COVID-19: Mechanisms of Vaccination and Immunity



Abstract
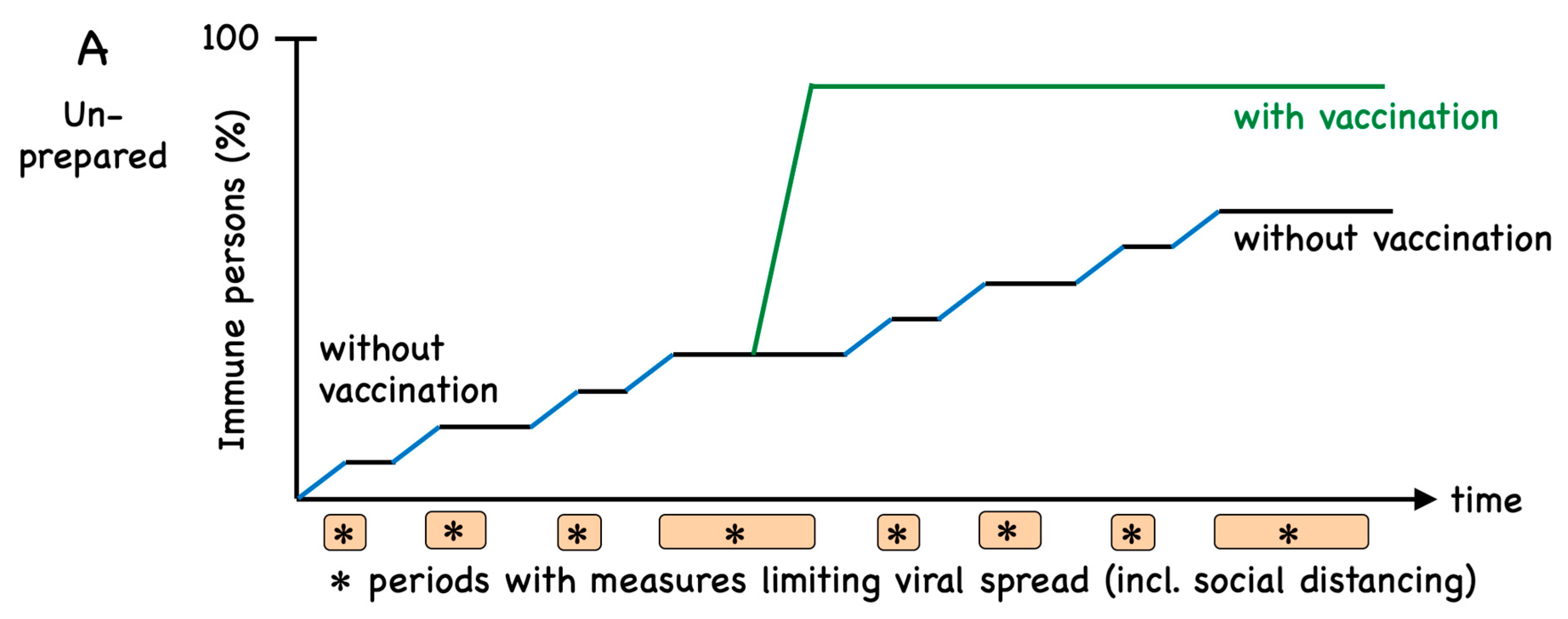
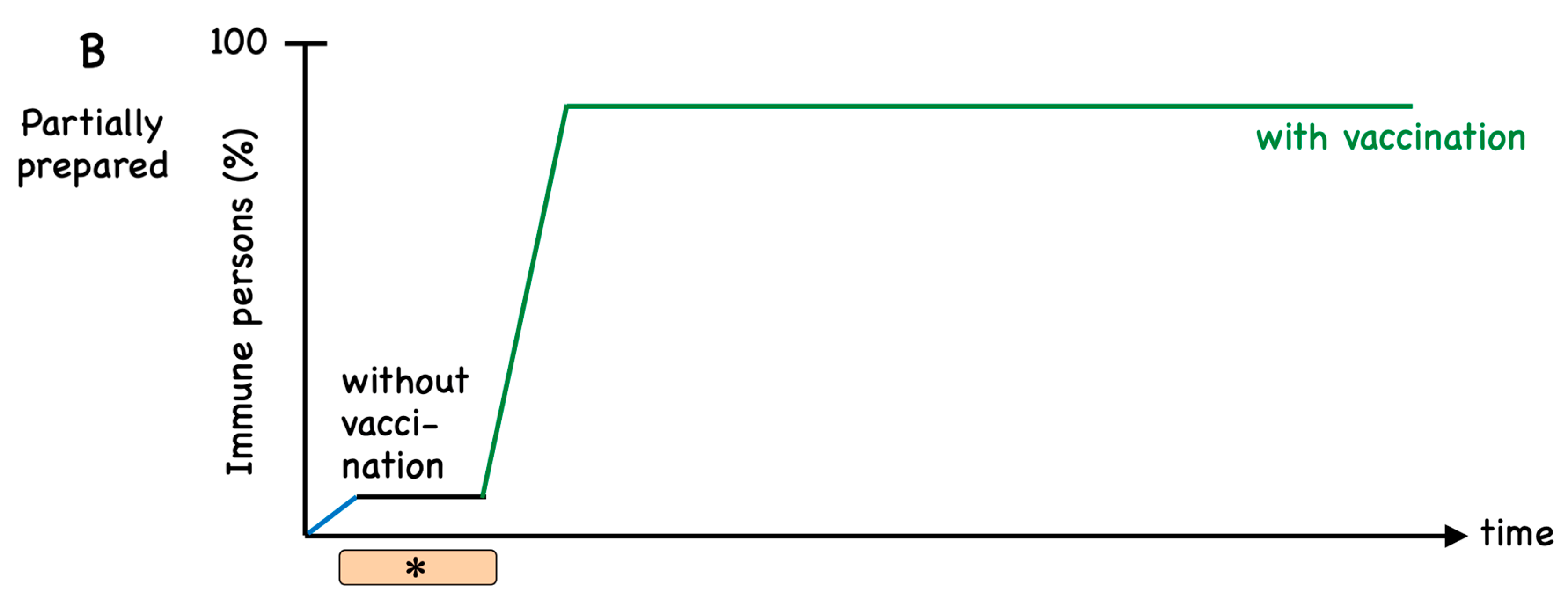
1. Introduction
2. Vaccine Development
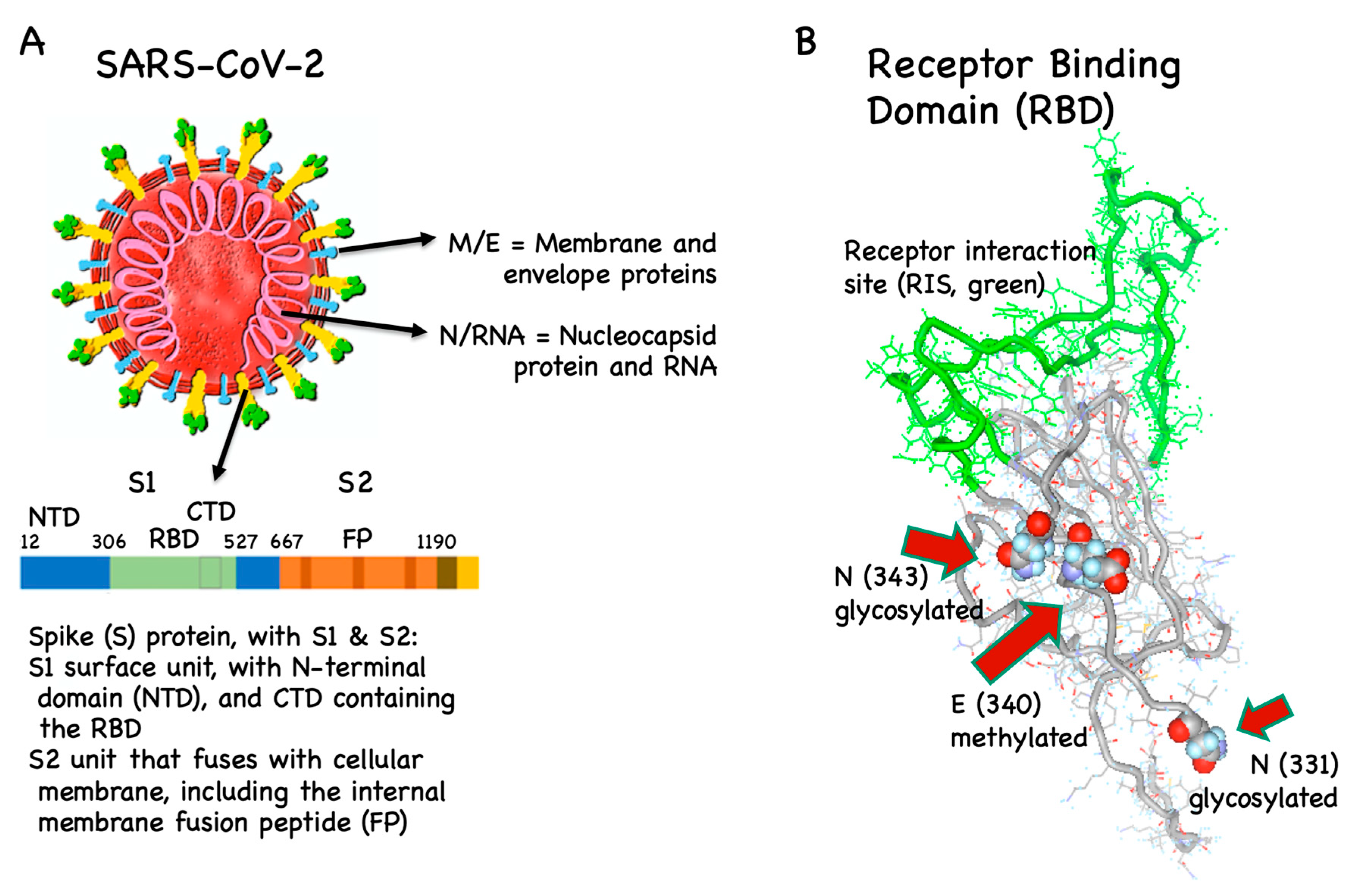
3. Vaccine Antigens
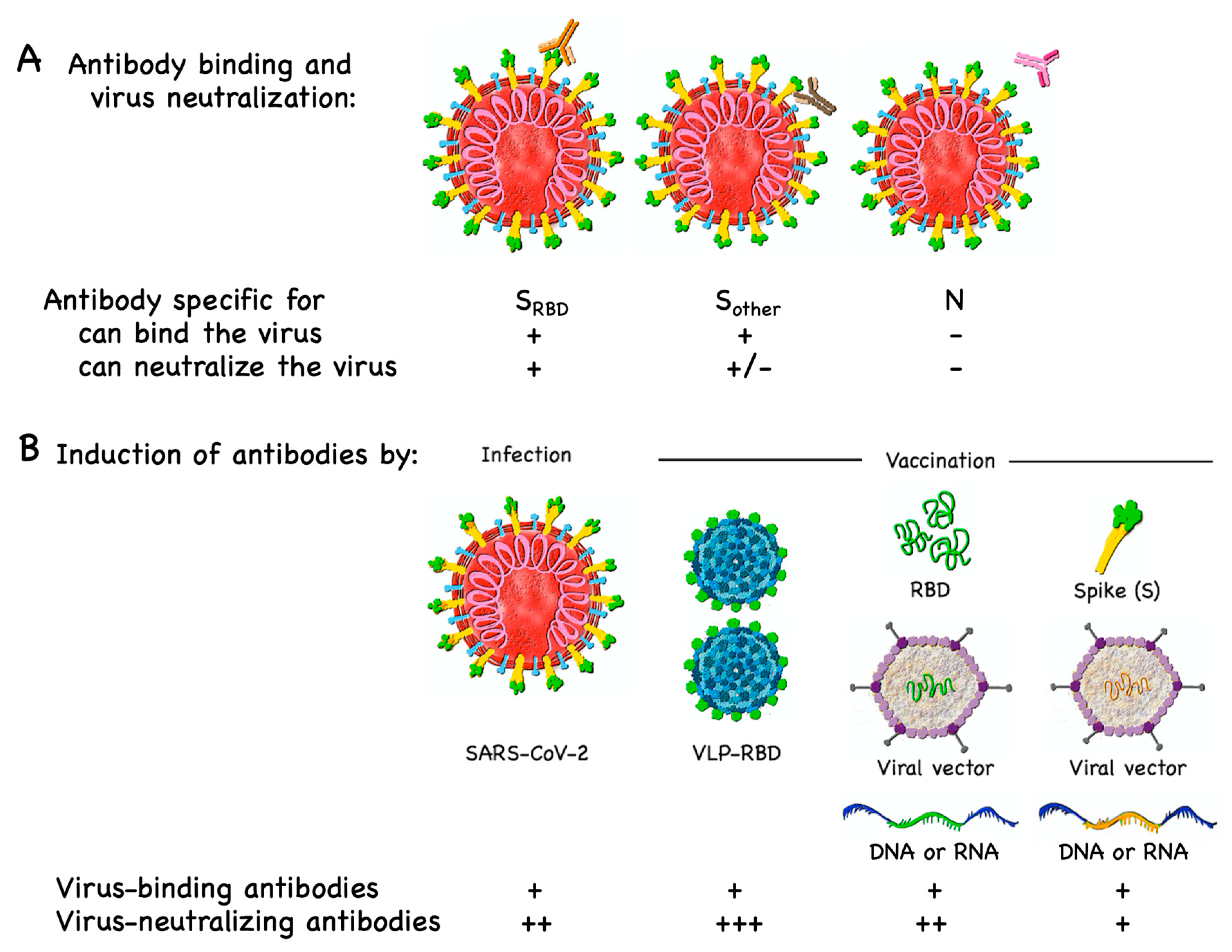
3.1. B Cell/Antibody Targets
3.2. T Cell Targets
4. Disease Enhancement
4.1. T Cell-Dependent Disease Enhancement
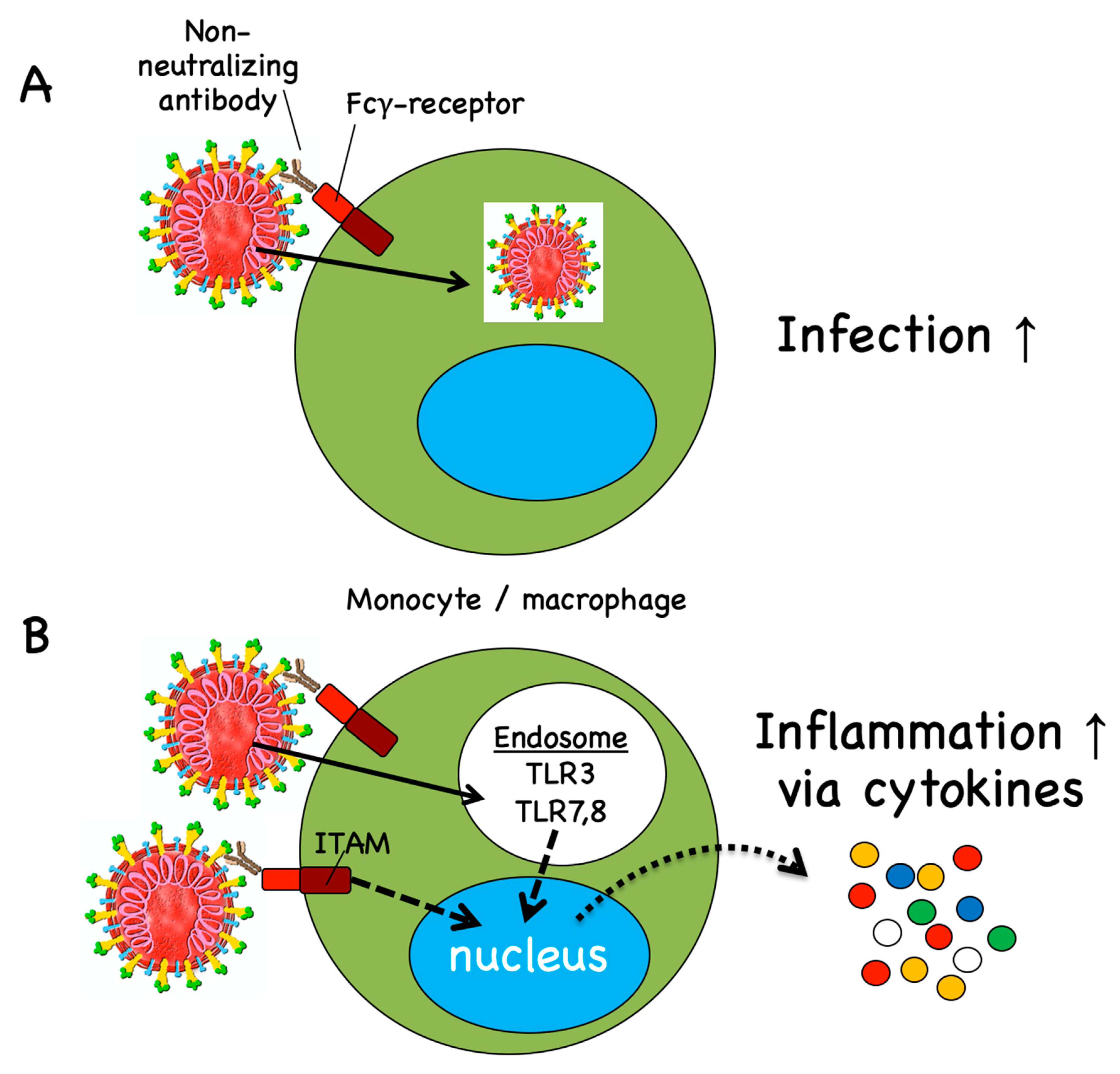
4.2. Antibody-Dependent Disease Enhancement
5. Assays for Measuring Coronavirus-Specific Immune Responses
5.1. Serology
5.2. Cytokine Measurements and T Cell Analysis
6. Immune Responses to Natural Infection Versus Vaccination
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gates, B. Responding to Covid-19—A Once-in-a-Century Pandemic? N. Engl. J. Med. 2020, 382, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Hoehl, S.; Rabenau, H.; Berger, A.; Kortenbusch, M.; Cinatl, J.; Bojkova, D.; Behrens, P.; Böddinghaus, B.; Götsch, U.; Naujoks, F.; et al. Evidence of SARS-CoV-2 Infection in Returning Travelers from Wuhan, China. N. Engl. J. Med. 2020, 382, 1278–1280. