 3, corresponds to the following coordinates:
 –H2 (calculated using the (harm, class) model),
–H2 (calculated using the (harm, class) model),  –H2 (calculated using the (Morse, class) model),
–H2 (calculated using the (Morse, class) model),  –H2 (experimental),
–H2 (experimental),  –O2 (calculated using the (harm, class) model),
–O2 (calculated using the (harm, class) model),  –O2 (calculated using the (Morse, class) model),
–O2 (calculated using the (Morse, class) model),  –O2 (experimental),
–O2 (experimental),  –Cl2 (calculated using the (harm, class) model),
–Cl2 (calculated using the (harm, class) model),  –Cl2 (calculated using the (Morse, class) model),
–Cl2 (calculated using the (Morse, class) model),  –Cl2 (experimental), and — all the molecules (calculated using the (class, class) model).
–Cl2 (experimental), and — all the molecules (calculated using the (class, class) model).



{kind=link}
{kind=link}
{kind=link}
{kind=link}