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Pei, S.; Chen, B.; Song, Y.; Zhang, T.; Yang, W.; Shaman, J. Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV-2). Science 2020, 368, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Salathé, M.; Althaus, C.L.; Neher, R.; Stringhini, S.; Hodcroft, E.; Fellay, J.; Zwahlen, M.; Senti, G.; Battegay, M.; Wilder-Smith, A.; et al. COVID-19 epidemic in Switzerland: On the importance of testing, contact tracing and isolation. Swiss Med. Wkly. 2020, 150, w20225. [Google Scholar] [CrossRef]
- Cheng, V.C.C.; Wong, S.-C.; Chen, J.H.K.; Yip, C.C.Y.; Chuang, V.W.M.; Tsang, O.T.Y.; Sridhar, S.; Chan, J.F.W.; Ho, P.-L.; Yuen, K.-Y. Escalating infection control response to the rapidly evolving epidemiology of the coronavirus disease 2019 (COVID-19) due to SARS-CoV-2 in Hong Kong. Infect. Control Hosp. Epidemiol. 2020, 41, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Heesterbeek, H.; Klinkenberg, D.; Hollingsworth, T.D. How will country-based mitigation measures influence the course of the COVID-19 epidemic? Lancet 2020, 395, 931–934. [Google Scholar] [CrossRef]
- Weissleder, R.; Lee, H.; Ko, J.; Pittet, M.J. COVID-19 diagnostics in context. Sci. Transl. Med. 2020, 12, eabc1931. [Google Scholar] [CrossRef]
- Tan, W.-T.; Lu, Y.; Zhang, J.; Wang, J.; Dan, Y.; Tan, Z.; He, X.; Qian, C.; Sun, Q.; Hu, Q.; et al. Viral Kinetics and Antibody Responses in Patients with COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Vogel, G. First antibody surveys draw fire for quality, bias. Science 2020, 368, 350–351. [Google Scholar]
- Thevarajan, I.; Nguyen, T.H.O.; Koutsakos, M.; Druce, J.; Caly, L.; Van De Sandt, C.E.; Jia, X.; Nicholson, S.; Catton, M.; Cowie, B.; et al. Breadth of concomitant immune responses prior to patient recovery: A case report of non-severe COVID-19. Nat. Med. 2020, 26, 453–455. [Google Scholar] [CrossRef]
- Lurie, N.; Saville, M.; Hatchett, R.; Halton, J. Developing Covid-19 Vaccines at Pandemic Speed. New Engl. J. Med. 2020, 382, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Gavi. The COVID-19 Vaccine Race. 2020. Available online: https://www.gavi.org/vaccineswork/covid-19-vaccine-race (accessed on 21 July 2020).
- Le, T.T.; Andreadakis, Z.; Kumar, A.; Román, R.G.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306. [Google Scholar] [CrossRef]
- WHO Draft Landscape of Covid-19 Candidate Vaccines. 2020. Available online: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines?utm_source=Human+Vaccines+Project+COVID+Report&utm_campaign=089a88336d-EMAIL_CAMPAIGN_2018_08_21_06_59_COPY_01&utm_medium=email&utm_term=0_89e0163bb8-089a88336d-180996954 (accessed on 21 July 2020).
- Yamey, G.; Schäferhoff, M.; Hatchett, R.; Pate, M.; Zhao, F.; McDade, K.K. Ensuring global access to COVID-19 vaccines. Lancet 2020, 395, 1405–1406. [Google Scholar] [CrossRef]
- Corey, B.L.; Mascola, J.R.; Fauci, A.S.; Collins, F.S. A strategic approach to COVID-19 vaccine R&D. Science 2020, 368, 948–950. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.S. Rapid COVID-19 vaccine development. Science 2020, 368, 945–946. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. World Report COVID-19 vaccine development pipeline gears up. Lancet 2020, 395, 1751–1752. [Google Scholar] [CrossRef]
- Callaway, E. The race for coronavirus vaccines: A graphical guide. Nature 2020, 580, 576–577. [Google Scholar] [CrossRef]
- Amanat, F.; Krammer, F. SARS-CoV-2 Vaccines: Status Report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef]
- With Record-Setting Speed, Vaccine Makers Take Their First Shots at the New Coronavirus. 2020. Available online: https://www.sciencemag.org/news/2020/03/record-setting-speed-vaccine-makers-take-their-first-shots-new-coronavirus (accessed on 21 July 2020).
- Tseng, C.-T.; Sbrana, E.; Iwata Yoshikawa, N.; Newman, P.C.; Garron, T.; Atmar, R.L.; Peters, C.J.; Couch, R.B. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS ONE 2012, 7, e35421. [Google Scholar] [CrossRef]
- Van Doremalen, N.; Lambe, T.; Spencer, A.; Belij-Rammerstorfer, S.; Purushotham, J.N.; Port, J.R.; Avanzato, V.; Bushmaker, T.; Flaxman, A.; Ulaszewska, M.; et al. ChAdOx1 nCoV-19 vaccination prevents SARS-CoV-2 pneumonia in rhesus macaques. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhu, F.-C.; Li, Y.-H.; Guan, X.-H.; Hou, L.-H.; Wang, W.-J.; Li, J.-X.; Wu, S.-P.; Wang, B.-S.; Wang, Z.; Wang, L.; et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: A dose-escalation, open-label, non-randomised, first-in-human trial. Lancet 2020, 395, 1845–1854. [Google Scholar] [CrossRef]
- Enjuanes, L.; DeDiego, M.L.; Alvarez, E.; Deming, D.; Sheahan, T.; Baric, R. Vaccines to prevent severe acute respiratory syndrome coronavirus-induced disease. Virus Res. 2008, 133, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Padron-Regalado, E. Vaccines for SARS-CoV-2: Lessons from Other Coronavirus Strains. Infect. Dis. Ther. 2020, 9, 255–274. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.-J.; Jiang, S. The spike protein of SARS-CoV—A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef]
- Urakami, A.; Sakurai, A.; Ishikawa, M.; Yap, M.L.; Flores-Garcia, Y.; Haseda, Y.; Aoshi, T.; Zavala, F.P.; Rossmann, M.G.; Kuno, S.; et al. Development of a Novel Virus-Like Particle Vaccine Platform That Mimics the Immature Form of Alphavirus. Clin. Vaccine Immunol. 2017, 24, e00090-17. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, J.-S.; Su, N.; Xu, J.; Wang, N.; Chen, J.-T.; Chen, X.; Liu, Y.-X.; Gao, H.; Jia, Y.-P.; et al. Safety and immunogenicity from a phase I trial of inactivated severe acute respiratory syndrome coronavirus vaccine. Antivir. Ther. 2007, 12, 1107–1113. [Google Scholar]
- Martin, J.E.; Louder, M.K.; Holman, L.A.; Gordon, I.J.; Enama, M.E.; Larkin, B.D.; Andrews, C.A.; Vogel, L.; Koup, R.A.; Roederer, M.; et al. A SARS DNA vaccine induces neutralizing antibody and cellular immune responses in healthy adults in a Phase I clinical trial. Vaccine 2008, 26, 6338–6343. [Google Scholar] [CrossRef]
- Zhao, L.; Seth, A.; Wibowo, N.; Zhao, C.-X.; Mitter, N.; Yu, C.; Middelberg, A.P.J. Nanoparticle vaccines. Vaccine 2014, 32, 327–337. [Google Scholar] [CrossRef]
- Modjarrad, K.; Roberts, C.C.; Mills, K.T.; Castellano, A.R.; Paolino, K.; Muthumani, K.; Reuschel, E.L.; Robb, M.L.; Racine, T.; Oh, M.-D.; et al. Safety and immunogenicity of an anti-Middle East respiratory syndrome coronavirus DNA vaccine: A phase 1, open-label, single-arm, dose-escalation trial. Lancet Infect. Dis. 2019, 19, 1013–1022. [Google Scholar] [CrossRef]
- Gao, Q.; Bao, L.; Mao, H.; Wang, L.; Xu, K.; Yang, M.; Li, Y.; Zhu, L.; Wang, N.; Lv, Z.; et al. Rapid development of an inactivated vaccine candidate for SARS-CoV-2. Science 2020, 369, 77–81. [Google Scholar] [CrossRef]
- Bolles, M.; Deming, M.E.; Long, K.; Agnihothram, S.; Whitmore, A.; Ferris, M.; Funkhouser, W.; Gralinski, L.; Totura, A.; Heise, M.; et al. A Double-Inactivated Severe Acute Respiratory Syndrome Coronavirus Vaccine Provides Incomplete Protection in Mice and Induces Increased Eosinophilic Proinflammatory Pulmonary Response upon Challenge. J. Virol. 2011, 85, 12201–12215. [Google Scholar] [CrossRef] [PubMed]
- Di Pietrantonj, C.; Rivetti, A.; Marchione, P.; Debalini, M.G.; Demicheli, V. Vaccines for measles, mumps, rubella, and varicella in children. Cochrane Database Syst. Rev. 2020, 4, CD004407. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Levin, M.J. Herpes Zoster and Its Prevention by Vaccination. Interdiscip. Top. Gerontol. Geriatr. 2020, 43, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Thao, T.T.N.; Labroussaa, F.; Ebert, N.; V’Kovski, P.; Stalder, H.; Portmann, J.; Kelly, J.; Steiner, S.; Holwerda, M.; Kratzel, A.; et al. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature 2020, 582, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Jegerlehner, A.; Maurer, P.; Bessa, J.; Hinton, H.J.; Kopf, M.; Bachmann, M.F. TLR9 signaling in B cells determines class switch recombination to IgG2a. J. Immunol. 2007, 178, 2415–2420. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Chang, S.-Y.; Sun, H.-Y.; Tsai, M.-S.; Liu, W.-C.; Su, Y.-C.; Wu, P.-Y.; Hung, C.; Chang, S.-Y. Long-Term Durability of Responses to 2 or 3 Doses of Hepatitis A Vaccination in Human Immunodeficiency Virus-Positive Adults on Antiretroviral Therapy. J. Infect. Dis. 2016, 215, 606–613. [Google Scholar] [CrossRef]
- Nakayama, T. Causal relationship between immunological responses and adverse reactions following vaccination. Vaccine 2019, 37, 366–371. [Google Scholar] [CrossRef] [PubMed]
- WHO. A Coordinated Global Research Roadmap. 2020, pp. 1–96. Available online: https://www.who.int/blueprint/priority-diseases/key-action/Roadmap-version-FINAL-for-WEB.pdf?ua=1 (accessed on 21 July 2020).
- Pardi, N.; Hogan, M.J.; Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 2020, 65, 14–20. [Google Scholar] [CrossRef]
- Francis, M.J. Recent Advances in Vaccine Technologies. Veter. Clin. N. Am. Small Anim. Pract. 2018, 48, 231–241. [Google Scholar] [CrossRef]
- Mohsen, M.O.; Zha, L.; Cabral-Miranda, G.; Bachmann, M.F. Major findings and recent advances in virus–like particle (VLP)-based vaccines. Semin. Immunol. 2017, 34, 123–132. [Google Scholar] [CrossRef]
- Minor, P. Live attenuated vaccines: Historical successes and current challenges. Virology 2015, 479–480, 379–392. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Zang, R.; Castro, M.F.G.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef]
- Qiu, M.; Shi, Y.; Guo, Z.; Chen, Z.; He, R.; Chen, R.; Zhou, D.; Dai, E.; Wang, X.; Si, B.; et al. Antibody responses to individual proteins of SARS coronavirus and their neutralization activities. Microbes Infect. 2005, 7, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.-C.D.; So, R.T.Y.; Lv, H.; Mok, C.K.; Wilson, I.A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.O.; West, A.P.; Huey-Tubman, K.E.; Hoffmann, M.A.; Sharaf, N.G.; Hoffman, P.R.; Koranda, N.; Gristick, H.B.; Gaebler, C.; Muecksch, F.; et al. Structures of Human Antibodies Bound to SARS-CoV-2 Spike Reveal Common Epitopes and Recurrent Features of Antibodies. Cell 2020, 182, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Åkerström, S.; Tan, Y.-J.; Mirazimi, A. Amino acids 15–28 in the ectodomain of SARS coronavirus 3a protein induces neutralizing antibodies. FEBS Lett. 2006, 580, 3799–3803. [Google Scholar] [CrossRef]
- Berry, J.D.; Hay, K.A.; Rini, J.M.; Yu, M.; Wang, L.; Plummer, F.A.; Corbett, C.R.; Andonov, A. Neutralizing epitopes of the SARS-CoV S-protein cluster independent of repertoire, antigen structure or mAb technology. mAbs 2010, 2, 53–66. [Google Scholar] [CrossRef]
- Zhang, J.; Zeng, H.; Gu, J.; Li, H.; Zheng, L.; Zou, Q.-M. Progress and Prospects on Vaccine Development against SARS-CoV-2. Vaccines 2020, 8, 153. [Google Scholar] [CrossRef]
- Sun, Z.; Ren, K.; Zhang, X.; Chen, J.; Jiang, Z.; Jiang, J.; Ji, F.; Ouyang, X.; Li, L.-J. Mass spectrometry analysis of newly emerging coronavirus HCoV-19 spike S protein and human ACE2 reveals camouflaging glycans and unique post-translational modifications. bioRxiv 2020. [Google Scholar] [CrossRef]
- Grant, O.C.; Montgomery, D.W.; Ito, K.; Woods, R.J. Analysis of the SARS-CoV-2 spike protein glycan shield: Implications for immune recognition. bioRxiv 2020, 9, 2754. [Google Scholar]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Yang, Y. The potential danger of suboptimal antibody responses in COVID-19. Nat. Rev. Immunol. 2020, 20, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Sun, H.; Gao, G.F.; Chen, J.; Sun, S.; Zhao, R.; Gao, G.; Hu, Y.; Zhao, G.; Chen, Y.; et al. Recombinant SARS-CoV-2 spike S1-Fc fusion protein induced high levels of neutralizing responses in nonhuman primates. Vaccine 2020. [Google Scholar] [CrossRef]
- Zha, L.; Zhao, H.; Mohsen, M.O.; Hong, L.; Zhou, Y.; Li, Z.; Yao, C.; Guo, L.; Chen, H.; Liu, X.; et al. Development of a COVID-19 vaccine based on the receptor binding domain displayed on virus-like particles. bioRxiv 2020. [Google Scholar] [CrossRef]
- He, Y.; Jiang, S. Vaccine Design for Severe Acute Respiratory Syndrome Coronavirus. Viral Immunol. 2005, 18, 327–332. [Google Scholar] [CrossRef]
- Bessa, J.; Bachmann, M.F. T Cell-dependent and -Independent IgA Responses: Role of TLR Signalling. Immunol. Investig. 2010, 39, 407–428. [Google Scholar] [CrossRef]
- Meiler, F.; Klunker, S.; Zimmermann, M.; Akdis, C.A.; Akdis, M. Distinct regulation of IgE, IgG4 and IgA by T regulatory cells and toll-like receptors. Allergy 2008, 63, 1455–1463. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef]
- Sridhar, S.; Begom, S.; Bermingham, A.; Hoschler, K.; Adamson, W.; Carman, W.; Bean, T.; Barclay, W.S.; Deeks, J.; Lalvani, A. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med. 2013, 19, 1305–1312. [Google Scholar] [CrossRef]
- Wilkinson, T.M.; Li, C.K.F.; Chui, C.S.C.; Huang, A.K.Y.; Perkins, M.; Liebner, J.C.; Lambkin-Williams, R.; Gilbert, A.S.; Oxford, J.; Nicholas, B.; et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat. Med. 2012, 18, 274–280. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef]
- Yang, Z.-Y.; Kong, W.-P.; Huang, Y.; Roberts, A.; Murphy, B.R.; Subbarao, K.; Nabel, G.J. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature 2004, 428, 561–564. [Google Scholar] [CrossRef]
- Colbert, J.D.; Cruz, F.M.; Rock, K.L. Cross-presentation of exogenous antigens on MHC I molecules. Curr. Opin. Immunol. 2020, 64, 1–8. [Google Scholar] [CrossRef]
- Johansen, P.; Storni, T.; Rettig, L.; Qiu, Z.; Der-Sarkissian, A.; Smith, K.A.; Manolova, V.; Lang, K.S.; Senti, G.; Müllhaupt, B.; et al. Antigen kinetics determines immune reactivity. Proc. Natl. Acad. Sci. USA 2008, 105, 5189–5194. [Google Scholar] [CrossRef]
- Zeltins, A.; West, J.; Zabel, F.; El-Turabi, A.; Balke, I.; Haas, S.; Maudrich, M.; Storni, F.; Engeroff, P.; Jennings, G.T.; et al. Incorporation of tetanus-epitope into virus-like particles achieves vaccine responses even in older recipients in models of psoriasis, Alzheimer’s and cat allergy. npj Vaccines 2017, 2, 30. [Google Scholar] [CrossRef]
- Oehen, S.; Hengartner, H.; Zinkernagel, R.M. Vaccination for disease. Science 1991, 251, 195–198. [Google Scholar] [CrossRef]
- Johnson, T.R.; Rao, S.; Seder, R.A.; Chen, M.; Graham, B.S. TLR9 agonist, but not TLR7/8, functions as an adjuvant to diminish FI-RSV vaccine-enhanced disease, while either agonist used as therapy during primary RSV infection increases disease severity. Vaccine 2009, 27, 3045–3052. [Google Scholar] [CrossRef]
- Yasui, F.; Kai, C.; Kitabatake, M.; Inoue, S.; Yoneda, M.; Yokochi, S.; Kase, R.; Sekiguchi, S.; Morita, K.; Hishima, T.; et al. Prior immunization with severe acute respiratory syndrome (SARS)-associated coronavirus (SARS-CoV) nucleocapsid protein causes severe pneumonia in mice infected with SARS-CoV. J. Immunol. 2008, 181, 6337–6348. [Google Scholar] [CrossRef]
- Jiang, S.; Bottazzi, M.E.; Du, L.; Lustigman, S.; Tseng, C.-T.K.; Curti, E.; Jones, K.; Zhan, B.; Hotez, P.J. Roadmap to developing a recombinant coronavirus S protein receptor-binding domain vaccine for severe acute respiratory syndrome. Expert Rev. Vaccines 2012, 11, 1405–1413. [Google Scholar] [CrossRef]
- Ramakrishnan, R.K.; Al Heialy, S.; Hamid, Q. Role of IL-17 in asthma pathogenesis and its implications for the clinic. Expert Rev. Respir. Med. 2019, 13, 1057–1068. [Google Scholar] [CrossRef]
- Tirado, S.M.C.; Yoon, K.-J. Antibody-Dependent Enhancement of Virus Infection and Disease. Viral Immunol. 2003, 16, 69–86. [Google Scholar] [CrossRef]
- Yang, Z.-Y.; Werner, H.C.; Kong, W.-P.; Leung, K.; Traggiai, E.; Lanzavecchia, A.; Nabel, G.J. Evasion of antibody neutralization in emerging severe acute respiratory syndrome coronaviruses. Proc. Natl. Acad. Sci. USA 2005, 102, 797–801. [Google Scholar] [CrossRef]
- Beltramello, M.; Williams, K.L.; Simmons, C.P.; Macagno, A.; Simonelli, L.; Quyen, N.T.H.; Sukupolvi-Petty, S.; Navarro-Sánchez, E.; Young, P.; De Silva, A.M.; et al. The Human Immune Response to Dengue Virus Is Dominated by Highly Cross-Reactive Antibodies Endowed with Neutralizing and Enhancing Activity. Cell Host Microbe 2010, 8, 271–283. [Google Scholar] [CrossRef]
- Katzelnick, L.C.; Gresh, L.; Halloran, M.E.; Mercado, J.C.; Kuan, G.; Gordon, A.; Balmaseda, A.; Harris, E. Antibody-dependent enhancement of severe dengue disease in humans. Science 2017, 358, 929–932. [Google Scholar] [CrossRef]
- Vennema, H.; De Groot, R.J.; Harbour, D.A.; Dalderup, M.; Gruffydd-Jones, T.; Horzinek, M.C.; Spaan, W.J. Early death after feline infectious peritonitis virus challenge due to recombinant vaccinia virus immunization. J. Virol. 1990, 64, 1407–1409. [Google Scholar] [CrossRef]
- Czub, M.; Weingartl, H.; Czub, S.; He, R.; Cao, J. Evaluation of modified vaccinia virus Ankara based recombinant SARS vaccine in ferrets. Vaccine 2005, 23, 2273–2279. [Google Scholar] [CrossRef]
- To, K.-F.; Tong, J.H.; Chan, P.K.; Au, F.W.; Chim, S.S.; Chan, K.A.; Cheung, J.L.; Liu, E.Y.; Tse, G.M.; Lo, A.W.I.; et al. Tissue and cellular tropism of the coronavirus associated with severe acute respiratory syndrome: An in-situ hybridization study of fatal cases. J. Pathol. 2004, 202, 157–163. [Google Scholar] [CrossRef]
- Shieh, W.-J.; Hsiao, C.-H.; Paddock, C.D.; Guarner, J.; Goldsmith, C.S.; Tatti, K.; Packard, M.; Mueller, L.; Wu, M.-Z.; Rollin, P.; et al. Immunohistochemical, in situ hybridization, and ultrastructural localization of SARS-associated coronavirus in lung of a fatal case of severe acute respiratory syndrome in Taiwan. Hum. Pathol. 2005, 36, 303–309. [Google Scholar] [CrossRef]
- Yip, M.S.; Leung, H.L.; Li, P.H.; Cheung, C.Y.; Dutry, I.; Li, D.; Daëron, M.; Bruzzone, R.; Peiris, J.S.; Jaume, M. Antibody-dependent enhancement of SARS coronavirus infection and its role in the pathogenesis of SARS. Hong Kong Med. J. 2016, 22, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-F.; Tseng, S.-P.; Yen, C.-H.; Yang, J.-Y.; Tsao, C.-H.; Shen, C.-W.; Chen, K.-H.; Liu, F.-T.; Liu, W.-T.; Chen, Y.-M.A.; et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem. Biophys. Res. Commun. 2014, 451, 208–214. [Google Scholar] [CrossRef]
- Martins, K.A.; Bavari, S.; Salazar, A.M. Vaccine adjuvant uses of poly-IC and derivatives. Expert Rev. Vaccines 2014, 14, 447–459. [Google Scholar] [CrossRef]
- Vasilakos, J.P.; Tomai, M.A. The use of Toll-like receptor 7/8 agonists as vaccine adjuvants. Expert Rev. Vaccines 2013, 12, 809–819. [Google Scholar] [CrossRef]
- Bode, C.; Zhao, G.; Steinhagen, F.; Kinjo, T.; Klinman, D.M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines 2011, 10, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Wölfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Müller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.R.; Anand, R.; Andersson, M.I.; Auckland, K.; Baillie, J.K.; Barnes, E.; Bell, J.; Berry, T.; Bibi, S.; Carroll, M.; et al. Evaluation of antibody testing for SARS-Cov-2 using ELISA and lateral flow immunoassays. medRxiv 2020. [Google Scholar] [CrossRef]
- Bryant, J.E.; Azman, A.S.; Ferrari, M.J.; Arnold, B.F.; Boni, M.F.; Boum, Y.; Hayford, K.; Luquero, F.J.; Mina, M.J.; Rodriguez-Barraquer, I.; et al. Serology for SARS-CoV-2: Apprehensions, opportunities, and the path forward. Sci. Immunol. 2020, 5, eabc6347. [Google Scholar] [CrossRef]
- Crawford, K.H.D.; Eguia, R.; Dingens, A.S.; Loes, A.N.; Malone, K.D.; Wolf, C.R.; Chu, H.Y.; Tortorici, M.A.; Veesler, D.; Murphy, M.; et al. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses 2020, 12, 513. [Google Scholar] [CrossRef]
- Schmidt, F.; Weisblum, Y.; Muecksch, F.; Hoffmann, H.-H.; Michailidis, E.; Lorenzi, J.C.C.; Mendoza, P.; Rutkowska, M.; Bednarski, E.; Gaebler, C.; et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. bioRxiv 2020. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor binding site of SARS-CoV-2. Nature 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Zhao, Q. Perspectives on therapeutic neutralizing antibodies against the Novel Coronavirus SARS-CoV-2. Int. J. Boil. Sci. 2020, 16, 1718–1723. [Google Scholar] [CrossRef] [PubMed]
- Sévajol, M.; Subissi, L.; Decroly, E.; Canard, B.; Imbert, I. Insights into RNA synthesis, capping, and proofreading mechanisms of SARS-coronavirus. Virus Res. 2014, 194, 90–99. [Google Scholar] [CrossRef]
- Shapshak, P.; Chiappelli, F.; Somboonwit, C.; Sinnott, J. The influenza pandemic of 2009: Lessons and implications. Mol. Diagn. Ther. 2011, 15, 63–81. [Google Scholar] [CrossRef]
- Moore, B.J.B.; June, C.H. Cytokine release syndrome in severe COVID-19. Science 2020, 368, 473–474. [Google Scholar] [CrossRef]
- Vardhana, S.A.; Wolchok, J.D. The many faces of the anti-COVID immune response. J. Exp. Med. 2020, 217, 166. [Google Scholar] [CrossRef]
- Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; Guan, L.; et al. Clinical course and risk factors for mortality of adult in patients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar]
- Chevalier, M.F.; Bobisse, S.; Costa-Nunes, C.; Cesson, V.; Jichlinski, P.; Speiser, D.E.; Harari, A.; Coukos, G.; Romero, P.; Nardelli-Haefliger, D.; et al. High-throughput monitoring of human tumor-specific T-cell responses with large peptide pools. Oncoimmunology 2015, 4, e1029702. [Google Scholar] [CrossRef]
- Liu, W.; Fontanet, A.; Zhang, P.-H.; Zhan, L.; Xin, Z.-T.; Baril, L.; Tang, F.; Lv, H.; Cao, W.-C. Two-year prospective study of the humoral immune response of patients with severe acute respiratory syndrome. J. Infect. Dis. 2006, 193, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Zhu, L.; Ni, Z.; Meng, H.; You, L. Duration of serum neutralizing antibodies for SARS-CoV-2: Lessons from SARS-CoV infection. J. Microbiol. Immunol. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.-C.; Liu, W.; Zhang, P.-H.; Zhang, F.; Richardus, J.H. Disappearance of antibodies to SARS-associated coronavirus after recovery. N. Engl. J. Med. 2007, 357, 1162–1163. [Google Scholar] [CrossRef]
- Lan, L.; Xu, D.; Ye, G.; Xia, C.; Wang, S.; Li, Y.; Xu, H. Positive RT-PCR Test Results in Patients Recovered From COVID-19. JAMA 2020, 323, 1502–1503. [Google Scholar] [CrossRef]
- Xiao, A.T.; Tong, Y.X.; Zhang, S. False-negative of RT-PCR and prolonged nucleic acid conversion in COVID-19: Rather than recurrence. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Ralph, R.; Lew, J.; Zeng, T.; Francis, M.; Xue, B.; Roux, M.; Toloue Ostadgavahi, A.; Rubino, S.; Dawe, N.J.; Al-Ahdal, M.N.; et al. 2019-nCoV (Wuhan virus), a novel Coronavirus: Human-to-human transmission, travel-related cases, and vaccine readiness. J. Infect. Dev. Ctries. 2020, 14, 3–17. [Google Scholar] [CrossRef]
- Hamre, D.; Beem, M. Virologic studies of acute respiratory disease in young adults. V. Coronavirus 229E infections during six years of surveillance. Am. J. Epidemiol. 1972, 96, 94–106. [Google Scholar] [CrossRef]
- Reed, S.E. The behaviour of recent isolates of human respiratory coronavirus in vitro and in volunteers: Evidence of heterogeneity among 229E-related strains. J. Med. Virol. 1984, 13, 179–192. [Google Scholar] [CrossRef]
- Callow, K.A.; Parry, H.F.; Sergeant, M.; Tyrrell, D.A. The time course of the immune response to experimental coronavirus infection of man. Epidemiol. Infect. 1990, 105, 435–446. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Zinkernagel, R.M. The influence of virus structure on antibody responses and virus serotype formation. Immunol. Today 1996, 17, 553–558. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Rohrer, U.H.; Kündig, T.M.; Bürki, K.; Hengartner, H.; Zinkernagel, R.M. The influence of antigen organization on B cell responsiveness. Science 1993, 262, 1448–1451. [Google Scholar] [CrossRef]
- Plotkin, S.A. Updates on immunologic correlates of vaccine-induced protection. Vaccine 2020, 38, 2250–2257. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vaccine Candidate | Platform, Route of Administration | Target (SARS-Cov-2) | Developer | Trial Phase, Registry Number, Study Start, Link |
|---|---|---|---|---|
| Synthetic minigene transfected APCs Covid-19/aAPC | Artificial antigen presenting cells (APCs) modified with lentiviral vector, s.c.. | Selected conserved structural and protease protein domains | Shenzhen Geno-immune Medical Institute, China | Phase 1/2, NCT04299724, 15 February 2020 http://szgimi.org/en/news.php |
| Synthetic minigene transfected APCs + cytotoxic T cells LV-SMENP-DC | Dendritic cells modified with lentiviral vector, s.c., plus i.v. infusion of cytotoxic T cells | Viral structural proteins and a polyprotein protease | Shenzhen Geno-immune Medical Institute, China | Phase 1/2, NCT04276896, 24 March 2020 http://szgimi.org/en/news.php |
| Recombinant adenovirus, Ad5-nCoV | Viral vector, Adenovirus 5, i.m. | Spike protein | CanSino Biologics, China | Phase 2, NCT04341389, 12 April 2020 http://www.cansinotech.com/homes/article/plist/56.html |
| Recombinant adenovirus, AZD1222 | Viral vector (non-replicating) Chimpanzee Adenovirus, i.m. | Spike protein | University of Oxford, UK, & AstraZeneca | Phase 2b/3, 2020-001228-32, 4 May 2020 https://www.ox.ac.uk/news-and-events/for-journalists |
| Recombinant adenovirus, Gam-COVID-Vac (Lyo) | Viral vector, Adenoviruses 5 and 26, i.m. | Spike protein | Gamaleya Research Institute, Russia | Phase 1, NCT04436471, 17 June 2020 http://gamaleya.org/ |
| Plasmid, INO-4800 | DNA, i.d., followed by electroporation | Spike protein | Inovio Pharmaceuticals USA, & CEPI | Phase 1, NCT04336410, 3 April 2020, and Phase 2, https://www.inovio.com/our-focus-serving-patients/covid-19/ |
| Plasmid + adjuvant, AG0301-COVID19 | DNA, i.m. | Spike protein | AnGes and Osaka University, Japan | Phase 1/2, NCT04463472, 29 June 2020 https://www.anges.co.jp/en/ |
| Plasmid, GX-19 | DNA, i.m. | Spike protein | Genexin Inc., Korea | Phase 1/2, NCT04445389, 17 June 2020 http://www.genexine.com/m62.php?cate=1 |
| Lipid nanoparticle encapsulated RNA, mRNA 1273 | mRNA, i.m. | Spike protein | Moderna and Natl Inst Allergy & Infectious Diseases (NIAID), USA | Phase 2, NCT04405076, 25 May 2020 https://www.niaid.nih.gov/clinical-trials/safety-immunogenicity-study-vaccine-covid-19 |
| Lipid nanoparticle encapsulated RNA, BNT162 | mRNA, i.m. | Various viral ags (4 vaccine candidates) | BioNTech, Germany, & Pfizer, USA | Phase 1/2, NCT04368728, 29 April 2020 https://investors.biontech.de/press-releases |
| Lipid nanoparticle encapsulated RNA. CVnCoV | mRNA, i.m. | Spike protein | CureVac, Germany | Phase 1, NCT04449276, 18 June 2020 https://www.curevac.com/covid-19 |
| COVAC1 (LNP-nCoVsaRNA) | mRNA in lipid nanoparticle, i.m. | Spike protein | Imperial College London, UK | Phase 1, ISRCTN17072692, 1 April 2020 http://www.imperial.ac.uk/news |
| Protein + adjuvant, NVX-CoV2373 | Protein subunit vaccine, i.m. | Spike protein and Matrix-M adjuvant | Novavax, USA | Phase 1/2, NCT04368988, 25 May 2020 http://ir.novavax.com/press-releases |
| Protein + adjuvant, SCB-2019 | Protein trimeric subunit vaccine, i.m. | Spike protein, AS03, CpG, alum adjuvant | Clover Biopharma, Australia, GSK, Dynavax | Phase 1, NCT04405908, 19 June 2020 http://www.cloverbiopharma.com/ |
| SARS-CoV-2 inactivated virus, PiCoVacc | Inactivated virus + alum adjuvant | Entire virus | Sinovac Research and Development Co, China | Phase 1/2, 16 April 2020, and Phase 3 http://www.sinovacbio.com/?optionid=754&auto_id=904 |
| SARS-CoV-2 inactivated virus | Inactivated virus | Entire virus | Chinese Academy of Medical Sciences | Phase 1/2, NCT04412538, 15 May 2020 http://english.cas.cn/newsroom/news/ |
| SARS-CoV-2 inactivated virus | Inactivated virus | Entire virus | Sinopharm | Phase 1/2, ChiCTR2000031809, 11 April 2020 http://www.chinacdc.cn/en/ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Speiser, D.E.; Bachmann, M.F. COVID-19: Mechanisms of Vaccination and Immunity. Vaccines 2020, 8, 404. https://doi.org/10.3390/vaccines8030404
Speiser DE, Bachmann MF. COVID-19: Mechanisms of Vaccination and Immunity. Vaccines. 2020; 8(3):404. https://doi.org/10.3390/vaccines8030404
Chicago/Turabian StyleSpeiser, Daniel E., and Martin F. Bachmann. 2020. "COVID-19: Mechanisms of Vaccination and Immunity" Vaccines 8, no. 3: 404. https://doi.org/10.3390/vaccines8030404
APA StyleSpeiser, D. E., & Bachmann, M. F. (2020). COVID-19: Mechanisms of Vaccination and Immunity. Vaccines, 8(3), 404. https://doi.org/10.3390/vaccines8030404